Snail Represses the Splicing Regulator Epithelial Splicing Regulatory Protein 1 to Promote Epithelial-Mesenchymal Transition (original) (raw)
Background: The Epithelial Splicing Regulatory Protein 1 (ESRP1) prevents CD44 splice isoform switching during epithelial-mesenchymal transition (EMT), a developmental process frequently reactivated in cancer progression.
Results: Snail represses ESRP1 transcription, thus promoting CD44 isoform switching during EMT.
Conclusion: Repression by Snail of ESRP1 transcription is required for EMT to occur.
Significance: Investigating mechanisms that regulate alternative splicing during EMT will facilitate our understanding of the EMT associated with cancer recurrence and metastasis.
Keywords: Alternative Splicing, CD44, EMT, Gene Regulation, RNA, ESRP1
Abstract
Epithelial-mesenchymal transition (EMT), a tightly regulated process that is critical for development, is frequently re-activated during cancer metastasis and recurrence. We reported previously that CD44 isoform switching is critical for EMT and showed that the splicing factor ESRP1 inhibits CD44 isoform switching during EMT. However, the mechanism by which ESRP1 is regulated during EMT has not been fully understood. Here we show that the transcription repressor Snail binds to E-boxes in the ESRP1 promoter, causing repression of the ESRP1 gene. Biochemically, we define the mechanism by which ESRP1 regulates CD44 alternative splicing: ESRP1 binds to the intronic region flanking a CD44 variable exon and causes increased variable exon inclusion. We further show that ectopically expressing ESRP1 inhibits Snail-induced EMT, suggesting that down-regulation of ESRP1 is required for function by Snail in EMT. Together, these data reveal how the transcription factor Snail mediates EMT through regulation of a splicing factor.
Introduction
Epithelial-mesenchymal transition (EMT),3 a process in which epithelial cells lose cell polarity and transition to a mesenchymal phenotype, plays a critical role during normal development. However, mounting evidence suggests that this process is often abnormally re-activated during cancer recurrence and metastasis (1–4). Key characteristics of EMT include a physical change from a cobblestone-like epithelial morphology to an elongated fibroblastic appearance, a cadherin switch in which epithelial E-cadherin is down-regulated and mesenchymal N-cadherin is up-regulated, cytoskeletal reorganization, increased resistance to cell death, and the acquisition of a migratory phenotype. Mediators of EMT include the transcription factors Snail and Twist, as well as cytokines such as TGF-β (3, 5–7).
Alternative splicing, a key mechanism for generating protein diversity, is another tightly regulated process that has been shown to be aberrantly altered in cancer (8–11). For example, our recent studies revealed that CD44 isoform switching plays a critical role in EMT and breast cancer progression (12). The CD44 gene is composed of nine or ten (in human or mouse, respectively) variable exons located between nine constitutive exons. Alternative splicing of CD44 produces two groups of protein isoforms: CD44 variants (CD44v) that contain diverse combinations of the variable exons and CD44 standard (CD44s), which is devoid of all variable exons. When cells undergo EMT, there is a gradual switch in CD44 isoform expression; whereas epithelial cells predominantly express CD44v, mesenchymal cells chiefly express CD44s. We have shown that the isoform switch from CD44v to CD44s is required in order for cells to undergo EMT (12).
We and others have shown that epithelial splicing regulatory protein 1 (ESRP1) plays a critical role in the regulation of CD44 alternative splicing (12, 13). ESRP1 is an epithelial-specific splicing factor that regulates the alternative splicing of several genes, including fibroblast growth factor receptor 2 and CD44 (12, 13). During EMT, ESRP1 expression drastically decreases, and this down-regulation facilitates the production of CD44s. Conversely, ectopic expression of ESRP1 prevents the CD44v to CD44s switch that would normally occur when cells are treated with EMT stimuli, and as a consequence, EMT is disrupted (12). These results indicated a novel mode of regulation by which EMT is controlled at the level of alternative splicing. However, the mechanism by which ESRP1 is regulated remained largely unexplored, and the precise mechanism underlying regulation by ESRP1 of CD44 alternative splicing was also unknown. In this study, we show that the transcription repressor Snail suppresses ESRP1 expression by binding to E-boxes located within the ESRP1 promoter. Overexpression of Snail led to marked changes in the alternative splicing of a number of additional ESRP1-regulated genes, including MAGI1, SCRIB, and ENAH. Furthermore, we identify ESRP1-binding sequences in the CD44 pre-mRNA that mediate action by ESRP1 in regulating CD44 alternative splicing and show direct association of ESRP1 with the CD44 mRNA. Biologically, we show that the decrease in ESRP1 expression is required in order for Snail-mediated EMT to occur. Restoration of the CD44s splice isoform rescued EMT, indicating that the inhibition of EMT by ESRP1 is directly mediated through regulation by ESRP1 of CD44 alternative splicing. Thus, our results reveal that the interplay among the transcription factor Snail, the splicing regulatory protein ESRP1, and its splicing target CD44 plays a key role in controlling EMT.
EXPERIMENTAL PROCEDURES
Cell Lines and EMT Induction
Maintenance of the immortalized human mammary epithelial cells HMLE/HA-ESRP1 and HMLE/Snail-estrogen receptor (ER), as well as tamoxifen induction of HMLE/Snail-ER cells, were performed as described previously (14). HCT116 colon carcinoma cells, human embryonic kidney 293 cells, and MDA-MB-231 cells were grown in DMEM supplemented with 10% fetal bovine serum and 1% l-glutamine.
Plasmids
ESRP1 promoter regions were PCR-amplified from genomic DNA isolated from human mammary MCF10A cells. Sequences of primers used are as follows: four E-boxes forward, AGT CGA GCT CTG GTT TGA AGG AGC CAA TG; four E-boxes reverse, GAC TAG ATC TCT TCC TTG CTA CTG CTA GTG C; two 5′ E-boxes forward, AGT CGA GCT CTG GTT TGA AGG AGC CAA TG; two 5′ E-boxes reverse, GAC TAG ATC TTC TGT TTT GGG ATG TGG CT; two 3′ E-boxes forward, AGT CGA GCT CCT CCA GGC TTT TTG CAT AGA CG; and two 3′ E-boxes reverse, GAC TAG ATC TCT TCC TTG CTA CTG CTA GTG C. PCR products were cloned into the SacI and BglII sites of the pGL3-Basic vector. E-box mutagenesis was conducted via PCR. The ESRP1 and CD44s plasmids were described previously (12).
Luciferase Assays
Luciferase reporter assays were performed using the Dual-Luciferase Reporter Assay System from Promega (Madison, WI) according to the manufacturer's instructions. Briefly, HCT116 or 293 cells were plated in 24-well plates 24 h prior to transfection using Lipofectamine 2000 (Invitrogen). HCT116 cells were co-transfected with ESRP1 promoter constructs and Snail or Twist along with a Renilla luciferase construct, which served as an internal control for transfection efficiency.
Quantitative RT-PCR
RNA was isolated from cells using the RNeasy Kit (Qiagen, Valencia, CA) or the E.Z.N.A. Total RNA Kit (Omega Bio-Tek, Norcross, GA). cDNA was generated by reverse transcription using Omniscript RT (Qiagen) or GoScript RT (Promega). qRT-PCR was performed using Power SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA) or GoTaq qPCR Master Mix (Promega), and mRNA levels were normalized to levels of TATA-binding protein.
RNA Pulldown Assays
Biotin-labeled CD44 RNA oligonucleotides wild-type I-8 (5′-Biotin-GCUUUGGUGGUGGAAUGGUGCUAUGUGG-3′) and mutated I-8 (5′-Biotin-GCUU_C_GAUCCUAGAACAGAGCUAUCUCG-3′; mutated nucleotides are underlined) were synthesized by Integrated DNA Technologies (Coralville, IA). Whole cell lysates from MDA-MB-231/HA-ESRP1 and HMLE/HA-ESRP1 cells were prepared in radioimmunoprecipitation assay buffer. Pulldown assays were performed as described elsewhere (15) with certain modifications. Briefly, biotin-labeled RNA oligonucleotides were prebound to 100-μl of immobilized streptavidin (bead conjugate; Pierce) for 2 h at 4 °C and then incubated with 200–400 μg of whole cell lysate for 2 h at 4 °C. Beads were washed three times, then resuspended in 20 μl of 2× SDS sample buffer, and boiled for 10 min at 95 °C. Proteins were analyzed by Western blotting. For input samples, 20 μg of whole cell lysate was added to 2× SDS sample buffer and boiled for 10 min at 95 °C.
Antibodies
Antibodies used for Western blotting were as follows: CD44 (R&D Systems, Minneapolis, MN, and Santa Cruz Biotechnology Inc., Santa Cruz, CA), E-cadherin (Cell Signaling Technology, Danvers, MA), N-cadherin (BD Transduction Laboratories, Franklin Lakes, NJ), vimentin (NeoMarkers), HA (Roche Applied Science), β-actin (Sigma-Aldrich), and GAPDH (Millipore, Billerica, MA).
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed essentially as described elsewhere (16) with certain modifications. Briefly, cells were cross-linked in 1% formaldehyde/PBS for 10 min at room temperature. Cross-linking was quenched with 0.125 m glycine. Cells were collected by centrifugation at 1,000 rpm for 2 min and washed with PBS. Nuclei were isolated by incubation in cell lysis buffer (10 mm Tris, 10 mm NaCl, 0.2% Nonidet P-40, 1× protease inhibitor (Roche), pH 8.0) for 10 min on ice, followed by centrifugation at 2,500 rpm for 5 min. Nuclei were lysed in nuclei lysis buffer (50 mm Tris, 10 mm EDTA, 1% SDS, 1× protease inhibitor (Roche), pH 8.1) for 10 min on ice. Lysates were sonicated using a Branson Sonifier 150 to obtain chromatin fragments with an average size of <500 bp. Sonicated chromatin was diluted with immunoprecipitation dilution buffer (20 mm Tris, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100, 0.01% SDS, 1× protease inhibitor (Roche), pH 8.1) and precleared by the addition of 15 μg of IgG antibody and 200 μl of protein-G beads. An aliquot of precleared chromatin was removed (input) and used in the subsequent quantitative PCR analysis. Remaining chromatin was immunoprecipitated using 5 μg of estrogen receptor (ER, from Thermo Scientific) or mouse IgG (The Jackson Laboratory, Bar Harbor, ME) antibody prebound to protein-G beads for 6 h at 4 °C. Beads were washed seven times, and the bound proteins were eluted into 100 mm NaHCO3 and 1% SDS. Samples were incubated overnight at 67 °C to reverse cross-links and then incubated with Proteinase K (0.3 mg/ml) for 2 h at 45 °C to digest protein. DNA was purified using the QIAquick PCR Purification Kit (Qiagen). Quantitative PCR was performed using GoTaq qPCR Master Mix. Sequences of primers used are as follows: ESRP1 E-boxes forward, AGT CGA GCT CCT CCA GGC TTT TTG CAT AGA CG; ESRP1 E-boxes reverse, GAC TAG ATC TCT TCC TTG CTA CTG CTA GTG C; intergenic forward, GGA GGG ACA GAG GGA AAC TC; and intergenic reverse: ACG GTG GAA ATC TTG GAC TG.
Statistical Analyses
Unless indicated otherwise, all data are presented as means ±S.E.
RESULTS AND DISCUSSION
The Transcriptional Repressor Snail Regulates ESRP1 Expression
To investigate the transcriptional regulation of the ESRP1 gene during EMT, we analyzed the ESRP1 promoter sequences in human, chimpanzee, rat, and mouse. Strikingly, we observed that there are four E-boxes (CANNTG) located in the proximal region of the ESRP1 promoter that are highly conserved among species (Fig. 1A). Because the EMT-inducer Snail is known to regulate transcription by binding to E-box sequences located within the promoters of its target genes (17, 18), our observation raised the possibility that Snail may directly repress ESRP1 transcription by binding to the evolutionarily conserved ESRP1 E-boxes. To test this hypothesis, we utilized an inducible-EMT system in which immortalized human mammary epithelial cells stably express a Snail-ER fusion protein (HMLE/Snail-ER). Upon treatment with tamoxifen, Snail-ER is translocated to the nucleus where it can act upon its target genes, causing the cells to undergo EMT in 12–14 days (14). We found that ESRP1 was highly expressed in the untreated HMLE/Snail-ER cells. Upon tamoxifen treatment that allows Snail to enter the nucleus and function as a transcriptional repressor, ESRP1 was gradually down-regulated by 2-fold during the first 6 days, followed by a sharper decrease after day 6, culminating in an 11.5-fold decrease in expression by day 14, when EMT was complete (Fig. 1B). These results indicate that Snail represses endogenous levels of ESRP1 during EMT.
FIGURE 1.

The transcriptional repressor Snail regulates ESRP1 expression. A, schematic of the ESRP1 promoter that contains four E-box sequences that are highly conserved among human, chimpanzee, rat, and mouse. The transcription start is indicated by a bent arrow. B, qRT-PCR analysis of ESRP1 levels in HMLE/Snail-ER cells during tamoxifen-induced EMT. Relative expression levels are normalized to TATA-binding protein at each time point, and the results are shown relative to day 0. Error bars indicate S.E.; n = 4. C, relative luciferase activity in HCT116 cells transfected with ESRP1 luciferase reporter constructs that include an E-box-containing fragment of the ESRP1 promoter upstream of luciferase. The ratios of Photinus to Renilla luciferase activities were normalized to cells transfected with a control luciferase reporter construct that lacks promoter and enhancer sequences. D, relative luciferase activities in HCT116 cells co-transfected with ESRP1 promoter luciferase reporter constructs and Snail. For each ESRP1 promoter construct, _Photinus_-to-Renilla luciferase activities were normalized to cells co-transfected with an empty vector control. E, relative luciferase activities in HCT116 cells co-transfected with a wild-type or mutant ESRP1 promoter luciferase reporter construct and Snail. C–E, bar graphs depict averages of at least three independent experiments; error bars indicate S.E.
Snail Represses the Promoter Activity of ESRP1
We next set out to determine whether Snail represses ESRP1 by binding to the E-boxes located in the ESRP1 promoter. First, we generated a luciferase reporter construct that contains the four putative ESRP1 E-boxes upstream of a luciferase gene. Transfecting HCT116 cells with this construct caused a 7.7-fold increase in luciferase activity compared with control (Fig. 1C), suggesting that this four E-boxes-containing region represents a functional segment of the ESRP1 promoter. We next separated the four E-boxes-containing regions into two smaller fragments: one encompassing the two 5′ E-boxes, and the other encompassing the two 3′ E-boxes. Transfecting HCT116 cells with either construct caused a marked increase in luciferase activity compared with control (Fig. 1C). In particular, transfecting the two 5′ E-boxes construct resulted in a nearly 6-fold increase in luciferase activity that was comparable with the increase in activity observed using the four E-boxes fragment, suggesting that the two 5′ E-boxes region mediates the core promoter activity of ESRP1.
To determine whether the transcriptional activity mediated by the ESRP1 promoter region could be repressed by Snail, we co-transfected Snail with each of the above ESRP1 promoter luciferase reporter constructs. Co-transfection of Snail resulted in a 3-fold repression in luciferase activity mediated by the four E-boxes region compared with control (Fig. 1D, left panel). Similarly, co-transfection of Snail with the two 5′ E-boxes and two 3′ E-boxes promoter constructs resulted in a 4.1-fold and 2.2-fold decrease in luciferase activity, respectively (Fig. 1D, center and right panels). These results demonstrate that Snail represses ESRP1 promoter activity. Importantly, luciferase activity mediated by the four E-boxes construct or either of the two E-boxes constructs was not diminished upon co-transfection of Twist, another transcriptional repressor known to promote EMT, suggesting that Snail specifically represses ESRP1 transcription (Fig. 1D).
We next determined whether mutating the E-boxes within the ESRP1 promoter region abolishes Snail-mediated repression. We first focused on the two 5′ E-boxes region because this region plays the most significant role in mediating ESRP1 promoter activity and because Snail was able to cause the most dramatic repression of luciferase activity with this region (Fig. 1, C and D). The consensus E-box sequences CANNTG were mutated to AANNTA in the two 5′ E-boxes construct, and these mutations resulted in a moderate but consistent 1.44-fold decrease in Snail-mediated repression of luciferase activity (Fig. 1E, left panel), suggesting that Snail represses ESRP1 transcription by binding to the E-boxes located in this promoter region. Similar to the results observed with the two 5′ E-boxes construct, mutating the E-boxes from CANNTG to AANNTA in the two 3′ E-boxes construct caused a 1.5-fold decrease in Snail-mediated repression (Fig. 1E, right panel). These findings further suggest that the E-boxes play an important role in the ability of Snail to repress transcriptional activity conferred by these regions. We next generated a mutant construct in which all four E-boxes were mutated to AANNTA. For reasons we do not fully understand, mutating all four E-boxes did not cause a decrease in the ability of Snail to repress ESRP1 transcription. It is possible that transcriptional activators also bind to this conserved ESRP1 promoter region, thus confounding our results that focus on analyzing transcriptional repression. Alternatively, these mutations may cause a change in the secondary structure of the promoter region that affects its accessibility such that transcriptional activators that are able to bind to the wild-type sequence are no longer able to bind to the mutated construct. Therefore, loss of base-line transcription activity may mask the decrease in Snail-mediated repression. Indeed, mutating the four E-boxes resulted in a 2-fold decrease in transcriptional activity compared with the wild-type four E-boxes region (data not shown). Collectively, the above ESRP1 promoter activity and mutation analyses suggested that Snail represses ESRP1 transcription through binding to E-boxes located in the ESRP1 promoter.
Snail Binds to an E-box-containing Region in the ESRP1 Promoter
To investigate whether Snail represses ESRP1 transcription by binding directly to the ESRP1 promoter, we conducted ChIP assays. These experiments were carried out using chromatin isolated from HMLE/Snail-ER cells and an ER antibody that recognizes an epitope within the region of the estrogen receptor that is present in the Snail-ER fusion protein. This approach therefore allowed us to assay for Snail occupancy on chromatin following tamoxifen-induced activation of the Snail-ER protein. We analyzed Snail occupancy of the ESRP1 promoter in HMLE/Snail-ER cells following 6 days of tamoxifen treatment, the time point at which ESRP1 expression started to decline more noticeably (Fig. 1B). Our ChIP studies revealed a 4.4-fold enrichment for Snail binding to this region compared with control (Fig. 2A). These results correlated well with our qRT-PCR data showing that the level of ESRP1 mRNA in tamoxifen-treated HMLE/Snail-ER cells decreased steadily as the cells transitioned to the mesenchymal state (Fig. 1B). As an additional negative control, ChIP assays were also conducted using the ER antibody and HMLE cells, which do not express the Snail-ER protein. As expected, we did not observe any enrichment at the ESRP1 promoter in HMLE cells (Fig. 2B), supporting that the pulldown detected using the ER antibody in HMLE/Snail-ER cells was specific to the Snail-ER protein. Collectively, these data indicate that Snail down-regulates ESRP1 during EMT by binding to E-boxes located within the ESRP1 promoter.
FIGURE 2.
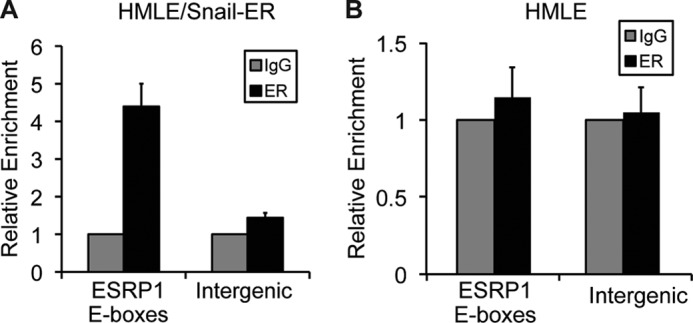
Snail regulates ESRP1 expression by binding to E-box sequences located in the ESRP1 promoter. A and B, quantitative ChIP analysis of the relative occupancy of Snail at the ESRP1 promoter in HMLE/Snail-ER cells treated with tamoxifen for 6 days (A) and in HMLE cells (B). As a negative control, enrichment at an amplicon located in an intergenic region was also assayed. Additionally, ChIP assays were performed with preimmune IgG. Bar graphs show averages of three independent ChIP experiments.
Snail Expression Alters the ESRP1-associated Splicing Signature
We and others have shown previously that ESRP1 regulates CD44 alternative splicing during EMT (12, 13). ESRP1 has also been shown to regulate the alternative splicing of a subset of genes that exhibit differential exon inclusion in epithelial versus mesenchymal cells (19). We hypothesized that if Snail represses ESRP1 expression, the alternative splicing pattern of genes regulated by ESRP1 should be affected upon Snail expression. Thus, we analyzed the alternative splicing of known targets of ESRP1 in HMLE cells that are mesenchymal because of stable expression of Snail (HMLE/Snail). As shown in Fig. 3A, overexpression of Snail in HMLE cells caused a 14.5-fold decrease in ESRP1 expression, further supporting our observation that Snail represses ESRP1. To assess the downstream effects of this decrease in ESRP1, we measured the inclusion-to-exclusion ratio of the alternatively spliced exon for each ESRP1-regulated gene in control versus Snail-overexpressing cells. If Snail represses ESRP1, ectopic expression of Snail should cause an increase in the inclusion-to-exclusion ratio for genes for which ESRP1 promotes exon exclusion. Similarly, Snail overexpression should promote a decrease in the inclusion-to-exclusion ratio for genes for which ESRP1 promotes exon inclusion. Quantitative RT-PCR analysis shows that Snail overexpression did indeed change the alternative splicing of several ESRP1-regulated genes. In two of the three genes for which ESRP1 promotes exon exclusion, overexpression of Snail caused an increase in the inclusion-to-exclusion ratio, indicating a reversion of the ESRP1 splicing signature. Similarly, in seven of the eight genes for which ESRP1 promotes exon inclusion, Snail expression resulted in a decrease in the inclusion-to-exclusion ratio. These results confirm that Snail represses ESRP1 expression, which in turn modulates the ESRP1-associated splicing signature (Fig. 3B).
FIGURE 3.

Snail expression alters the ESRP1-associated splicing signature. A, qRT-PCR analysis of ESRP1 levels in control HMLE cells versus HMLE cells that stably express Snail (HMLE/Snail). B, qRT-PCR analysis indicating that Snail overexpression in HMLE cells altered the ESRP1-associated splicing signature. qRT-PCR assays were conducted using primer pairs that specifically detect exon inclusion or exclusion for each of the 11 ESRP1-regulated genes indicated. Exon inclusion-to-exclusion ratios were calculated for each gene in control and Snail-overexpressing cells. Bar graphs indicate -fold change in the inclusion-to-exclusion ratio in Snail-overexpressing cells compared with control cells. Error bars indicate S.E.; n = 4.
ESRP1 Interacts with GU-rich Elements Located in the CD44 mRNA
The above data showed that the Snail-mediated decrease in ESRP1 expression alters splicing globally, highlighting the importance of ESRP1 in maintaining an epithelial phenotype. This prompted us to investigate the underlying mechanism by which ESRP1 modulates alternative splicing, focusing on CD44 as an example of a critical ESRP1 target gene. Carstens and co-workers have identified previously ESRP1 recognition sequences that contain UGG or GGU repeats (19, 20). Our analysis of CD44 variable exons and flanking intronic sequences identified putative ESRP1-binding sites located in introns downstream of CD44 variable exons. This is in accordance with the previous finding that ESRP1 binding to its recognition motif in an intron located downstream of an alternatively spliced exon promotes inclusion of that exon (19). In addition, a previous study identified intronic UGG repeats downstream of the CD44 v8 exon that promoted variable exon inclusion (21). However, the factors that interact with this element to confer an epithelial-specific splicing pattern remained unknown. To investigate whether ESRP1 directly interacts with the CD44 pre-mRNA, we therefore used the CD44 v8 exon as an example and investigated the role of the downstream intronic sequence that contains putative ESRP1-binding motifs, which we termed the CD44 I-8 (Fig. 4A). First, we examined whether ESRP1 promotes v8 inclusion by using a CD44 splicing reporter construct that contains the v8 exon and its flanking introns. After transfecting cells with this construct and harvesting RNA, qRT-PCR analysis was used to measure the amounts of v8 exon inclusion and exclusion. When co-transfecting 293 cells with ESRP1 and the CD44 minigene, we found that ESRP1 caused a 4.3-fold increase in the ratio of v8 inclusion to exclusion (Fig. 4, B and C), indicating that ESRP1 promotes v8 exon inclusion. These results are in agreement with our previous findings using a mouse CD44 v5 minigene construct (12), indicating that ESRP1 promotes inclusion of CD44 variable exons.
FIGURE 4.
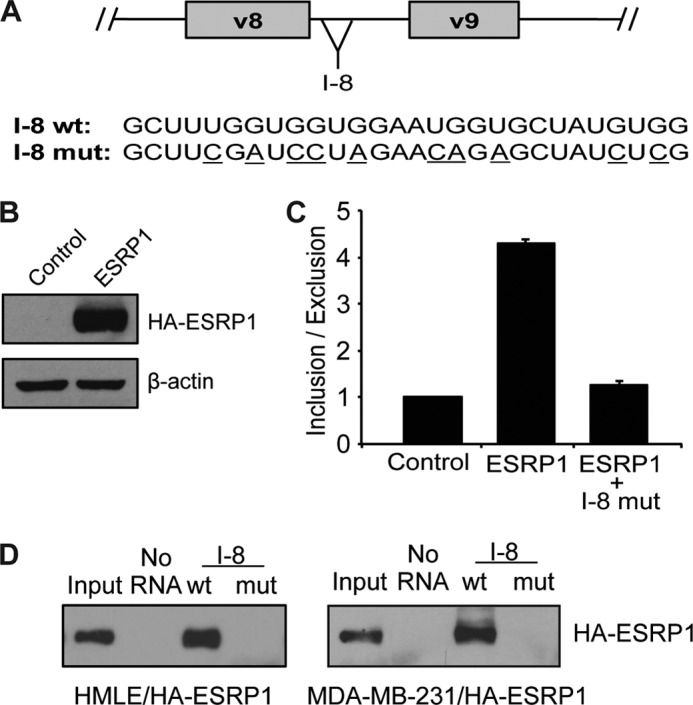
ESRP1 promotes CD44 exon inclusion by interacting with GU-rich elements in the CD44 pre-mRNA. A, schematic shows the location of the CD44 I-8 region. Sequences of the wild-type and mutated biotin-labeled RNA probes are shown; mutated nucleotides are underlined. B, immunoblot analysis shows overexpression of HA-tagged ESRP1 in 293 cells used in splicing reporter assays. C, quantitative RT-PCR analysis in 293 cells co-transfected with ESRP1 and a CD44v8 splicing reporter construct demonstrates that ESRP1-promoted inclusion of the CD44 v8 exon. When the putative ESRP1-binding motifs in the I-8 region are mutated, the ability of ESRP1 to promote inclusion of the CD44 v8 exon is diminished. _Bar graph_s depict averages of three independent experiments; error bars indicate S.E. D, immunoblot analysis using an HA antibody shows that ESRP1 interacted with the wild-type, but not the mutated, CD44 I-8 RNA probe (compare lane 3 with lane 4 in each panel). Assays were performed using whole cells lysates from HA-ESRP1-overexpressing HMLE cells (left panel) and HA-ESRP1-overexpressing MDA-MB-231 cells (right panel).
To investigate the importance of the putative ESRP1-binding motifs in the CD44 I-8 in promoting v8 exon inclusion, we next generated a CD44 v8 splicing reporter construct that contained several point mutations in the I-8 region that disrupted the UGG or GGU repeats (Fig. 4A). We hypothesized that these mutations would effectively disrupt the ESRP1-binding motifs, thus abolishing the ESRP1-mediated increase in v8 exon inclusion. Indeed, qRT-PCR analysis of v8 inclusion-to-exclusion ratios showed that mutating the CD44 I-8 inhibited the ability of ESRP1 to promote v8 inclusion (Fig. 4C); that is, when co-transfecting ESRP1 and the CD44 v8 splicing reporter construct containing I-8 mutations, ESRP1 was no longer able to promote an increase in v8 inclusion compared with the results obtained using the wild-type v8 splicing reporter construct. These results indicate that the GU-rich ESRP1-binding motifs located within the CD44 I-8 are required for ESRP1 to promote CD44 v8 exon inclusion.
To determine whether ESRP1 directly binds to the CD44 I-8 region, we next conducted RNA pulldown assays using wild-type or mutated biotin-labeled I-8 RNA probes. The sequence of the wild-type probe directly corresponded to a 28-nucleotide sequence located within the CD44 I-8 element, whereas the mutated probe contained the same point mutations that were present in the mutant splicing reporter construct (Fig. 4A). In pulldown assays using lysates from epithelial HMLE cells that overexpress HA-tagged ESRP1, we detected an interaction between ESRP1 and the wild-type, but not the mutated, CD44 I-8 probe (Fig. 4D, left panel, lane 3 compared with lane 4). These results were recapitulated in breast cancer MDA-MB-231 cells that overexpress HA-tagged ESRP1 (Fig. 4D, right panel, lane 3 compared with lane 4). Taken together with our results from the splicing reporter assays, these findings suggest that ESRP1 promotes CD44 v8 exon inclusion by directly binding to its consensus-binding motifs located in the intron downstream of this exon.
ESRP1 Overexpression Inhibits Snail-induced EMT by Regulating CD44 Alternative Splicing
Although it was well established that Snail induces EMT, it was not known whether ESRP1 down-regulation is required for Snail-mediated EMT. To this end, we ectopically expressed HA-tagged ESRP1 in HMLE/Snail-ER cells to determine whether ESRP1 overexpression impairs Snail-induced EMT (HA-ESRP1 expression is shown in the left two lanes in Fig. 5D, top panel). As shown in Fig. 5, A and B, following 14 days of tamoxifen treatment, control HMLE/Snail-ER cells underwent EMT. In contrast, ESRP1 overexpression prevented the cells from undergoing EMT, as indicated by the preservation of a high level of the epithelial marker E-cadherin, impaired up-regulation of the mesenchymal marker N-cadherin, and the maintenance of a cobblestone-like epithelial morphology (Fig. 5, A and B). These results indicate that ESRP1 expression prevents Snail-induced EMT.
FIGURE 5.
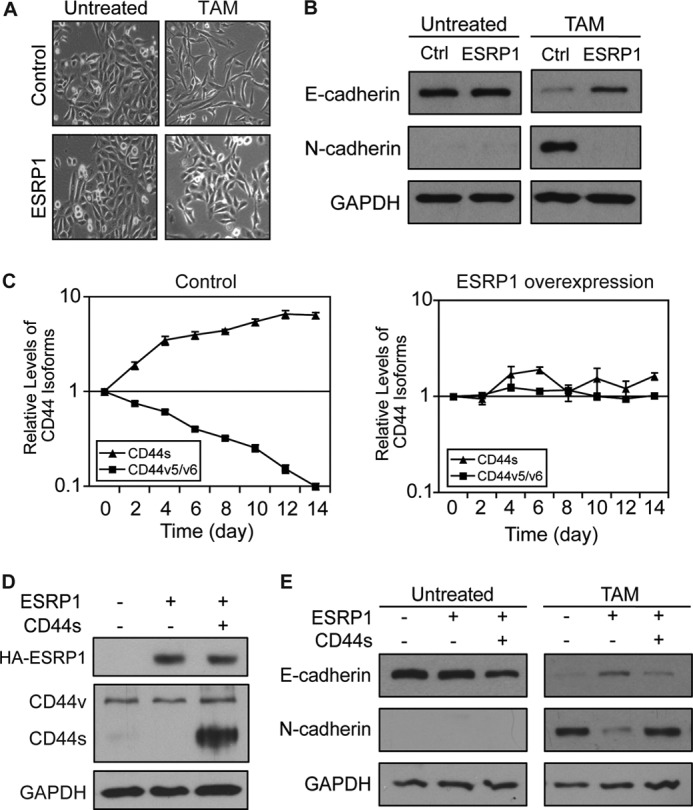
ESRP1 inhibits Snail-induced EMT by modulating CD44 alternative splicing. A, phase contrast images (10×) illustrate that ESRP1-overexpressing HMLE/Snail-ER cells exhibited impaired morphological changes following 14 days of tamoxifen (TAM) treatment compared with control cells. B, immunoblot analysis of E-cadherin and N-cadherin demonstrates that the control cells underwent a complete EMT, whereas the ESRP1-overexpressing cells maintained E-cadherin expression and failed to up-regulate N-cadherin following tamoxifen treatment. GAPDH served as a loading control. C, qRT-PCR analysis of mRNA levels of CD44 isoforms using primers that specifically detect either CD44s or CD44v that contain the v5 and v6 exons in TAM-treated control (left panel) or ESRP1-overexpressing (right panel) cells shows that ESRP1 overexpression prevents the CD44 isoform switch during EMT. Relative expression levels are normalized to TATA-binding protein at each time point, and the results are shown relative to day 0. Error bars indicate S.E.; n = 4. D, immunoblot analysis shows protein levels of HA-ESRP1 and CD44 isoforms in HMLE/Snail-ER cell lines that express ESRP1 or both ESRP1 and CD44s. GAPDH served as a loading control. E, immunoblot analysis of E-cadherin and N-cadherin shows that overexpression of the CD44s isoform in the ESRP1-overexpressing cells rescued the impaired EMT phenotype observed in the ESRP1-overexpressing cells. GAPDH served as a loading control.
We have shown previously that ESRP1 regulates CD44 alternative splicing and that the decline of ESRP1 expression during EMT facilitates the shift in isoform expression from CD44v to CD44s that is required for EMT to occur. Thus, we examined the levels of CD44 isoforms during Snail-induced EMT in both control and ESRP1-overexpressing HMLE/Snail-ER cells. As predicted, during the course of Snail-induced EMT, the control HMLE/Snail-ER cells exhibited a 10-fold decrease in CD44v and a 6.5-fold increase in CD44s (Fig. 5C, left panel). In contrast, the isoform switch from CD44v to CD44s did not occur in the ESRP1-overexpressing cells (Fig. 5C, right panel).
Given our findings that the isoform switch from CD44v to CD44s is required for EMT and that ESRP1 inhibits Snail-induced EMT, we next set out to determine whether CD44 is a major splicing target of ESRP1 during Snail-induced EMT. We hypothesized that ESRP1 exerts its inhibitory effect on Snail-mediated EMT by regulating CD44 alternative splicing and preventing the production of CD44s. If so, ectopic expression of the CD44s isoform in the ESRP1-overexpressing HMLE/Snail-ER cells (Fig. 5D, middle panel) should rescue the impaired EMT phenotype observed in the ESRP1-overexpressing cells. Indeed, as shown in Fig. 5E, EMT was no longer impaired in the cells in which CD44s had been re-expressed. Rather, these cells were able to down-regulate E-cadherin and up-regulate N-cadherin in a manner that was comparable with that seen in the control HMLE/Snail-ER cells. These results demonstrate that ESRP1 prevents Snail-induced EMT by inhibiting the isoform switch from CD44v to CD44s that is essential for the transition to the mesenchymal state.
In conclusion, this study has shown that the transcription repressor Snail interacts directly with E-boxes in the ESRP1 promoter, thus identifying the mechanism by which ESRP1 is repressed during Snail-induced EMT. Furthermore, we defined the downstream effects of ESRP1 down-regulation by showing that ESRP1 normally promotes inclusion of CD44 variable exons via an interaction with its consensus-binding motif in adjacent CD44 introns. Snail-mediated ESRP1 repression thus results in a decrease in the levels of CD44v and an increase in production of the CD44s isoform, which drives EMT. To confirm this sequence of events, we further showed that ectopic expression of ESRP1 inhibited Snail-induced EMT and that re-expression of the CD44s splice isoform in these cells rescued the impaired EMT phenotype. Our findings thus demonstrated that CD44 is a major splicing target downstream of ESRP1 during Snail-induced EMT. Furthermore, we showed that overexpression of Snail modulated the ESRP1-associated splicing signature, highlighting the fact that whereas CD44 is a critical target of ESRP1, the Snail-mediated decrease in ESRP1 expression alters splicing globally.
In this study, we also noted that the transcriptional repressor Twist does not directly repress ESRP1 promoter activity (Fig. 1D). Because we have shown previously that ESRP1 is also down-regulated during Twist-induced EMT (12), this raised the possibility that ESRP1 expression is regulated by different transcription factors during Twist-induced EMT. In accordance with this, a recent publication showed that Zeb1 and Zeb2 repress ESRP1 during TGF-β-induced EMT (22). Taken together with our results that Snail regulates ESRP1 promoter activity, these findings indicate that ESRP1 is regulated by different pathways that mediate EMT because of the critical role that ESRP1 plays in promoting the epithelial state.
The importance of splicing factors in carcinogenesis has been increasingly recognized in recent years. For example, the SFRS1 gene, which encodes the splicing factor SF2/ASF, was shown to be a proto-oncogene (23). SF2/ASF regulates the alternative splicing of several genes that are involved in transformation and apoptosis, including the tumor suppressor BIN1, the transcription factor TEF-1, and the kinases MNK2, S6K1, and RON (23, 24). Similarly, the splicing factor Sam68 has been shown to regulate Cyclin D1 alternative splicing, such that it promotes the production of the Cyclin D1b isoform, which exhibits higher oncogenic potential than the Cyclin D1a isoform (25). The growing body of evidence demonstrating a critical role for splicing factors in cancer progression stresses the importance of identifying such _trans_-acting factors, as well as characterizing the pathways that control their expression and activation. Our present study revealed how Snail facilitates EMT by regulating ESRP1 and identified ESRP1 as a critical factor in maintaining an epithelial phenotype and serving as a barrier to the transition to the mesenchymal state. Given that EMT is frequently re-activated during cancer metastasis and recurrence, defining the role of splicing factors such as ESRP1 in regulating EMT may advance our efforts to prevent these deadly aspects of cancer progression.
Acknowledgments
We thank Sendurai Mani, Alex Minella, and Timothy Chlon for reagents and materials; Lou Dore for technical assistance; and members in the Cheng laboratory and John Crispino for discussion and critical reading of this manuscript.
*
This work was supported, in whole or in part, by grants from the American Cancer Society, American Association for Cancer Research, the Department of Defense Breast Cancer Research Program, the Schweppe Foundation, and the Lynn Sage Foundation (to C. C.).
3
The abbreviations used are:
EMT
epithelial-mesenchymal transition
ESRP1
Epithelial Splicing Regulatory Protein 1
ER
estrogen receptor
qRT-PCR
quantitative reverse transcription-PCR.
REFERENCES
- 1.Moody S. E., Perez D., Pan T. C., Sarkisian C. J., Portocarrero C. P., Sterner C. J., Notorfrancesco K. L., Cardiff R. D., Chodosh L. A. (2005) The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8, 197–209 [DOI] [PubMed] [Google Scholar]
- 2.Thiery J. P., Sleeman J. P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 [DOI] [PubMed] [Google Scholar]
- 3.Yang J., Mani S. A., Donaher J. L., Ramaswamy S., Itzykson R. A., Come C., Savagner P., Gitelman I., Richardson A., Weinberg R. A. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 [DOI] [PubMed] [Google Scholar]
- 4.Yang J., Weinberg R. A. (2008) Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829 [DOI] [PubMed] [Google Scholar]
- 5.Batlle E., Sancho E., Francí C., Domínguez D., Monfar M., Baulida J., García De Herreros A. (2000) The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat. Cell Biol. 2, 84–89 [DOI] [PubMed] [Google Scholar]
- 6.Cano A., Pérez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., Nieto M. A. (2000) The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 [DOI] [PubMed] [Google Scholar]
- 7.Miettinen P. J., Ebner R., Lopez A. R., Derynck R. (1994) TGF-β-induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J. Cell Biol. 127, 2021–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brickell P. M., Latchman D. S., Murphy D., Willison K., Rigby P. W. (1983) Activation of a Qa/Tla class I major histocompatibility antigen gene is a general feature of oncogenesis in the mouse. Nature 306, 756–760 [DOI] [PubMed] [Google Scholar]
- 9.Sharma S., Lichtenstein A. (2009) Aberrant splicing of the E-cadherin transcript is a novel mechanism of gene silencing in chronic lymphocytic leukemia cells. Blood 114, 4179–4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shtivelman E., Lifshitz B., Gale R. P., Roe B. A., Canaani E. (1986) Alternative splicing of RNAs transcribed from the human ABL gene and from the BCR-ABL-fused gene. Cell 47, 277–284 [DOI] [PubMed] [Google Scholar]
- 11.Venables J. P. (2006) Unbalanced alternative splicing and its significance in cancer. Bioessays 28, 378–386 [DOI] [PubMed] [Google Scholar]
- 12.Brown R. L., Reinke L. M., Damerow M. S., Perez D., Chodosh L. A., Yang J., Cheng C. (2011) CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J. Clin. Invest. 121, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warzecha C. C., Sato T. K., Nabet B., Hogenesch J. B., Carstens R. P. (2009) ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cel. 33, 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Brooks M., Reinhard F., Zhang C. C., Shipitsin M., Campbell L. L., Polyak K., Brisken C., Yang J., Weinberg R. A. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia R., Liu X., Tao M., Kruhlak M., Guo M., Meyers C., Baker C. C., Zheng Z. M. (2009) Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J. Virol. 83, 167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dore L. C., Amigo J. D., Dos Santos C. O., Zhang Z., Gai X., Tobias J. W., Yu D., Klein A. M., Dorman C., Wu W., Hardison R. C., Paw B. H., Weiss M. J. (2008) A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 105, 3333–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mauhin V., Lutz Y., Dennefeld C., Alberga A. (1993) Definition of the DNA-binding site repertoire for the Drosophila transcription factor Snail. Nucleic Acids Res. 21, 3951–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakayama H., Scott I. C., Cross J. C. (1998) The transition to endoreduplication in trophoblast giant cells is regulated by the mSNA zinc finger transcription factor. Dev. Biol. 199, 150–163 [DOI] [PubMed] [Google Scholar]
- 19.Warzecha C. C., Jiang P., Amirikian K., Dittmar K. A., Lu H., Shen S., Guo W., Xing Y., Carstens R. P. (2010) An ESRP-regulated splicing program is abrogated during the epithelial-mesenchymal transition. EMBO J. 29, 3286–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmar K. A., Jiang P., Park J. W., Amirikian K., Wan J., Shen S., Xing Y., Carstens R. P. (2012) Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high throughput sequencing. Mol. Cell. Biol. 32, 1468–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galiana-Arnoux D., Del Gatto-Konczak F., Gesnel M. C., Breathnach R. (2005) Intronic UGG repeats coordinate splicing of CD44 alternative exons v8 and v9. Biochem. Biophys. Res. Commun. 336, 667–673 [DOI] [PubMed] [Google Scholar]
- 22.Horiguchi K., Sakamoto K., Koinuma D., Semba K., Inoue A., Inoue S., Fujii H., Yamaguchi A., Miyazawa K., Miyazono K., Saitoh M. (2012) TGF-β drives epithelial-mesenchymal transition through δEF1-mediated down-regulation of ESRP. Oncogene 31, 3190–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karni R., de Stanchina E., Lowe S. W., Sinha R., Mu D., Krainer A. R. (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 14, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghigna C., Giordano S., Shen H., Benvenuto F., Castiglioni F., Comoglio P. M., Green M. R., Riva S., Biamonti G. (2005) Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 20, 881–890 [DOI] [PubMed] [Google Scholar]
- 25.Paronetto M. P., Cappellari M., Busà R., Pedrotti S., Vitali R., Comstock C., Hyslop T., Knudsen K. E., Sette C. (2010) Alternative splicing of the Cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res. 70, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]