Calpains, mitochondria, and apoptosis (original) (raw)
Abstract
Mitochondrial activity is critical for efficient function of the cardiovascular system. In response to cardiovascular injury, mitochondrial dysfunction occurs and can lead to apoptosis and necrosis. Calpains are a 15-member family of Ca2+-activated cysteine proteases localized to the cytosol and mitochondria, and several have been shown to regulate apoptosis and necrosis. For example, in endothelial cells, Ca2+ overload causes mitochondrial calpain 1 cleavage of the Na+/Ca2+ exchanger leading to mitochondrial Ca2+ accumulation. Also, activated calpain 1 cleaves Bid, inducing cytochrome c release and apoptosis. In renal cells, calpains 1 and 2 promote apoptosis and necrosis by cleaving cytoskeletal proteins, which increases plasma membrane permeability and cleavage of caspases. Calpain 10 cleaves electron transport chain proteins, causing decreased mitochondrial respiration and excessive activation, or inhibition of calpain 10 activity induces mitochondrial dysfunction and apoptosis. In cardiomyocytes, calpain 1 activates caspase 3 and poly-ADP ribose polymerase during tumour necrosis factor-α-induced apoptosis, and calpain 1 cleaves apoptosis-inducing factor after Ca2+ overload. Many of these observations have been elucidated with calpain inhibitors, but most calpain inhibitors are not specific for calpains or a specific calpain family member, creating more questions. The following review will discuss how calpains affect mitochondrial function and apoptosis within the cardiovascular system.
Keywords: Calpains, Apoptosis, Mitochondria, Cardiovascular system
1. Calpain family
Calpains are Ca2+-activated non-lysosomal cysteine proteases1 and the first calpain discovered and purified by Dayton et al.2,3 in 1976 was calpain 2. The calpain family is conserved in many different species, from fungi to humans.1 In mammals, there are 14 large subunit members, one small subunit member, and one endogenous inhibitor. Some calpains are ubiquitously expressed—calpains 1, 2, 4, 5, 7, and 101,4–9—whereas others are found in specific tissues: calpain 3 (skeletal muscle),9 calpain 6 (placenta),10 calpain 8 (smooth muscle),11 calpain 9 (stomach),11 calpain 11 (testes),12,13 calpain 12 (skin after birth),14 and calpain 13 (testes and lung).12 Additionally, calpains are divided into two groups based on domain IV structure (Figure 1).1 Typical calpains (1, 2, 3, 8, 9, 11, 12, and 14) contain a penta-EF hand in domain IV that can bind Ca2+, the calpain small subunit (only calpains 1, 2, and 9 have been shown to dimerize), or calpastatin. Atypical calpains (5, 6, 7, 10, 13, and 15) lack a penta-EF hand in domain IV and are unable to bind the calpain small subunit or calpastatin.15–17
Figure 1.
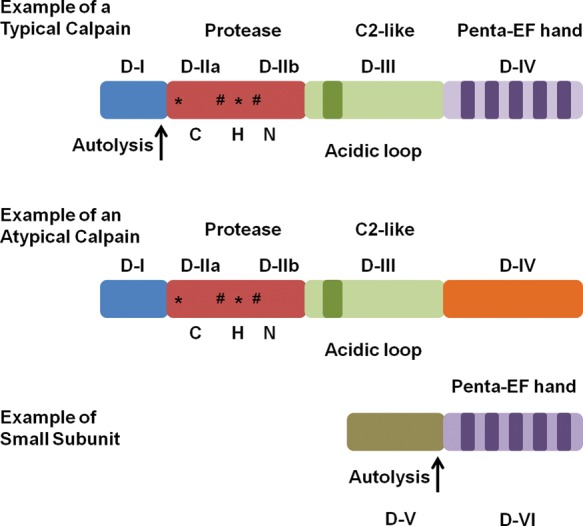
Diagram of a typical calpain, an atypical calpain, and small calpain subunit detailing different structural features. *Ca2+-binding sites. #Phosphorylation sites. CHN represents the Cys/His/Asn catalytic triad that is conserved throughout the family. Modified from Goll et al.1
Most calpains contain four structural domains. In calpains 1, 2, and 9, domain I is cleaved after Ca2+ activation (autolysis) (Figure 1),1 but whether other typical calpains undergo autolysis of domain I is not clear. In atypical calpains, domain I is not cleaved and the function of domain 1 is unknown for many of these calpains, except for calpain 10 which contains a mitochondrial targeting sequence.18 Domain II contains the active site, with the catalytic triad of cysteine, asparagine, and histidine, and this catalytic triad is conserved throughout the entire family, except for calpain 6 which lacks proteolytic activity.19 In addition to containing the catalytic triad, crystallographic studies revealed that domain II can bind two atoms of Ca2+ and assist in calpain activation.16,20 Domain III contains two Ca2+-binding sites and a phospholipid-binding motif in the C2-like area.21 These Ca2+ and phospholipid-binding residues are conserved throughout the family, except for calpain 10.16 Also, domain III is believed to regulate calpain activity through specific electrostatic interactions and be involved in substrate recognition.16,22,23 Domain IV contains the penta-EF hand that can bind Ca2+, calpastatin, or the small subunit (calpain 4).1 These penta-EF hands are thought to be one of the most important features in calpain activation.24
Calpain 4 (small calpain subunit) is a 28 kDa protein that dimerizes with typical calpains.1 It only contains two domains, V and VI, and the only known function for domain V is to bind to the C-terminus region of domain IV in large calpain subunits.25 Domain VI is identical to domain IV, in that it has a penta-EF hand that is available for Ca2+-binding and heterodimer formation.26 Recently, calpain small subunit 2 (CSS2) was discovered and it can dimerize with calpain 2,27 but CSS2 is not completely redundant with calpain 4 because knockdown of calpain 4 is embryonic lethal.28
Atypical calpains 5, 6, and 10 have the same general structure as typical calpains for the first three domains, but lack the penta-EF hand in domain IV.16,29 Instead, atypical calpains contain a divergent domain IV, which presently has an unknown function. Since atypical calpains do not contain a penta-EF hand there are only several Ca2+-binding residues available, suggesting that large concentrations of Ca2+ are not necessary for activation and that Ca2+ modulates the activity.18 Such differences in domain IV suggest different activation and inhibition patterns for atypical calpains.
Calpastatin, a protein that specifically inhibits calpains, has eight splice variants, ranging from 18.7 to 85 kDa.30–32 The full-length calpastatin has six domains (XL, L, I, II, III, and IV) and domains I–IV contain subdomains A–C that are essential for calpain inhibition (Figure 2).1 Of the four domains, the order of inhibition effectiveness is: I > IV > III > II.33 Little is known about the XL domain other than the three protein kinase A phosphorylation sites,34 and the function of the L domain is still unclear. Because calpastatin must bind domain II and domain IV or VI to inhibit calpains, it seems unlikely that atypical calpains are inhibited. Therefore, atypical calpains must have other regulatory mechanisms. Further reading on the calpain family can be found by reviewing articles by Goll et al.1 and Suzuki et al.16
Figure 2.
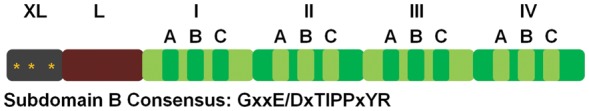
Diagram of the domain structure of calpastatin with a consensus sequence for subdomain B. *PKA phosphorylation site. Modified from Wendt et al.112
2. Endothelial and smooth muscle cells
Endothelial cells are an essential part of the cardiovascular system through their functions in blood vessel formation, cell barrier, coagulation, vascular tone, inflammation and angiogenesis.35 Many of these processes require cytoskeletal rearrangements, and numerous cytoskeletal proteins are known calpain substrates.1,36 Indeed, calpains have been discovered in rabbit, bovine, and human endothelium.37 More recently, Fujitani et al. purified calpains 1 and 2 and calpastatin from human umbilical vein endothelial cells and detected active calpains 1 and 2 by measuring the cleavage of talin and filamin.38 Calpains 1 and 2 have been shown to be important in cell migration and proliferation in many mammalian cell types,1,39–41 including endothelial cells.42–49 A review focusing on the regulation and physiological roles of calpains can be reviewed in this same journal issue.
Recent research revealed that calpains can affect mitochondrial function and regulate apoptosis, but there has been limited research involving endothelial cells. Vindis et al.50 showed that calpains are essential for apoptosis in human microvascular endothelial cells. Specifically, during oxidized-LDL-stimulated apoptosis, intracellular Ca2+ increases and induces calpain activation, leading to BH3-interacting domain death agonist (Bid) cleavage, cytochrome c release, apoptosome formation, and caspase 3 activation (Figure 3). Using EGTA and calpeptin, Vindis' group was able to prevent apoptosis. Calpain cleavage of Bid can lead to several outcomes: (i) tBid-mediated Bak and Bax oligomerization, (ii) formation of tBid homodimers, and (iii) tBid-induced mitochondrial permeability transition pore opening and remodelling of the inner mitochondrial membrane,51–56 which all lead to apoptosis. These data are in agreement with Walter et al.57 and Pörn-Ares et al.58 Under oxidized-LDL-stimulated apoptosis and calpain cleavage of Bid, calpain-independent apoptosis-inducing factor (AIF) release occurred.59 From a pathological perspective, the increase in oxidized-LDL activation of calpain and induction of apoptosis in atherosclerotic areas60 suggests that this type of apoptosis is important in atherothrombotic events.60,61 Also, in lysophosphatidylcholine-induced apoptosis of endothelial cells, luteolin treatment reduced Ca2+ influx which decreased calpain activation and prevented cytochrome c release.62 This further implicates calpains as pro-apoptotic proteases.
Figure 3.
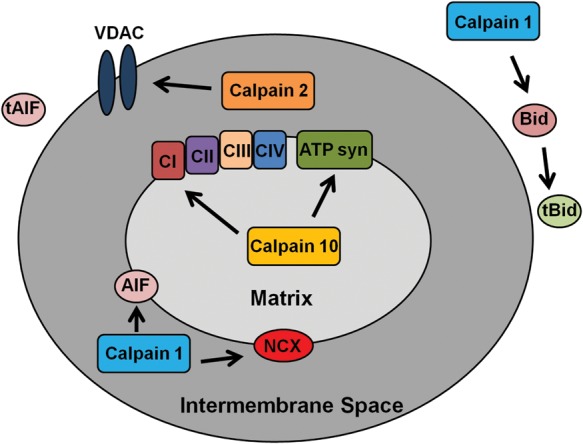
Diagram of the possible effects of calpains on the mitochondria. Calpain 10 cleaves electron transport chain proteins, calpain 1 cleaves BH3-interacting domain death agonist (Bid),50 AIF,108,109 and NCX,66 and calpain 2 cleaves voltage-dependent anion channel (VDAC).113
Several researchers have reported roles for mitochondrial calpain 1 in endothelial cells. In rat heart microvascular endothelial cells, Moshal et al. reported that hyperhomocysteinemia (Hcy)-induced extracellular matrix remodelling by matrix metalloproteinase 9 (MMP-9) is caused by calpain 1 activation.63 Hcy is defined as excess homocysteine in the blood, which leads to increased reactive oxygen species (ROS) production and vascular damage.64 In this model, Hcy increased Ca2+ flux, thereby activating calpain 1, which translocates from the cytosol to the mitochondria. Once in the mitochondria, calpain 1 increased ROS production, which activates extracellular-signal-regulated kinases 1/2 and MMP-9.63 MMP-9 activation leads to the cleavage of the extracellular matrix causing vascular dysfunction. Kar et al.65 reported that in bovine pulmonary artery smooth muscle mitochondria, calpain 1 and calpastatin are bound to the mitochondrial inner membrane and treatment with the Ca2+ ionophore, A23187, which increases mitochondrial Ca2+ concentration, induced calpain 1 cleavage of the mitochondrial Na+/Ca2+ exchanger (NCX) (Figure 3).66 Kar's group suggested that this is the main cause of Ca2+ accumulation in the mitochondria after injury.
Another important aspect of calpain biology is its role in ischaemia/reperfusion injury. Hoang et al. used calpain inhibitors (MDL28170, PD150606, and ALLN) in an in vivo model of ischaemic retinopathy to demonstrate that inhibition of calpain activity improves neovascular structure and function.67 In this model, hypoxia causes calpain activation leading to new dysfunctional vessels that have a disrupted actin cytoskeleton. Interestingly, calpain inhibition increased stability and function in new vessels reducing retinal damage. However, several articles discussing ischaemia/reperfusion damage in endothelial cells report that cathepsin B is more important than calpains in apoptotic cell death.68–70 Additional information about ischaemia/reperfusion injury and calpains can be learned from other review articles in this issue.
3. Kidney
The kidney plays an important role in the cardiovascular system by maintaining ion, water, and metabolic substrate homeostasis. Our laboratory has studied calpains in renal proximal tubular cell (RPTC) function and viability. We showed in various RPTC injury models that calpains 1 and 2 degrade cytoskeletal proteins and contribute to increased plasma membrane permeability, with Ca2+, Cl−, and water influx leading to necrosis.71–75
Calpains 1 and 2 have a more direct role in apoptosis. Some research has shown that calpains are important for the inactivation of caspases,76,77 whereas other reports suggest that calpains are necessary for caspase activation.78,79 Specifically, Lee et al.78 showed that calpains activate caspases in cadmium-induced apoptosis in RPTC because calpain inhibitors reduced caspase 3 activity and cell death.79
In 2006, Arrington et al.18 were the first to discover calpain 10 residing inside the mitochondria. Using rabbit mitochondria, calpain 10 was found in all of the mitochondrial compartments, with the majority of the activity in the mitochondrial matrix. Further analysis revealed that the first 15 amino acids on the N-terminus act as a mitochondrial targeting sequence. Calpain 10 has eight splice variants, ranging from 15 to 75 kDa in size, and it was shown using zymography that the 75, 56, and 50 kDa variants are in the mitochondria. Immunoblots for calpains 1 and 2 on mitochondrial fractions revealed calpain 10 to be the only calpain in the mitochondria. This was confirmed in rat and mouse renal mitochondria.80 Interestingly, even though mouse, rat, and rabbit all contain the same splice variants, they did not migrate similarly during zymography, suggesting that there are different binding partners in each species. Additionally, mitochondrial calpain 10 did not require Ca2+ for activity, but Ca2+ increased its activity moderately.
We used PEST sequence analysis to elucidate mitochondrial calpain 10 substrates, proteins that contain a large number of proline, glutamate, serine, and threonine residues signal rapid degradation by either calpains or the proteasome.81–83 NDUFV2, NDUFB8, ORP150, and ATP synthase-β were confirmed to be calpain 10 substrates (Figure 3). After Ca2+ overload, mitochondrial calpain 10 cleaves these proteins (and possibly other unknown substrates), causing a decrease in state 3 respiration. Thus, mitochondrial calpain 10 plays an important role in degradation of electron transport chain proteins.
Recently, calpain 10 was shown to be important in renal ageing, and kidneys regress in size and function with ageing by an unknown mechanism.84 Covington et al.85 discovered that calpain 10 protein and mRNA levels are reduced in aged rat, mouse, and human kidneys and caloric restriction prevented the decrease. Caloric restriction also prevented degeneration of the rat proximal tubules with age.86 Immunoblot analysis of the kidney and liver revealed that age did not affect calpain 1 and 2 protein or mRNA levels.85 Additionally, liver calpain 10 was unaffected at any age. Using our primary RPTC model, we explored the effect of acute reduction in calpain 10 by administration of adenoviral-delivered calpain 10 shRNA.85 Three days after adenovirus treatment when mitochondrial calpain 10 was ∼80% decreased with no effect on cytosolic calpain 10, we detected increased apoptosis. This suggests that renal mitochondrial calpain 10 is important in maintaining cellular viability and provides a possible reason why renal function decreases with age. Interestingly, when calpain 10 is overexpressed, there is mitochondrial swelling and cell death.18 Thus, calpain 10 protein must be maintained at a specific level or cell death will occur.
Because calpain 10 has been implicated in type 2 diabetes,87 we explored the effects of high glucose (17 mM) on primary RPTC.88 Interestingly, mitochondrial calpain 10 protein levels increased from 3 to 12 h after incubation in high glucose and then returned to normal until 48 h and decreased until at least 120 h. Cytosolic calpain 10 was unaffected until 120 h. We detected decreased basal and uncoupled respiration, an accumulation of mitochondrial calpain 10 substrates (NDUFB8 and ATP synthase-β), and increased apoptosis 96 h after high-glucose incubation. Using the streptozotocin-induced rat diabetic nephropathy model, rats had decreased renal calpain 10 protein and mRNA, with no effects on renal calpains 1 and 2 after 10 weeks.88 Accumulations of mitochondrial calpain 10 substrates and apoptosis were detected. Finally, calpain 10 siRNA knockdown of rat renal calpain 10 resulted in apoptosis and kidney dysfunction.88 These data demonstrate that diabetes results in decreased renal calpain 10 mRNA and protein, apoptosis, and decreased renal function and that a direct knockdown of renal calpain 10 in non-diabetic rats results in similar renal pathology, providing evidence that loss of renal calpain 10 may be a critical component of diabetic nephropathy.
Calpain inhibitors are promiscuous among calpain isoforms and other protease families. Thus, the use of calpain inhibitors to determine the role of calpains in physiology and pathology is difficult. Calpastatin is specific for typical calpains and the development of PD150606 provided a tool to inhibit typical calpains, because it binds the penta-EF hand in domain IV, but PD150606 can still inhibit multiple typical calpains.89,90 Our laboratory developed a calpain 10-specific peptide inhibitor with an IC50 of ∼100 nM that prevented Ca2+-induced reduction in state 3 respiration and mitochondrial calpain 10 substrate cleavage.91 While this inhibitor was not efficacious in cells, current research is focused on optimizing CYGAK to improve efficacy in cellular models.
4. Heart
Myocardial infarction can result in mitochondrial damage and apoptosis of cardiomyocytes.92 Numerous studies have documented that calpains are important in ischaemia/reperfusion injury in the heart,93–96 mitochondrial permeability transition,97,98 and necrotic/apoptotic cell death.99,100 Additional information about this type of injury can be found in another review article in this journal issue.
Trumbeckaite et al.101 used isolated rabbit hearts and performed 45 min of ischaemia followed by 60 min of reperfusion with and without pretreatment of BSF 409425, a calpain inhibitor, and showed that BSF 409425 blunted the decrease in state 3 and the increase in state 4 respiration. Several other studies have demonstrated that calpain inhibition during ischaemia/reperfusion reduces apoptosis and infarct size.102–104 These results suggest that excessive calpain activity plays a role in damaging mitochondria and oxidative phosphorylation during cardiac ischaemia/reperfusion injury.
One of the cytokines released after ischaemia/reperfusion injury is tumour necrosis factor-α (TNF-α).105 TNF-α is known to induce apoptosis and inflammation.106 Bajaj and Sharma showed the importance of calpains in TNF-α-induced apoptosis in the cardiac muscle cell line from the AT-1 mouse atrial cardiomyocyte tumor lineage cardiomyocyte cell line by pretreating cells with Z-LLY-fmk, a calpain inhibitor, for 30 min prior to TNF-α exposure for up to 12 h.107 Over the time course, they demonstrated that Z-LLY-fmk decreased cleaved pro-caspase 3 and poly-ADP ribose polymerase (PARP), suggesting that calpains are important in the activation of PARP and caspase 3, and ultimately apoptosis.
Calpains have been reported to be pro-apoptotic proteases by mediating the cleavage of AIF after Ca2+ overload (Figure 3).108,109 AIF is bound to the inner mitochondrial membrane, and when there is mitochondrial damage, AIF can be cleaved allowing it to translocate to the nucleus and induce DNA degradation.110 Chen et al.108 showed in isolated heart mitochondria that the addition of exogenous Ca2+ causes AIF release and that pretreatment with MDL28170, a calpain inhibitor, prevents this release. Therefore, it appears that the inhibition of mitochondrial calpain 1 prevents the release of AIF resulting in less cardiac cell death. However, there is some controversy because Ozaki et al. reported that liver mitochondrial calpain 1 mediated the release of AIF after Ca2+ overload and pretreatment with MDL28170 prevented this release.109 Joshi et al.111 reported that calpastatin and PD150606 pretreatment prior to Ca2+ overload in liver mitochondria did not prevent AIF release, suggesting that calpain 1 or 2 is not involved in this process. Furthermore, these researchers confirmed that AIF release was calpain-independent in isolated brain mitochondria. Much research has been performed in non-cardiac tissue mitochondria regarding calpain 1 cleavage of AIF. Further research is needed to determine whether mitochondrial calpain 1 cleaves AIF in cardiac tissue.
5. Summary and conclusions
Calpains have been discovered in the mitochondria. Much of the mitochondrial calpain research performed to date has focused on its pro-apoptotic role after Ca2+ overload, in particular, the cleavage of caspase 3 and/or AIF. There remains limited research on the physiological functions of mitochondrial calpains. However, calpain 10 regulates electron transport chain proteins and overexpression or knockdown of calpain 10 leads to mitochondrial dysfunction and cell death. Thus, mitochondrial calpain 10 protein must be tightly regulated for mitochondrial and renal cell function, but the underlying mechanisms need to be determined. Throughout the literature, many discrepancies exist concerning the role of calpains in physiology and different pathologies. Unfortunately, because the calpain active site is conserved throughout the entire family, creation of calpain isoform-specific inhibitors is difficult. Additionally, many popular calpain inhibitors also inhibit cathepsins, proteasome, and Lon protease (a mitochondrial matrix protease). Thus, many results must be confirmed with a knockdown model to ensure that calpain is the target and to identify the specific calpain isoform.
Conflict of interest: The article content does represent the views of the Department of Veterans Affairs of the US Government.
Funding
This study was supported by NIH Grant (GM 084147), the NIH Grant (ES-012239), the NIH/NIEHS Training Program in Environmental Stress Signaling (T32ES012878-05), and by the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs. Animal facilities were funded by NIH grant (C06 RR-015455).
References
- 1.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 2.Dayton WR, Reville WJ, Goll DE, Stromer MH. A calcium(2+) ion-activated protease possibly involved in myofibrillar protein turnover. Partial characterization of the purified enzyme. Biochemistry. 1976;15:2159–2167. doi: 10.1021/bi00655a020. [DOI] [PubMed] [Google Scholar]
- 3.Dayton WR, Goll DE, Zeece MG, Robson RM, Reville WJ. A Ca2+ ion-activated protease possibly involved in myofibrillar protein turnover. Purification from porcine muscle. Biochemistry. 1976;15:2150–2158. doi: 10.1021/bi00655a019. [DOI] [PubMed] [Google Scholar]
- 4.Futai E, Kubo T, Sorimachi H, Suzuki K, Maeda T. Molecular cloning of PalBH, a mammalian homologue of the Aspergillus atypical calpain PalB. Biochim Biophys Acta. 2001;1517:316–319. doi: 10.1016/s0167-4781(00)00256-6. [DOI] [PubMed] [Google Scholar]
- 5.Lee H-J, Tomioka S, Kinbara K, Masumoto H, Jeong S-Y, Sorimachi H, et al. Characterization of a human digestive tract-specific calpain, nCL-4, expressed in the baculovirus system. Arch Biochem Biophys. 1999;362:22–31. doi: 10.1006/abbi.1998.1021. [DOI] [PubMed] [Google Scholar]
- 6.Liu K, Li L, Cohen SN. Antisense RNA-mediated deficiency of the calpain protease, nCL-4, in NIH3T3 cells is associated with neoplastic transformation and tumorigenesis. J Biol Chem. 2000;275:31093–31098. doi: 10.1074/jbc.M005451200. [DOI] [PubMed] [Google Scholar]
- 7.Ma H, Fukiage C, Kim YH, Duncan MK, Reed NA, Shih M, et al. Characterization and expression of calpain 10. J Biol Chem. 2001;276:28525–28531. doi: 10.1074/jbc.M100603200. [DOI] [PubMed] [Google Scholar]
- 8.Matena K, Boehm T, Dear TN. Genomic organization of MouseCapn5andCapn6Genes confirms that they are a distinct calpain subfamily. Genomics. 1998;48:117–120. doi: 10.1006/geno.1997.5133. [DOI] [PubMed] [Google Scholar]
- 9.Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, et al. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J Biol Chem. 1989;264:20106–20111. [PubMed] [Google Scholar]
- 10.Dear N, Matena K, Vingron M, Boehm T. A new subfamily of vertebrate calpains lacking a calmodulin-like domain: implications for calpain regulation and evolution. Genomics. 1997;45:175–184. doi: 10.1006/geno.1997.4870. [DOI] [PubMed] [Google Scholar]
- 11.Sorimachi H, Ishiura S, Suzuki K. A novel tissue-specific calpain species expressed predominantly in the stomach comprises two alternative splicing products with and without Ca(2+)-binding domain. J Biol Chem. 1993;268:19476–19482. [PubMed] [Google Scholar]
- 12.Dear TN, Boehm T. Identification and characterization of two novel calpain large subunit genes. Gene. 2001;274:245–252. doi: 10.1016/s0378-1119(01)00599-6. [DOI] [PubMed] [Google Scholar]
- 13.Dear TN, Möller A, Boehm T. CAPN11: a calpain with high mRNA levels in testis and located on chromosome 6. Genomics. 1999;59:243–247. doi: 10.1006/geno.1999.5859. [DOI] [PubMed] [Google Scholar]
- 14.Dear TN, Meier NT, Hunn M, Boehm T. Gene structure, chromosomal localization, and expression pattern of Capn12, a new member of the calpain large subunit gene family. Genomics. 2000;68:152–160. doi: 10.1006/geno.2000.6289. [DOI] [PubMed] [Google Scholar]
- 15.Kamei M, Webb GC, Young IG, Campbell HD. SOLH, a human homologue of the Drosophila melanogaster small optic lobes gene is a member of the calpain and zinc-finger gene families and maps to human chromosome 16p13.3 near CATM (Cataract with Microphthalmia) Genomics. 1998;51:197–206. doi: 10.1006/geno.1998.5395. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes. 2004;53:S12–S18. doi: 10.2337/diabetes.53.2007.s12. [DOI] [PubMed] [Google Scholar]
- 17.Zatz M, Starling A. Calpains and disease. N Engl J Med. 2005;352:2413–2423. doi: 10.1056/NEJMra043361. [DOI] [PubMed] [Google Scholar]
- 18.Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol. 2006;291:C1159–C1171. doi: 10.1152/ajpcell.00207.2006. [DOI] [PubMed] [Google Scholar]
- 19.Reverter D, Braun M, Fernandez-Catalan C, Strobl S, Sorimachi H, Bode W. Flexibility analysis and structure comparison of two crystal forms of calcium-free human m-calpain. Biol Chem. 2002;383:1415–1422. doi: 10.1515/BC.2002.160. [DOI] [PubMed] [Google Scholar]
- 20.Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca2+ switch aligns the active site of calpain. Cell. 2002;108:649–660. doi: 10.1016/s0092-8674(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 21.Tompa P, Emori Y, Sorimachi H, Suzuki K, Friedrich P. Domain III of calpain is a Ca2+-regulated phospholipid-binding domain. Biochem Biophys Res Commun. 2001;280:1333–1339. doi: 10.1006/bbrc.2001.4279. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi M, Suzuki H, Kawashima S, Saido TC, Inomata M. The behavior of calpain-generated N- and C-terminal fragments of talin in integrin-mediated signaling pathways. Arch Biochem Biophys. 1999;371:133–141. doi: 10.1006/abbi.1999.1427. [DOI] [PubMed] [Google Scholar]
- 23.Strobl S, Fernandez-Catalan C, Braun M, Huber R, Masumoto H, Nakagawa K, et al. The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc Natl Acad Sci USA. 2000;97:588–592. doi: 10.1073/pnas.97.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorimachi H, Ishiura S, Suzuki K. Structure and physiological function of calpains. Biochem J. 1997;328:721–732. doi: 10.1042/bj3280721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinbara K, Sorimachi H, Ishiura S, Suzuki K. Muscle-specific calpain, p94, interacts with the extreme C-terminal region of connectin, a unique region flanked by two immunoglobulin C2 motifs. Arch Biochem Biophys. 1997;342:99–107. doi: 10.1006/abbi.1997.0108. [DOI] [PubMed] [Google Scholar]
- 26.Friedrich P, Papp H, Halasy K, Farkas A, Farkas B, Tompa P, et al. Differential distribution of calpain small subunit 1 and 2 in rat brain. Eur J Neurosci. 2004;19:1819–1825. doi: 10.1111/j.1460-9568.2004.03313.x. [DOI] [PubMed] [Google Scholar]
- 27.Ma H, Nakajima E, Shih M, Azuma M, Shearer TR. Expression of calpain small subunit 2 in mammalian tissues. Curr Eye Res. 2004;29:337–347. doi: 10.1080/02713680490516242. [DOI] [PubMed] [Google Scholar]
- 28.Zimmerman U-JP, Boring L, Pak UH, Mukerjee N, Wang KKW. The calpain small subunit gene is essential: its inactivation results in embryonic lethality. IUBMB Life. 2000;50:63–68. doi: 10.1080/15216540050176610. [DOI] [PubMed] [Google Scholar]
- 29.Sorimachi H, Suzuki K. The structure of calpain. J Biochem. 2001;129:653–664. doi: 10.1093/oxfordjournals.jbchem.a002903. [DOI] [PubMed] [Google Scholar]
- 30.Geesink GH, Nonneman D, Koohmaraie M. An improved purification protocol for heart and skeletal muscle calpastatin reveals two isoforms resulting from alternative splicing. Arch Biochem Biophys. 1998;356:19–24. doi: 10.1006/abbi.1998.0747. [DOI] [PubMed] [Google Scholar]
- 31.Parr T, Sensky PL, Bardsley RG, Buttery PJ. Calpastatin expression in porcine cardiac and skeletal muscle and partial gene structure. Arch Biochem Biophys. 2001;395:1–13. doi: 10.1006/abbi.2001.2546. [DOI] [PubMed] [Google Scholar]
- 32.Takano J, Watanabe M, Hitomi K, Maki M. Four types of calpastatin isoforms with distinct amino-terminal sequences are specified by alternative first exons and differentially expressed in mouse tissues. J Biochem. 2000;128:83–92. doi: 10.1093/oxfordjournals.jbchem.a022733. [DOI] [PubMed] [Google Scholar]
- 33.Kawasaki H, Emori Y, Imajoh-Ohmi S, Minami Y, Suzuki K. Identification and characterization of inhibitory sequences in four repeating domains of the endogenous inhibitor for calcium-dependent protease. J Biochem. 1989;106:274–281. doi: 10.1093/oxfordjournals.jbchem.a122844. [DOI] [PubMed] [Google Scholar]
- 34.Cong M, Thompson VF, Goll DE, Antin PB. The bovine calpastatin gene promoter and a new N-terminal region of the protein are targets for cAMP-dependent protein kinase activity. J Biol Chem. 1998;273:660–666. doi: 10.1074/jbc.273.1.660. [DOI] [PubMed] [Google Scholar]
- 35.Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430–443. doi: 10.1002/jcp.10333. [DOI] [PubMed] [Google Scholar]
- 36.Rodgers G, Cong J, Goll D, Kane W. Activation of coagulation factor V by calcium-dependent proteinase. Biochim Biophys Acta. 1987;929:263–270. doi: 10.1016/0167-4889(87)90252-7. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi M, Kasai Y, Kawashima S. Preferential localization of calcium-activated neutral protease in epithelial tissues. Biochem Biophys Res Commun. 1987;148:567–574. doi: 10.1016/0006-291x(87)90914-4. [DOI] [PubMed] [Google Scholar]
- 38.Fujitani K, Kambayashi J-i, Sakon M, Ohmi SI, Kawashima S-i, Yukawa M, et al. Identification of μ-, m-calpains and calpastatin and capture of μ-calpain activation in endothelial cells. J Cell Biochem. 1997;66:197–209. doi: 10.1002/(sici)1097-4644(19970801)66:2<197::aid-jcb7>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 39.Carragher NO, Frame MC. Calpain: a role in cell transformation and migration. Int J Biochem Cell Biol. 2002;34:1539–1543. doi: 10.1016/s1357-2725(02)00069-9. [DOI] [PubMed] [Google Scholar]
- 40.Glading A, Lauffenburger DA, Wells A. Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol. 2002;12:46–54. doi: 10.1016/s0962-8924(01)02179-1. [DOI] [PubMed] [Google Scholar]
- 41.Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–250. doi: 10.1091/mbc.E05-06-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiu K, Su Y, Block E. Use of recombinant calpain-2 siRNA adenovirus to assess calpain-2 modulation of lung endothelial cell migration and proliferation. Mol Cell Biochem. 2006;292:69–78. doi: 10.1007/s11010-006-9219-2. [DOI] [PubMed] [Google Scholar]
- 43.Ma H, Tochigi A, Shearer TR, Azuma M. Calpain inhibitor SNJ-1945 attenuates events prior to angiogenesis in cultured human retinal endothelial cells. J Ocul Pharmacol Ther. 2009;25:409–414. doi: 10.1089/jop.2009.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mo X-G, Chen Q-W, Li X-S, Zheng M-M, Ke D-Z, Deng W, et al. Suppression of NHE1 by small interfering RNA inhibits HIF-1α-induced angiogenesis in vitro via modulation of calpain activity. Microvasc Res. 2011;81:160–168. doi: 10.1016/j.mvr.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Letavernier B, Zafrani L, Nassar D, Perez J, Levi C, Bellocq A, et al. Calpains contribute to vascular repair in rapidly progressive form of glomerulonephritis: potential role of their externalization. Arterioscler Thromb Vasc Biol. 2012;32:335–342. doi: 10.1161/ATVBAHA.111.240242. [DOI] [PubMed] [Google Scholar]
- 46.Hoang MV, Nagy J, Fox JEB, Senger DR. Moderation of calpain activity promotes neovascular integration and lumen formation during VEGF-induced pathological angiogenesis. PLoS One. 2010;5:e13612. doi: 10.1371/journal.pone.0013612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonscherowski V, Becker BF, Moroder L, Motrescu E, Gil-Parrado S, Gloe T, et al. Calpains: a physiological regulator of the endothelial barrier? Am J Physiol Heart Circ Physiol. 2006;290:H2035–H2042. doi: 10.1152/ajpheart.00772.2004. [DOI] [PubMed] [Google Scholar]
- 48.Scalia R, Gong Y, Berzins B, Freund B, Feather D, Landesberg G, et al. A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling/novelty and significance. Circ Res. 2011;108:1102–1111. doi: 10.1161/CIRCRESAHA.110.229393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang H, Kwak H-I, Kaunas R, Bayless KJ. Fluid shear stress and sphingosine 1-phosphate activate calpain to promote membrane type 1 matrix metalloproteinase (MT1-MMP) membrane translocation and endothelial invasion into three-dimensional collagen matrices. J Biol Chem. 2011;286:42017–42026. doi: 10.1074/jbc.M111.290841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vindis C, Elbaz M, Escargueil-Blanc I, Augé N, Heniquez A, Thiers J-C, et al. Two distinct calcium-dependent mitochondrial pathways are involved in oxidized LDL-induced apoptosis. Arterioscler Thromb Vasc Biol. 2005;25:639–645. doi: 10.1161/01.ATV.0000154359.60886.33. [DOI] [PubMed] [Google Scholar]
- 51.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 52.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 53.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 54.Bernardi P, Petronilli V, Di Lisa F, Forte M. A mitochondrial perspective on cell death. Trends Biochem Sci. 2001;26:112–117. doi: 10.1016/s0968-0004(00)01745-x. [DOI] [PubMed] [Google Scholar]
- 55.Henry-Mowatt J, Dive C, Martinou J-C, James D. Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene. 2004;23:2850–2860. doi: 10.1038/sj.onc.1207534. [DOI] [PubMed] [Google Scholar]
- 56.Zhao Y, Ding W-x, Qian T, Watkins S, Lemasters JJ, Yin X-m. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology. 2003;125:854–867. doi: 10.1016/s0016-5085(03)01066-7. [DOI] [PubMed] [Google Scholar]
- 57.Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S. Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation. 1998;98:1153–1157. doi: 10.1161/01.cir.98.12.1153. [DOI] [PubMed] [Google Scholar]
- 58.Pörn-Ares MI, Saido TC, Andersson T, Ares MPS. Oxidized low-density lipoprotein induces calpain-dependent cell death and ubiquitination of caspase 3 in HMEC-1 endothelial cells. Biochem J. 2003;374:403–411. doi: 10.1042/BJ20021955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 60.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130:947–962. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geng Y-J, Libby P. Progression of atheroma. Arterioscler Thromb Vasc Biol. 2002;22:1370–1380. doi: 10.1161/01.atv.0000031341.84618.a4. [DOI] [PubMed] [Google Scholar]
- 62.Song J, Liu K, Yi J, Zhu D, Liu G, Liu B. Luteolin inhibits lysophosphatidylcholine-induced apoptosis in endothelial cells by a calcium/mitocondrion/caspases-dependent pathway. Planta Med. 2010;76:433–438. doi: 10.1055/s-0029-1186197. [DOI] [PubMed] [Google Scholar]
- 63.Moshal KS, Singh M, Sen U, Rosenberger DSE, Henderson B, Tyagi N, et al. Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol. 2006;291:H2825–H2835. doi: 10.1152/ajpheart.00377.2006. [DOI] [PubMed] [Google Scholar]
- 64.Papatheodorou L, Weiss N. Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxid Redox Signal. 2007;9:1941–1958. doi: 10.1089/ars.2007.1750. [DOI] [PubMed] [Google Scholar]
- 65.Kar P, Chakraborti T, Samanta K, Chakraborti S. Submitochondrial localization of associated μ-calpain and calpastatin. Arch Biochem Biophys. 2008;470:176–186. doi: 10.1016/j.abb.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 66.Kar P, Chakraborti T, Samanta K, Chakraborti S. μ-Calpain mediated cleavage of the Na+/Ca2+ exchanger in isolated mitochondria under A23187 induced Ca2+ stimulation. Arch Biochem Biophys. 2009;482:66–76. doi: 10.1016/j.abb.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 67.Hoang MV, Smith LEH, Senger DR. Calpain inhibitors reduce retinal hypoxia in ischemic retinopathy by improving neovascular architecture and functional perfusion. Biochim Biophys Acta. 2011;1812:549–557. doi: 10.1016/j.bbadis.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsubokawa T, Yamaguchi-Okada M, Calvert JW, Solaroglu I, Shimamura N, Yata K, et al. Neurovascular and neuronal protection by E64d after focal cerebral ischemia in rats. J Neurosci Res. 2006;84:832–840. doi: 10.1002/jnr.20977. [DOI] [PubMed] [Google Scholar]
- 69.Yadav SS, Sindram D, Perry DK, Clavien P-A. Ischemic preconditioning protects the mouse liver by inhibition of apoptosis through a caspase-dependent pathway. Hepatology. 1999;30:1223–1231. doi: 10.1002/hep.510300513. [DOI] [PubMed] [Google Scholar]
- 70.Seyfried DM, Veyna R, Han Y, Li K, Tang N, Betts RL, et al. A selective cysteine protease inhibitor is non-toxic and cerebroprotective in rats undergoing transient middle cerebral artery ischemia. Brain Res. 2001;901:94–101. doi: 10.1016/s0006-8993(01)02289-2. [DOI] [PubMed] [Google Scholar]
- 71.Liu X, Rainey JJ, Harriman JF, Schnellmann RG. Calpains mediate acute renal cell death: role of autolysis and translocation. Am J Physiol Renal Physiol. 2001;281:F728–F738. doi: 10.1152/ajprenal.2001.281.4.F728. [DOI] [PubMed] [Google Scholar]
- 72.Waters SL, Sarang SS, Wang KKW, Schnellmann RG. Calpains mediate calcium and chloride influx during the late phase of cell injury. J Pharmacol Exp Ther. 1997;283:1177–1184. [PubMed] [Google Scholar]
- 73.Harriman JF, Waters-Williams S, Chu D-L, Powers JC, Schnellmann RG. Efficacy of novel calpain inhibitors in preventing renal cell death. J Pharmacol Exp Ther. 2000;294:1083–1087. [PubMed] [Google Scholar]
- 74.Harriman J, Liu X, Aleo M, Machaca K, Schnellmann R. Endoplasmic reticulum Ca2+ signaling and calpains mediate renal cell death. Cell Death Differ. 2002;9:734–741. doi: 10.1038/sj.cdd.4401029. [DOI] [PubMed] [Google Scholar]
- 75.Liu X, Schnellmann RG. Calpain mediates progressive plasma membrane permeability and proteolysis of cytoskeleton-associated paxillin, talin, and vinculin during renal cell death. J Pharmacol Exp Ther. 2003;304:63–70. doi: 10.1124/jpet.102.043406. [DOI] [PubMed] [Google Scholar]
- 76.Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem. 2003;278:14162–14167. doi: 10.1074/jbc.M212255200. [DOI] [PubMed] [Google Scholar]
- 77.Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- 78.Lee W-K, Abouhamed M, Thévenod F. Caspase-dependent and -independent pathways for cadmium-induced apoptosis in cultured kidney proximal tubule cells. Am J Physiol Renal Physiol. 2006;291:F823–F832. doi: 10.1152/ajprenal.00276.2005. [DOI] [PubMed] [Google Scholar]
- 79.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giguere CJ, Covington MD, Schnellmann RG. Mitochondrial calpain 10 activity and expression in the kidney of multiple species. Biochem Biophys Res Commun. 2008;366:258–262. doi: 10.1016/j.bbrc.2007.11.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shumway SD, Maki M, Miyamoto S. The PEST domain of IκBα is necessary and sufficient for in vitro degradation by μ-calpain. J Biol Chem. 1999;274:30874–30881. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- 82.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 83.Spencer ML, Theodosiou M, Noonan DJ. NPDC-1, a novel regulator of neuronal proliferation, is degraded by the ubiquitin/proteasome system through a PEST degradation motif. J Biol Chem. 2004;279:37069–37078. doi: 10.1074/jbc.M402507200. [DOI] [PubMed] [Google Scholar]
- 84.Coca SG. Acute kidney injury in elderly persons. Am J Kidney Dis. 2010;56:122–131. doi: 10.1053/j.ajkd.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Covington MD, Arrington DD, Schnellmann RG. Calpain 10 is required for cell viability and is decreased in the aging kidney. Am J Physiol Renal Physiol. 2009;296:F478–F486. doi: 10.1152/ajprenal.90477.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jung K-Y, Dean D, Jiang J, Gaylor S, Griffith WH, Burghardt RC, et al. Loss of N-cadherin and α-catenin in the proximal tubules of aging male Fischer 344 rats. Mech Ageing Dev. 2004;125:445–453. doi: 10.1016/j.mad.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 87.Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–175. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- 88.Covington MD, Schnellmann RG. Chronic high glucose downregulates mitochondrial calpain 10 and contributes to renal cell death and diabetes-induced renal injury. Kidney Int. 2012;81:391–400. doi: 10.1038/ki.2011.356. [DOI] [PubMed] [Google Scholar]
- 89.Lin G-d, Chattopadhyay D, Maki M, Wang KKW, Carson M, Jin L, et al. Crystal structure of calcium bound domain VI of calpain at 1.9 A resolution and its role in enzyme assembly, regulation, and inhibitor binding. Nat Struct Mol Biol. 1997;4:539–547. doi: 10.1038/nsb0797-539. [DOI] [PubMed] [Google Scholar]
- 90.Todd B, Moore D, Deivanayagam CCS, Lin G-d, Chattopadhyay D, Maki M, et al. A Structural model for the inhibition of calpain by calpastatin: crystal structures of the native domain VI of calpain and its complexes with calpastatin peptide and a small molecule inhibitor. J Mol Biol. 2003;328:131–146. doi: 10.1016/s0022-2836(03)00274-2. [DOI] [PubMed] [Google Scholar]
- 91.Rasbach KA, Arrington DD, Odejinmi S, Giguere C, Beeson CC, Schnellmann RG. Identification and optimization of a novel inhibitor of mitochondrial calpain 10. J Med Chem. 2008;52:181–188. doi: 10.1021/jm800735d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leonardo B. Changing patterns of ST elevation myocardial infarction epidemiology. Am Heart J. 2010;160:S1–S3. doi: 10.1016/j.ahj.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 93.Gao W, Atar D, Liu Y, Perez N, Murphy A, Marban E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res. 1997;80:393–399. [PubMed] [Google Scholar]
- 94.Iwamoto H, Miura T, Okamura T, Shirakawa K, Iwatate M, Kawamura S, et al. Calpain inhibitor-1 reduces infarct size and DNA fragmentation of mycardium in ischemia/reperfused rat heart. J Cardiovasc Pharmacol. 1999;33:580–586. doi: 10.1097/00005344-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 95.Urthaler F, Wolkowicz PE, Digerness SB, Harris KD, Walker AA. MDL-28170, a membrane-permeant calpain inhibitor, attenuates stunning and PKCɛ proteolysis in reperfused ferret hearts. Cardiovasc Res. 1997;35:60–67. doi: 10.1016/s0008-6363(97)00099-0. [DOI] [PubMed] [Google Scholar]
- 96.Gao G, Dou QP. N-terminal cleavage of Bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes Bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem. 2001;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 97.Aguilar HI, Botla R, Arora AS, Bronk SF, Gores GJ. Induction of the mitochondrial permeability transition by protease activity in rats: A mechanism of hepatocyte necrosis. Gastroenterology. 1996;110:558–566. doi: 10.1053/gast.1996.v110.pm8566604. [DOI] [PubMed] [Google Scholar]
- 98.Gores GJ, Miyoshi H, Botla R, Aguilar HI, Bronk SF. Induction of the mitochondrial permeability transition as a mechanism of liver injury during cholestasis: a potential role for mitochondrial proteases. Biochim Biophys Acta. 1998;1366:167–175. doi: 10.1016/s0005-2728(98)00111-x. [DOI] [PubMed] [Google Scholar]
- 99.Kohli V, Madden J, Bentley R, Clavien P. Calpain mediates ischemic injury of the liver through modulation of apoptosis and necrosis. Gastroenterology. 1999;116:168–178. doi: 10.1016/s0016-5085(99)70241-6. [DOI] [PubMed] [Google Scholar]
- 100.Wang KKW, Posmantur R, Nadimpalli R, Nath R, Mohan P, Nixon RA, et al. Caspase-mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch Biochem Biophys. 1998;356:187–196. doi: 10.1006/abbi.1998.0748. [DOI] [PubMed] [Google Scholar]
- 101.Trumbeckaite S, Neuhof C, Zierz S, Gellerich FN. Calpain inhibitor (BSF 409425) diminishes ischemia/reperfusion-induced damage of rabbit heart mitochondria. Biochem Pharmacol. 2003;65:911–916. doi: 10.1016/s0006-2952(02)01610-6. [DOI] [PubMed] [Google Scholar]
- 102.Khalil PN, Neuhof C, Huss R, Pollhammer M, Khalil MN, Neuhof H, et al. Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. Eur J Pharmacol. 2005;528:124–131. doi: 10.1016/j.ejphar.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 103.Perrin C, Escarnot-Laubriet A, Vergely C, Rochette L. Calpain and caspase-3 inhibitors reduce infarct size and post-ischemic apoptosis in rat heart without modifying contractile recovery. Cell Mol Biol. 2003;49:497–505. [PubMed] [Google Scholar]
- 104.Kakkar R, Wang X, Radhi JM, Rajala RVS, Wang R, Sharma RK. Decreased expression of high-molecular-weight calmodulin-binding protein and its correlation with apoptosis in ischemia-reperfused rat heart. Cell Calcium. 2001;29:59–71. doi: 10.1054/ceca.2000.0157. [DOI] [PubMed] [Google Scholar]
- 105.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. New Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 106.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 107.Bajaj G, Sharma RK. TNF-α-mediated cardiomyocyte apoptosis involves caspase-12 and calpain. Biochem Biophys Res Commun. 2006;345:1558–1564. doi: 10.1016/j.bbrc.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 108.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, et al. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia–reperfusion. Biochem Biophys Res Commun. 2011;415:533–538. doi: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ozaki T, Yamashita T, Ishiguro S-i. ERp57-associated mitochondrial μ-calpain truncates apoptosis-inducing factor. Biochim Biophys Acta. 2008;1783:1955–1963. doi: 10.1016/j.bbamcr.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 110.Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, et al. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Mol Biol. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- 111.Joshi A, Bondada V, Geddes JW. Mitochondrial μ-calpain is not involved in the processing of apoptosis-inducing factor. Exp Neurol. 2009;218:221–227. doi: 10.1016/j.expneurol.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wendt A, Thompson V, Goll D. Interaction of calpastatin with calpain: a review. Biol Chem. 2004;385:465–472. doi: 10.1515/BC.2004.054. [DOI] [PubMed] [Google Scholar]
- 113.Ozaki T, Yamashita T, Ishiguro S-i. Mitochondrial m-calpain plays a role in the release of truncated apoptosis-inducing factor from the mitochondria. Biochim Biophys Acta. 2009;1793:1848–1859. doi: 10.1016/j.bbamcr.2009.10.002. [DOI] [PubMed] [Google Scholar]