Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene (original) (raw)
Abstract
The human fragile X mental retardation 1 (FMR1) gene contains a (CGG)n trinucleotide repeat in its 5′ untranslated region (5′UTR). Expansions of this repeat result in a number of clinical disorders with distinct molecular pathologies, including fragile X syndrome (FXS; full mutation range, greater than 200 CGG repeats) and fragile X–associated tremor/ataxia syndrome (FXTAS; premutation range, 55–200 repeats). Study of these diseases has been limited by an inability to sequence expanded CGG repeats, particularly in the full mutation range, with existing DNA sequencing technologies. Single-molecule, real-time (SMRT) sequencing provides an approach to sequencing that is fundamentally different from other “next-generation” sequencing platforms, and is well suited for long, repetitive DNA sequences. We report the first sequence data for expanded CGG-repeat FMR1 alleles in the full mutation range that reveal the confounding effects of CGG-repeat tracts on both cloning and PCR. A unique feature of SMRT sequencing is its ability to yield real-time information on the rates of nucleoside addition by the tethered DNA polymerase; for the CGG-repeat alleles, we find a strand-specific effect of CGG-repeat DNA on the interpulse distance. This kinetic signature reveals a novel aspect of the repeat element; namely, that the particular G bias within the CGG/CCG-repeat element influences polymerase activity in a manner that extends beyond simple nearest-neighbor effects. These observations provide a baseline for future kinetic studies of repeat elements, as well as for studies of epigenetic and other chemical modifications thereof.
The human fragile X mental retardation 1 (FMR1) gene contains a (CGG)n trinucleotide repeat that is responsible for a number of heritable disorders affecting both early neurodevelopment and late-onset neurodegeneration (Willemsen et al. 2011; Leehey and Hagerman 2012). The repeat element is located in the 5′ untranslated region (5′UTR) of the gene and is thus transcribed into mRNA but not translated into the FMR1 protein product (FMRP). Expanded alleles in the premutation range (55–200 CGG repeats) result in elevated FMR1 mRNA expression (Tassone et al. 2000) and are associated with a number of disorders including the adult-onset neurodegenerative disorder, fragile X–associated tremor/ataxia syndrome (FXTAS) (Leehey and Hagerman 2012), fragile X–associated premature ovarian insufficiency (FXPOI) (Wittenberger et al. 2007; Sullivan et al. 2011), as well as learning disabilities, autism spectrum disorders, ADHD, and seizures (Farzin et al. 2006; Clifford et al. 2007; Chonchaiya et al. 2012). The molecular pathology of premutation expansion disorders is generally considered to be a toxic RNA gain of function resulting from the expanded CGG-repeat region in the mRNA (Garcia-Arocena and Hagerman 2010; Ross-Inta et al. 2010; Sellier et al. 2010). Alleles in this range also show a propensity to expand beyond 200 repeats (full mutation range) upon maternal transmission, in which case the FMR1 CpG-island promoter generally becomes hypermethylated and transcriptionally silenced (Willemsen et al. 2011). The resultant loss of FMRP expression disrupts early neurodevelopment and leads to fragile X syndrome (FXS), the most common heritable form of cognitive impairment and the most common single-gene mutation associated with autism (Willemsen et al. 2011; Hagerman et al. 2012).
CGG-repeat expansions have been the focus of intense research since identification of the gene in 1991 (Verkerk et al. 1991); however, the inability to sequence repeat-expansion alleles in the disease-relevant size range has limited their complete genetic and epigenetic characterization. Indeed, investigators of the original gene-discovery study noted their inability to sequence the CGG repeats, and other early attempts to use sequencing to characterize the repeats describe the inability to fully traverse the region (Hornstra et al. 1993). Whereas PCR and Southern blotting are capable of genotyping repeat expansion alleles on the basis of DNA fragment size (Nolin et al. 2003; Saluto et al. 2005; Filipovic-Sadic et al. 2010), and even identify methylation status and AGG-repeat interruptions (Chen et al. 2010, 2011; Yrigollen et al. 2012), such methods lack the single-nucleotide resolution obtained with DNA sequencing and, more importantly, are severely limited in their ability to detect the presence of minor alleles. Furthermore, because dideoxyribose sequencing strategies (Sanger et al. 1977) and most “next-generation” sequencing technologies (Metzker 2010) rely on reading signal from bulk DNA populations, they are limited by the loss of sequence phase coherence—a particular problem for GC-rich sequence—as well as decreasing size resolution with increasing DNA length. As a consequence, it is generally not possible to sequence FMR1 alleles in excess of ∼100 CGG repeats, a limit that falls well short of the full mutation range that is responsible for fragile X syndrome.
A fundamentally different sequencing approach, single-molecule, real-time (SMRT) sequencing, uses zero-mode waveguide (ZMW) nanowells to determine DNA sequence from individual DNA templates (Fig. 1; Eid et al. 2009). This is accomplished through real-time observation of individual nucleotide incorporation events catalyzed by a single DNA polymerase. This approach bypasses critical limitations of previous technologies in the context of highly repetitive sequences such as trinucleotide expansions. In particular, measurement of the signal from isolated molecules overcomes the problems of sample heterogeneity (phase-coherence) and diminishing resolution inherent in bulk sequencing approaches. Since the SMRT sequencing reads are limited only by loss of activity of individual polymerase molecules, single-molecule readlengths approaching 15 kb (average readlengths approaching 3 kb) (Rasko et al. 2011; Sebra et al. 2012) can be attained, with improved sequence accuracy achieved by iteratively sequencing the same SMRTbell circular sequencing template (circular consensus sequencing [CCS]) (Fig. 1; Travers et al. 2010).
Figure 1.

Schematic representation of SMRT sequencing. DNA polymerase synthesizes a nascent strand complementary to a closed-circular SMRTbell DNA template. Fluorescent phospholinked nucleotides produce real-time fluorescent pulse data for both basecalls and incorporation kinetics. Inter-pulse distance (IPD) is defined as the time gap between two consecutive pulses and includes the time taken to move the DNA polymerase between base positions on the DNA template. Pulse width (PW) is the duration of a given pulse and represents the residence time of the nucleotide at the active site of DNA polymerase, up to the point of incorporation and cleavage of the fluorescent tag. Closed SMRTbell sequencing templates yield multiple, overlapping subreads of both forward and reverse strands of the insert, which are then assembled in silico with the primary consensus core algorithm into circular consensus sequence (CCS), eliminating randomly distributed pulse-read sequencing errors (Supplemental Table S1). Note also that although a single enzymatic misincorporation is indicated for completeness, such errors occur during polymerization with a frequency that is several orders of magnitude less than photonic miscall errors and therefore do not contribute to the error profile.
By utilizing the SMRT sequencing approach for the analysis of the CGG-repeat region of the FMR1 gene, we have demonstrated that it is possible to generate sequence data for FMR1 alleles in excess of 750 CGG repeats, which translates to over 2.25 kb of 100% CGG-repeat DNA. We show that this method produces repeat-size distributions reflective of the distributions expected from the input (e.g., cloned or PCR-amplified) DNA. We also demonstrate that by using CCS reads, we are able to identify AGG “interruptions” within the CGG-repeat tract that are of direct medical relevance (Yrigollen et al. 2012). Our approach should be broadly applicable to the analysis of other repetitive elements, particularly those containing high CG content and possessing short homopolymer runs (e.g., the GGGGCC motif in 9p-linked amyotrophic lateral sclerosis-frontal temporal dementia [ALS-FTLD]) (DeJesus-Hernandez et al. 2011; Renton et al. 2011). Finally, SMRT sequencing possesses the unique capability of analyzing the kinetics of individual DNA polymerase molecules, and we show clear, strand-specific transitions within the CGG-repeat region, which should facilitate future studies of differential methylation.
Results
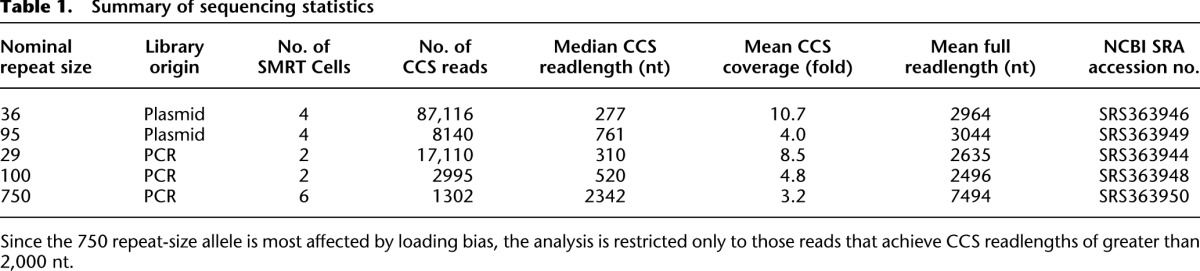
To examine the ability of SMRT technology in sequencing trinucleotide repeats, we created and sequenced SMRTbell libraries of FMR1 DNA directly from plasmids harboring alleles of nominally 36 and 95 CGG repeats, and by PCR-amplification from genomic DNAs, with nominally 29, 100, and 750 CGG repeats (Table 1). Each library was sequenced at least in duplicate, with the number of sequencing runs indicated as the number of SMRT cells. The following analyses were run on the pooled data across all sequencing runs, filtered for quality. Overlay of size distributions produced by each of four individual SMRT cells shows little variability across sequencing replicates of the same library (Supplemental Fig. S1).
Table 1.
Summary of sequencing statistics
SMRT sequencing of short CGG repeats
CCS reads of the 36 CGG-repeat plasmid show clear subpopulations of 34, 35, and 36 repeats (Fig. 2A). Comparing the size distribution of the flanking regions to the repeat regions (Fig. 2B) indicates that length heterogeneity originates entirely from the repeat region. We used alignment to a 36 CGG FMR1 reference sequence to determine the accuracy of each single-molecule consensus read. As previously demonstrated, increasing the number of intramolecular subreads—that is, the number of subreads used to assemble a single CCS read—results in higher accuracy of the resulting sequence (Fig. 2C; Travers et al. 2010). Relative to the high accuracy achieved in the flanking regions, the lower apparent accuracy for the repeat region is in part an artifact of using a single reference (Fig. 2C). By comparing the accuracy achieved using a single reference to the accuracy resulting from using a reference whose repeat section length is matched per molecule, a 3%–4% improvement in the measured accuracy of the repeat section can be achieved (Supplemental Fig. S2).
Figure 2.

SMRT sequencing of short CGG repeats. (A) Sequence alignment of representative reads from a library of plasmid-derived FMR1 sequence with nominally 36 CGG repeats. Three major CGG-repeat size species are observed. Flanking and CGG-repeat regions are delineated by vertical tick marks. (B) Frequency of sequence lengths in the top 1000 reads (by predicted quality) plotted by region as indicated. Three major peaks observed in the repeats (red) correspond to 34, 35, and 36 repeats as seen in A. Both the left (green broken line) and right (blue) flanking sequence regions are uniform. (C) Accuracy by alignment to reference of each region of the insert increases with each successive pass of consensus coverage, saturating after four subreads for the flanking regions. Accuracy of the reads within the CGG-repeat region has improved through the use of reference sequences corresponding to the individual lengths within the distribution (see Supplemental Fig. S2).
SMRT sequencing of mid-size CGG repeats
CCS reads of the 95 CGG-repeat plasmid reflect the biology underlying the library construction with sequenced populations of full-length 95 CGG repeats, as well as smaller molecular species resulting from deletion of the repeats in Escherichia coli (Fig. 3A). A close-up comparison of the flanking sequence to the repeat sequence shows an increase in sporadic single-base insertions and deletions, mostly within the CGG-repeat region (Fig. 3B). As with the 36 CGG sequence, length heterogeneity originates entirely from the repeat region (Fig. 3C). Despite the sample heterogeneity, we are able to clearly detect the presence of a single AGG polymorphism 15 repeats into the CGG region; this observation was independently confirmed by Sanger sequencing (Supplemental Fig. S3).
Figure 3.

SMRT sequencing of a mid-premutation CGG-repeat expansion (approximately 95 CGG repeats). (A) Sequence alignment of representative CCS reads from a library of plasmid-generated FMR1 sequence; note that the original construct was generated from PCR-amplified genomic DNA followed by bacterial clonal selection. Sporadic single-base additions and deletions result from comparatively lower CCS coverage than the smaller repeat library; upper and lower sets represent sample CCS reads from the main peak at ∼280 nucleotides and from the smaller, broad distribution, respectively; horizontal lines indicate the CGG-repeat regions. (B) Expanded view of the transition from flanking sequence into the CGG repeats. An AGG repeat (boxed) is unambiguously recognized in all reads, demonstrating the utility in genotyping polymorphic CGG-repeat interruptions. (C) Frequency distribution of sequence lengths in the top 1000 reads plotted by region. A major peak is observed in the repeats (red), with minor peaks generally corresponding to units of single repeats, and a spread of shorter fragments produced by bacterial deletion of the CGG repeats. Both left (green broken line) and right (blue) flanking sequence regions are uniform.
SMRT sequencing of discrete CGG-repeat alleles within full-mutation allelic distributions
To determine if our approach would have the ability to sequence alleles that are well into the full-mutation range, we sequenced a library from a PCR-amplified allele, size-genotyped at ∼750 CGG repeats (well beyond what can be cloned in bacteria) (Fig. 4). We generated more than 1000 CCS reads >2 kb in length (Fig. 4B; Table 1), each assembled from raw readlengths in excess of 6 kb, containing at least three passes through over 2000 bases of 100% GC repetitive sequence. The observed distribution of loading-bias–corrected readlengths distribution (Supplemental Fig. S4; Supplemental Table S2) has a mode of 2425 bp, equivalent to 720 CGG repeats plus flanking sequence (Fig. 4B). This CGG-repeat mean differs from the gel electrophoresis–sizing of the PCR fragment used to make the library, although the difference falls well within the 5%–10% tolerance limits for such size measurements.
Figure 4.

SMRT sequencing of a full mutation allele of (nominally) 750 CGG repeats. (A) Representative CCS sequences from a library of PCR-amplified FMR1 genomic DNA. These reads are the result of reading through >2 kb of CGG repeats at least three times. (B) Size-corrected distribution of sequence lengths in the sequenced library plotted by region. PCR amplification creates a broad distribution of repeat sizes with a mode at 720 repeats.
The lower accuracy of the sequence of this library (Fig. 4A) is a result of several factors. First, the average number of passes that contribute to each consensus read for the 750 CGG library is less than for the shorter libraries (Figs. 2, 3) due to a much larger insert size dividing into the same average raw readlength distribution (Table 1). Specifically, the 10.7 average number of passes obtained from the 36-repeat plasmid sample is expected to produce a mean accuracy of >99% (Supplemental Table S3), whereas the average of only 3.2 passes for the 750-repeat sample would translate into a mean accuracy of ∼98% for the flanking sequence and ∼92% for the repeats. Second, since this repeat expansion size has the widest distribution of lengths, comparison to a single reference will yield greater alignment discrepancies for this sample. Finally, these observations also underscore the fact that PCR is in general—and in particular for repetitive sequence—prone to far more replication errors than those introduced in vivo by bacterial DNA replication (Fig. 5B,C).
Figure 5.

Analysis of the SD in read length for CGG-repeat-containing FMR1 alleles. (A) Mean SD comparison between inter- and intramolecular subreads for the two plasmid- (underlined tick labels) and PCR-generated libraries. Higher intermolecular (versus intramolecular) SDs are apparent only for the repeat region, consistent with the presence of complex populations of repeat sizes. (B,C) Comparisons of CGG-repeat size distributions for plasmid- and PCR-generated CGG-repeat–containing DNA for (B) normal (∼30 CGG repeats) and (C) premutation (∼100 CGG repeats). Broader distributions for PCR-generated fragments reflect errors associated with PCR amplification of CGG-repeat elements.
Quantifying the readlength heterogeneity
To further bolster the evidence that indeed the majority of the readlength heterogeneity is contributed by the CGG-repeat region and to determine if this heterogeneity is due to an underlying population heterogeneity, we compared the standard deviation of subread lengths within the same CCS read (intramolecular) to the standard deviation of subread lengths sampled across different CCS reads (intermolecular) (Fig. 5A). Consistent with an intrinsic sample heterogeneity that is localized to the repeat region, the standard deviation of that section, calculated across molecules (intermolecular) shows the largest values, especially for larger allele sizes (nominally 95 and 100 repeats). The subreads from two examples of CCS reads (95 CGG-repeat sample) illustrate the general intramolecular agreement relative to intermolecular readlength diversity (Supplemental Fig. S5).
Comparison of the CGG-repeat readlength distributions for PCR- and plasmid-derived libraries at 30 (Fig. 5B) and 100 (Fig. 5C) CGG repeats clearly reflects the biology underlying the source of each library. PCR-generated DNA produces a broader size distribution than plasmid-cloned DNA for both size ranges (Fig. 5B,C).
Polymerase kinetics within the CGG-repeat region reveal strand-specific and position-specific differences in polymerization rates
By measuring rates of individual nucleoside–polymerase association events during the process of DNA synthesis within ZMWs, our data reveal strand-specific differences in the rates of polymerization both within the CGG-repeat region and in the region of transition between the flanking sequence and the repeats (Fig. 6). The principal outcome measure for the time-domain measurements is the interpulse distance (IPD), which represents the time delay from a nucleotide incorporation event (loss of fluorescent probe from the nucleoside) and the association of the next (incoming) nucleotide triphosphate (Fig. 1). The other outcome measure is the pulse width (PW), which reflects the dwell time of the fluorescently labeled nucleoside prior to its incorporation. Such SMRT kinetic measurements have been described previously as reporting on secondary structure and base modification (Eid et al. 2009; Flusberg et al. 2010). For the kinetics analysis, we used SMRTbell templates generated from the 36 and 95 CGG-repeat plasmid DNAs; these species permitted a sufficient number of passes to provide well-defined repeat size distributions and statistics for the IPDs for each base position.
Figure 6.

Time-domain analysis with mean IPD values faceted by base, colored according to the strand being synthesized (CGG, red; GCC, blue), and synchronized by aligned template position. (Error bars) SD from 200 reads. Vertical black lines demarcate the start and end of the repeat region per sample. The IPD, as illustrated in Figure 1, is the time interval from the end of the previous incorporation pulse to the start of the current incorporation pulse. (A,C) 36-mer sample shows an increased G IPD inside the repeat region only for the GCC strand. (B,D) 95-mer sample with the same increased G IPD only for the GCC strand, with a dip localized to an AGG interruption (arrow). (C,D) Expanded view of the start of the repeat region for both 36-mer (C) and 95-mer (D) reveals that the IPD increase begins at the fourth CGG repeat.
Analysis of the IPD data for the normal alleles and mid-range premutation alleles reveals several distinct features. First, the variation in IPDs within the flanking sequences, even within short CGG-repeat or G-stretches, is much broader than within the CGG-repeat region itself, which is a reflection of the previously described sequence context effect on polymerase kinetics (Fig. 6A,B; Flusberg et al. 2010). Second, within the CGG-repeat region itself, there is an increase in the IPD prior to G incorporations (when using the CGG strand as a template) that commences at the fourth repeat from the flank in both short (Fig. 6A) and mid-size (Fig. 6B) CGG repeats. This observation likely reflects a context-dependent local DNA conformation experienced by the polymerase as it moves into the CGG-repeat region; the upward IPD shift at the fourth repeat is consistent with the previously observed kinetic footprint of the enzyme (Flusberg et al. 2010). There is no corresponding shift in G incorporations in the CGG read (synthesizing) sequence (GCC template). Finally, there is a slight diminution of the IPD for the second C residue following a G incorporation in each trinucleotide repeat of the synthesized GCC strand (i.e., CGG template strand).
Interestingly, while the G IPD shift is uniform within the 36 CGG-repeat sample, the 95 CGG sample shows two perturbations: a perturbation in both C (increased IPDs) and G (reduced IPDs) kinetics that colocalizes with the previously described AGG polymorphism (Fig. 6B upward arrow), and an increased C IPD at the location of a TGG variant. Further analysis is required to determine what role these perturbations might play in the molecular mechanisms underlying the described effect of AGG interruptions on CGG-repeat expansion (Yrigollen et al. 2012), since the current kinetics are set within an artificial context.
Discussion
We demonstrate the ability of SMRT sequencing technology to sequence expanded CGG repeats of the FMR1 gene, including the first demonstration of sequencing CGG-repeat elements extending well into the full mutation repeat range. This sequencing methodology does not appear to be adversely influenced by CGG-repeat expansions that exceed 750 repeats (>2 kb of 100% CG), suggesting that the upper limit of productive sequence is limited only by those factors that govern the productive lifetime of the polymerase, itself a function of light intensity and other factors within the ZMWs, and the desired number of subreads within individual CCSs (Rasko et al. 2011; Sebra et al. 2012). These results demonstrate that FMR1 alleles can be readily sequenced through the threshold region separating premutation and full mutation alleles (∼200 CGG repeats), and well into the full mutation region that gives rise to FXS through methylation-coupled gene silencing. More broadly, the current work serves as a proof of concept for the study of additional repeat expansions that are associated with other diseases, such as myotonic dystrophy (Lee and Cooper 2009; Sicot et al. 2011), Huntington's disease (Reiner et al. 2011), Friedreich's ataxia (Koeppen 2011), and the recently discovered hexanucleotide (GGGGCC) repeat disorder ALS-FTLD (Braida et al. 2010; Hannan 2010; DeJesus-Hernandez et al. 2011).
Accuracy determines the utility of sequence data in a manner that depends on the goal of a particular study (e.g., trinucleotide-expansion genotyping versus bisulfite mapping of methylation events). CCS reads produce high-accuracy consensus sequences; however, if there is repeat-size heterogeneity, then comparisons of CCS reads to a single reference will give the appearance of lower accuracy within the repeat region that does not match the intermolecular consistency of the flanking regions. Errors resulting from PCR amplification or plasmid replication used to generate our libraries lead to accurate sequence that would appear erroneous in comparison to an idealized reference sequence. Additionally, dispersion in the exact repeat number within each library complicates intermolecular alignments. The superior intramolecular versus intermolecular subread agreement (Fig. 5A) supports the conclusion that CCS reads are more accurate than is apparent in the estimates of accuracy based on agreement to a single reference. It is especially important to note that the sensitivity of this sequencing approach to substitution variants (Fig. 3B), and the ability to penetrate long-repeat regions, will also facilitate AGG-interruption genotyping and the identification of other sequence variants deep within this repeat region and others (Braida et al. 2010).
The accuracy observed in the sequence flanking the CGG-repeat region (Fig. 2C) at threefold and greater single-molecule coverage produces sufficiently accurate sequence (median approaching 100%) to make variant determinations, and we are able to achieve this level of coverage with even the largest CGG repeat in this study (Fig. 4). We demonstrate the ability to sequence through very large repeat expansions, but mechanistic studies are likely to focus around the ∼200 CGG-repeat threshold for promoter methylation and gene silencing. For CGG-repeat alleles around this threshold, one would need a raw readlength of ∼2.4 kb, which is shorter than the average raw readlength reported here (Table 1) and is thus easily attained in future studies to map methylation patterns at the promoter and inside the CGG repeats.
The different repeat-size distributions observed for different methods (plasmid versus PCR) to generate the DNA for sequencing libraries (Fig. 5B,C) demonstrates the utility of the SMRT sequencing platform to characterize the size distribution of the input DNA samples. This issue is of particular importance for those genetic elements where there is a high degree of allele-size mosaicism with multiple alleles present in individual males (Nolin et al. 1994), or where the complexity of the sequence is very low (e.g., microsatellite regions). Our observations also underscore the need for better methods for initial generation of DNA species for downstream functional studies, that is, for highly repetitive sequence elements such as the CGG-repeat alleles studied herein. Errors introduced through PCR and/or the limitations of cloning repetitive sequences obscure the original population of size species. To define true size distributions from patients, we will need to sequence chromosomal DNA directly, which presents a separate set of technical hurdles. Otherwise, this technology is immediately useful in validating exogenous CGG model constructs (i.e., expression plasmids or transgenic animal models) and in several other PCR-based methods previously not possible, like chemical footprinting inside the CGG repeats. Additionally, the use of unique-sequence identifiers (“barcodes”) and sample multiplexing could eventually allow for high-throughput genotyping for population screening at a fraction of the current cost.
Passive loading of the SMRT cell results in a selection bias for the loading of smaller alleles into the ZMWs and, consequently, a length bias of the counts of alleles (Supplemental Fig. S3). To address this bias in the short term, we have used equimolar mixtures of size standards as a means for normalization of broad allele distributions; a longer-term solution, currently in development, would involve an active loading approach that significantly reduces this bias. This issue is important both for improving counts and sequence information for the longest alleles in a size distribution (e.g., mosaicism) and for accurate estimates for allele-size distributions.
In generating sequence data for large CGG-repeat expansions, we have simultaneously produced the first single-molecule DNA polymerase kinetic data within this repeat sequence. This time-domain (IPD) information reports on the DNA conformational landscape experienced by the polymerase as it traverses the sequence, albeit in a manner that we have not analyzed, and is clearly sensitive both to local and more regional sequence elements (e.g., Fig. 6C,D). In particular, we have observed that IPD increases at positions of G incorporation that coincide with the template C residue of the CGG template strand and that the increases are independent of the length of the CGG-repeat region. Moreover, the relative constancy of the IPD within the repeat region relative to the broad variation of IPD in the flanking regions is further evidence of the previously described context effect on polymerase kinetics (Flusberg et al. 2010). These observations thus establish a clear foundation for the detection of epigenetic modifications within the CGG-repeat region, which has not been possible at the nucleotide level for full mutation alleles that are epigenetically silenced in FXS. It is also possible that the kinetic differences between the two strands of the CGG-repeat sequence are related to the poorly understood mechanism of repeat instability (Yrigollen et al. 2012), especially in light of the observed drop in IPD at the position of an AGG-repeat polymorphism.
In summary, we have presented a novel approach to the characterization of expanded CGG-repeat alleles of the FMR1 gene, utilizing a new sequencing technology (SMRT sequencing) that is not intrinsically limited by tracts of 100% CG repetitive DNA that exceed 2 kb. This sequencing methodology is applicable to many other sequence elements of low complexity or expanded-repeat elements, many of which are associated with neurodegenerative disorders for expanded-repeat alleles. With the development of more advanced library preparation methods, this approach is expected to fill a critical need for screening large populations for expanded-repeat alleles—in the current instance, for expanded CGG-repeat alleles of the FMR1 gene that are associated with neurodevelopmental, reproductive, and neurodegenerative disorders. Research in the 20 years since the discovery of the FMR1 gene has been limited by the overall intractability of expanded CGG repeats to otherwise standard molecular techniques (PCR, cloning, sequencing). SMRT sequencing, as we have applied here, finally allows the potential for a complete genetic and epigenetic characterization of this, and other repeat-expansion/microsatellite genes, through massively parallel base-resolution sequencing studies.
Methods
Generation of subcloned fragments
FMR1 sequence containing 36 CGG repeats was cloned using pSmart (Lucigen); the FMR1 sequence containing 95 CGG repeats was cloned according to the method described previously (Chen et al. 2003). Fragments containing the repeats and flanking FMR1 sequence were digested with BlpI and NheI (36 CGG) and PstI (95 CGG), gel-purified (0.8% Agarose), post-stained with SYBR Gold (Life Technologies Corporation), and further purified using QIAQuick Gel Extraction columns (Qiagen).
Generation of PCR fragments
Genomic DNA containing FMR1 alleles with either 29, 100, or ∼750 CGG repeats was harvested from cultured, human dermal fibroblasts. Individual FMR1 alleles were amplified with AmplideX FMR1 PCR reagents (Asuragen) using primers identical to those specified in the commercial kit but lacking a fluorescent tag. The 750 CGG-repeat allele was Agarose gel-purified (0.8% Agarose) as described for cloned fragments.
Construction and sequencing of SMRTbell libraries
SMRTbell sequencing libraries were constructed, as previously described (Travers et al. 2010), using the DNA Template Prep Kit 1.0 (Pacific Biosciences). Purified, closed circular SMRTbell libraries were annealed with an RNA sequencing primer complementary to a portion of the single-stranded region of the hairpin and with the sequence 5′-AmAmCmGmGmAmGmGmAmGGAGGA-3′; mN denotes a 2′OMe modification to the ribose backbone to enhance primer stability. The size-standard ladder (see Supplemental Methods) was formed by separately annealing equimolar amounts of each SMRTbell template (50 nM each, 200 bp, 650 bp, 1 kb, 2 kb) with a 20-fold molar excess (1 μM) of the sequencing primer. For all other SMRTbell libraries, annealing was performed at a final template concentration between 40 and 60 nM, with a 20-fold molar excess of sequencing primer. All annealing reactions were carried out for 2 min at 80°C, with a slow cool to 25°C. Annealed templates were stored at −20°C until polymerase binding.
DNA polymerase enzymes were stably bound to the primed sites of the annealed SMRTbell templates using the DNA Polymerase Binding Kit (Pacific Biosciences). SMRTbell templates (3 nM) were incubated with 9 nM of polymerase in the presence of phospholinked (Pacific Biosciences) nucleotides for 4 h at 30°C. Following incubation, samples were stored at 4°C. Sequencing was performed within 36 h of binding. Each sample was sequenced as previously described (Rasko et al. 2011) using commercial sequencing chemistry. Sequencing data collection was performed on the PacBio RS (Pacific Biosciences) for 75 min in each case.
Readlength histogram analysis
The standard output from a PacBio RS run includes both a raw FASTQ file and a CCS, FASTQ file. The raw FASTQ file contains all of the photonic events judged to be bases during data acquisition for every ZMW (75k per movie). The CCS FASTQ file, on the other hand, contains the subset of the raw reads that achieve at least four hits to the adaptor sequence that comprises the two ends of the SMRTbell template (Fig. 1). For each such read, the adaptors are stripped and the best consensus sequence is constructed from the resulting set of repeated passes through the insert region of the SMRTbell (both the forward and reverse strands are used) (see Supplemental Fig. S4). The predicted per-base quality values, QVb, contained in the CCS FASTQ file are used to create a single, per-CCS-read accuracy by converting to a linear scale and taking the average (Accuracy = 1 − <10^(−QVb/10)>). By performing local-global alignments of the reference flanking regions to the CCS reads, the start and end of the repeat region can be determined, which allows the lengths and regional accuracies of the separate regions to be determined. All of the readlength histograms shown in Figures 2 through 4 include the top 1000 reads for each sample type as determined by the overall predicted quality value of the read.
Standard deviation analysis
An analysis at the raw read level was performed in order to assess the inter- versus intramolecular variation. Again, a local-global alignment of the reference flanks to the raw read was carried out (including both strand directions) in order to obtain all of the start and end coordinates of the flanking and repeat regions. The intramolecular variation was obtained by taking the standard deviation in the lengths of a single molecule's subreads. The intermolecular variation is calculated by taking the standard deviation of the lengths of 10 randomly sampled subreads across the molecules. The number 10 was chosen to yield equivalent levels of confidence between the inter- and intramolecular calculations. The number of intermolecular samples was taken to match the 200 intramolecular values (one standard deviation calculation for each molecule) of the top-quality reads included in the readlength histograms.
Analysis of enzyme kinetics
To perform the kinetics analyses, the pls.h5 output file was used to link alignments to pulse parameters. First, a loose pattern-matching alignment (with 30% indel allowance) to the flanks was performed on the raw reads, with requirements on separation-distance consistency between different passes present in the raw read. The entire region between the start of one flank and the end of the subsequent flank was aligned using a local-global alignment approach to the putative reference sequence. Then, the PW and IPD of each pulse associated with the alignment were tabulated, allowing averages of kinetics to be taken for each alignment position across multiple molecules or within one molecule.
Data access
Raw sequence data have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession no. SRP015790, as well as the PacBio developer's network (DevNet) website (http://www.smrtcommunity.com/Share/Datasets/Fragile-X).
Competing interest statement
E.W.L. and J.Y. have no conflicts of interest to declare. P.P., J.S.E., D.R., S.M., and L.H. are employees of Pacific Biosciences, Inc., which is a for-profit, publicly traded company. They are also stock holders through the employee stock options program and stock purchasing plan. R.J.H. receives funding from Novartis, Roche, Curemark, Seaside Therapeutics and Forest for clinical trials in fragile X syndrome or autism. She has also consulted with Novartis to guide treatment of fragile X syndrome. F.T. holds a patent with Paul Hagerman: PCR-based assay for rapid detection of expanded CGG-repeat alleles. PCR Assay, US Patent no. 7,855,053; 12/21/2010. No personal revenue is generated. P.J.H. is a nonpaid consultant for Asuragen, Inc., and holds two patents from which no personal revenue is generated. US Patent no. 7,855,053; 12/21/2010. PCR-based assay for rapid detection of expanded CGG-repeat alleles. PCR Assay; P Hagerman, F Tassone. US Patent no. 8,084,220; 12/27/2011. An ELISA method for quantification of FMRP. ELISA Assay; P Hagerman, C Iwahashi.
Acknowledgments
This work was supported by an Interdisciplinary Research Consortium (NIH Roadmap) grant (DE019583, AG032119, P.J.H.) and NIH grants HD040661 (P.J.H.) and HD036071 (R.J.H.). We thank Jackie Yen and John Major for their assistance in an earlier phase of project development, Chris Raske for his advice in culturing expanded-CGG repeat plasmids, Sean Roenspie for providing materials from a clonal, full mutation fibroblast line, Lisa Makhoul for editorial assistance in manuscript preparation, and the UC Davis Genome Center DNA Technologies Core for their assistance. Finally, we appreciate the families who have provided support for this work.
Footnotes
[Supplemental material is available for this article.]
References
- Braida C, Stefanatos RK, Adam B, Mahajan N, Smeets HJ, Niel F, Goizet C, Arveiler B, Koenig M, Lagier-Tourenne C, et al. 2010. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet 19: 1399–1412 [DOI] [PubMed] [Google Scholar]
- Chen LS, Tassone F, Sahota P, Hagerman PJ 2003. The (CGG)n repeat element within the 5′ untranslated region of the FMR1 message provides both positive and negative cis effects on in vivo translation of a downstream reporter. Hum Mol Genet 12: 3067–3074 [DOI] [PubMed] [Google Scholar]
- Chen L, Hadd A, Sah S, Filipovic-Sadic S, Krosting J, Sekinger E, Pan R, Hagerman PJ, Stenzel TT, Tassone F, et al. 2010. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn 12: 589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hadd AG, Sah S, Houghton JF, Filipovic-Sadic S, Zhang W, Hagerman PJ, Tassone F, Latham GJ 2011. High-resolution methylation polymerase chain reaction for fragile X analysis: Evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet Med 13: 528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Au J, Schneider A, Hessl D, Harris SW, Laird M, Mu Y, Tassone F, Nguyen DV, Hagerman RJ 2012. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet 131: 581–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ 2007. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord 37: 738–747 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138 [DOI] [PubMed] [Google Scholar]
- Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R 2006. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr 27: S137–S144 [DOI] [PubMed] [Google Scholar]
- Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, Hagerman PJ, Stenzel TT, Hadd AG, Latham GJ, et al. 2010. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem 56: 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Arocena D, Hagerman PJ 2010. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet 19: R83–R89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R, Lauterborn J, Au J, Berry-Kravis E 2012. Fragile X syndrome and targeted treatment trials. In Modeling fragile X syndrome, Vol. 54 (ed. RB Denman), pp. 297–335. Springer, Berlin/Heidelberg, Germany [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ 2010. TRPing up the genome: Tandem repeat polymorphisms as dynamic sources of genetic variability in health and disease. Discov Med 10: 314–321 [PubMed] [Google Scholar]
- Hornstra IK, Nelson DL, Warren ST, Yang TP 1993. High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome. Hum Mol Genet 2: 1659–1665 [DOI] [PubMed] [Google Scholar]
- Koeppen AH 2011. Friedreich's ataxia: Pathology, pathogenesis, and molecular genetics. J Neurol Sci 303: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Cooper TA 2009. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans 37: 1281–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leehey MA, Hagerman PJ 2012. Fragile X-associated tremor/ataxia syndrome. Handb Clin Neurol 103: 373–386 [DOI] [PubMed] [Google Scholar]
- Metzker ML 2010. Sequencing technologies: The next generation. Nat Rev Genet 11: 31–46 [DOI] [PubMed] [Google Scholar]
- Nolin SL, Glicksman A, Houck GE Jr, Brown WT, Dobkin CS 1994. Mosaicism in fragile X affected males. Am J Med Genet 51: 509–512 [DOI] [PubMed] [Google Scholar]
- Nolin SL, Dobkin C, Brown WT 2003. Molecular analysis of fragile X syndrome. Curr Protoc Hum Genet Chapter 9: 9.5.1–9.5.12 [DOI] [PubMed] [Google Scholar]
- Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, et al. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 365: 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Dragatsis I, Dietrich P 2011. Genetics and neuropathology of Huntington's disease. Int Rev Neurobiol 98: 325–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman RJ, Hagerman PJ, Giulivi C 2010. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J 429: 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluto A, Brussino A, Tassone F, Arduino C, Cagnoli C, Pappi P, Hagerman P, Migone N, Brusco A 2005. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn 7: 605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci 74: 5463–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebra R, Ashby M, Bhamidipati A, Bjornson K, Cutliff C, Dalal R, Eid J, Fedorov A, Gray J, Hanes J, et al. 2012. Emerging SMRT sequencing technologies facilitating extended readlength. In Advances in Genome Biology & Technology Workshop. Pacific Biosciences, Marco Island, FL [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. 2010. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29: 1248–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicot G, Gourdon G, Gomes-Pereira M 2011. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: New findings and future challenges. Hum Mol Genet 20: R116–R123 [DOI] [PubMed] [Google Scholar]
- Sullivan SD, Welt C, Sherman S 2011. FMR1 and the continuum of primary ovarian insufficiency. Semin Reprod Med 29: 299–307 [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ 2000. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 66: 6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW 2010. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res 38: e159 doi: 10.1093/nar/gkq543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914 [DOI] [PubMed] [Google Scholar]
- Willemsen R, Levenga J, Oostra BA 2011. CGG repeat in the FMR1 gene: Size matters. Clin Genet 80: 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, Corrigan EC, Simpson JL, Nelson LM 2007. The FMR1 premutation and reproduction. Fertil Steril 87: 456–465 [DOI] [PubMed] [Google Scholar]
- Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, Tassone F 2012. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med 14: 729–736 [DOI] [PMC free article] [PubMed] [Google Scholar]