Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange (original) (raw)
. Author manuscript; available in PMC: 2013 Mar 1.
Published in final edited form as: Nat Struct Mol Biol. 2012 Aug 26;19(9):884–892. doi: 10.1038/nsmb.2312
Abstract
Set2-mediated methylation of histone H3 Lys36 (H3K36) is a mark associated with the coding sequences of actively transcribed genes, yet plays a negative role during transcription elongation. It prevents trans-histone exchange over coding regions and signals for histone deacetylation in the wake of RNA polymerase II (RNAPII) passage. We have found that in Saccharomyces cerevisiae the Isw1b chromatin-remodeling complex is specifically recruited to open reading frames (ORFs) by H3K36 methylation through the PWWP domain of its Ioc4 subunit in vivo and in vitro. Isw1b acts in conjunction with Chd1 to regulate chromatin structure by preventing trans-histone exchange from taking place over coding regions and thus maintains chromatin integrity during transcription elongation by RNA polymerase II.
Introduction
Under physiological conditions chromatin represents a strong barrier to transcription by RNA polymerase II that has to be overcome as well as restored in its wake. Modulation of chromatin structure is achieved by the concerted actions of chromatin remodelers, histone modifying enzymes and histone chaperones. One such enzyme, the lysine methyltransferase Set2, is associated with the elongating form of RNAPII and methylates Lys36 on histone H3 over coding sequences1,2. H3K36 methylation is required for efficient deacetylation of histones by the Rpd3S histone deacetylase complex3–5. Rpd3S also associates with RNAPII6,7 and thus maintains coding regions in a hypoacetylated state in the wake of transcription elongation. Loss of Set2 or Rpd3S activities leads to an accumulation of acetylated histones over coding regions and disruption of chromatin organization as evidenced by the accumulation of cryptic transcripts, the result of inappropriate transcription initiation from inside open reading frames3,8. Recent studies from our laboratory have shown that Set2-mediated H3K36 methylation furthermore maintains chromatin integrity by preventing histone exchange over coding regions, and thus limits the incorporation of new, acetylated histones9. In the current study we sought to identify mechanisms by which H3K36 methylation achieves and perpetuates the stable organization of transcribed chromatin.
Chromatin remodeling factors use the energy generated by ATP hydrolysis to slide or evict nucleosomes, or to alter their composition, thus affecting chromatin organization. They play important roles in a number of cellular processes, including gene transcription, replication and recombination. ISWI (_I_mitation _S_witch) and CHD (_C_hromodomain _H_elicase _D_NA-binding) represent two families of remodeling enzymes that are conserved from yeast to humans, although substantial diversity exists with respect to the subunit composition of individual complexes (reviewed in Clapier and Cairns10).
The Iswi remodeler was initially identified in Drosophila melanogaster11 and has two homologs in S. cerevisiae12, Isw1 and Isw2. Isw1 associates with Ioc3 (_I_sw _O_ne _C_omplex protein 3) or Ioc2 and Ioc4 to form two distinct remodeling complexes, Isw1a and Isw1b respectively13. Although Isw1 can interact with nucleosomes through its SANT and SLIDE domains14,15, it is thought that the associated Ioc proteins are involved in targeting the remodeling complexes to different genomic locations. Indeed previous experiments found Ioc3 associated with the promoter of MET16, while Ioc2 and Ioc4 localized to the MET16 ORF16.
Chd1 is the sole representative of the CHD family in S. cerevisiae and contains a characteristic double chromodomain at its N-terminus. Chromodomain mutations result in dissociation of yeast Chd1 from chromatin17, while no large-scale effects are observed for the fly mutant homolog18. However, Chd1 has been shown to interact with several known elongation factors such as the PAF complex, Spt5 and FACT19–21, as well as to co-localize with RNAPII19,22,23, thus linking Chd1 to transcription elongation.
Iswi and Chd1 chromatin remodelers fulfill partially redundant functions. An isw1Δ isw2Δ chd1Δ strain displays synthetic phenotypes12 as well as wide-spread disruption of nucleosome positioning throughout the yeast genome24,25. In vitro both Iswi and Chd1 remodelers are particularly effective in repositioning nucleosomes and generating spaced arrays12,26,27, a feature of remodeling enzymes often associated with transcriptional repression rather than activation. Deletion of ISW1 or CHD1 in a yeast reporter strain resulted in increased cryptic transcription at the FLO8 locus17,28, further underlining a repressive function for both remodeling factors.
In this study we determined that recruitment of Isw1b to ORFs is mediated by interaction of its Ioc4 subunit with H3K36 methylated nucleosomes both in vivo and in vitro. Furthermore, chromatin remodelers Isw1 and Chd1 were found to act synergistically within the Set2 pathway by antagonizing histone exchange and preventing increased incorporation of acetylated histones over coding regions. These results suggest a novel mechanism by which the chromatin remodeling activities of Isw1 and Chd1 maintain chromatin integrity in the wake of transcription elongation by RNAPII.
Results
Isw1 and Chd1 associate with H3K36me3 nucleosomes in vivo
Trimethylated H3K36 (H3K36me3) is a histone mark deposited on the ORFs of actively transcribed genes and signals for deacetylation of nucleosomes by the Rpd3S complex3–5. We were interested to determine what other factors were recruited either directly or indirectly by histone H3 K36me3. To this end we prepared a chromatin fraction from wildtype (WT) yeast cells and obtained mononucleosomes using micrococcal nuclease (MNase) digestion. Yeast mononucleosomes were immunoprecipitated using either H3K36me3 or a control IgG antibody and the eluates were analyzed by MudPIT mass spectrometry (Fig. 1a). In addition to histones, substantial amounts of RNAPII were found to be associated, as expected for nucleosomes representative of actively transcribed chromatin. Furthermore, we also identified all subunits of the Rpd3S complex as well as several histone chaperones and chromatin remodeling factors. Among the most abundant were the Isw1 remodeling complexes and Chd1. In particular we focused on the Isw1b remodeler, consisting of the catalytic Isw1 subunit complexed with Ioc2 and Ioc4, as this complex has two potential methyl-lysine interaction domains. Ioc2 contains a PHD-like domain, while Ioc4 features an N-terminal PWWP domain (Fig. 1b,c).
Figure 1. Proteins associated with H3K36me3-containing mononucleosomes.

(a) MudPIT mass spectrometry analysis of proteins co-immunoprecipitated with H3K36me3-containing mononucleosomes from wildtype yeast. IgG was used as a negative control. a Spectral count, total spectra matching peptides detected by tandem mass spectrometry for the indicated protein. b Sequence coverage, percentage of protein sequence represented in peptides identified by tandem mass spectrometry. (b) Subunit composition of Isw1 and Chd1 remodelers. Isw1 is the catalytic subunit of two chromatin-remodeling complexes: It associates with Ioc3 to form Isw1a, while Ioc2 and Ioc4 are part of the Isw1b complex. Chd1 is thought to exist largely as a monomer. (c) Schematic representations of the domain organizations for the Isw1 and Chd1 chromatin remodelers.
Ioc4 interacts directly with H3K36me3 nucleosomes
Next we wanted to determine whether Ioc2 and Ioc4 can interact directly with H3K36me3. TAP-purified Isw1b complex as well as purified recombinant Ioc2, Ioc4 and Isw1 proteins were used in histone peptide pull-down assays, but did not interact preferentially with histone H3 peptides methylated on the Lys36 residue (data not shown). However, nucleosomes might be required for binding. Thus, we used reconstituted, recombinant H3K36 methyl-lysine analogue (MLA) mononucleosomes. Histone H3 used for nucleosome reconstitutions contained a K36C mutation that was chemically modified29 to mimic either unmethylated or trimethylated Lys36 (Fig. 2). Electromobility gel shift assays (EMSA) showed that Ioc4 had a higher affinity for trimethylated than unmethylated mononucleosomes (Fig. 2a,c). This preference is dependent on the presence of the Ioc4 PWWP domain, as Ioc4 bearing an N-terminal deletion (Ioc4ΔPWWP) displayed equal binding to both unmethylated and trimethylated nucleosomes (Fig. 2b,c). Overall affinity of Ioc4ΔPWWP for nucleosomes was also reduced, suggesting that the PWWP domain represents a major interface for nucleosome binding. Analogous experiments for Ioc2 revealed no binding of purified, recombinant Ioc2 to either unmethylated or trimethylated mononucleosomes (Supplementary Fig. 1a). Recombinant Ioc3 also did not interact preferentially with peptides methylated on Lys36 (data not shown), while the Ioc3-containing Isw1a complex exhibited similar affinities for both unmethylated and trimethylated H3K36 MLA mononucleosomes (Supplementary Fig. 1b). Thus we conclude that Ioc4 interacts directly with H3K36me3-containing nucleosomes through its PWWP domain.
Figure 2. Ioc4 preferentially binds H3K36me3 nucleosomes.

(a,b) Recombinant, reconstituted H3K36 MLA nucleosomes (5 fmol) were incubated with increasing concentrations (0, 3, 7, 10, 13, 17, 20 and 27 nM) of Ioc4 (a) or Ioc4 with its N-terminal PWWP domain deleted (Ioc4ΔPWWP) (b) and analyzed using EMSAs. Mononucleosome bands are indicated. (c) Quantitation of Ioc4 and Ioc4ΔPWWP binding to tri- and unmethylated H3K36 MLA mononucleosomes. Mononucleosome bands were quantitated for each lane. Lanes 2–8 were normalized against input lane 1, while lanes 10–16 were normalized to input lane 9. The percentage of nucleosomes bound by Ioc4 or Ioc4ΔPWWP was expressed as 100 – % mononucleosomes for each lane. Mean values ± SEM were plotted for at least three independent experiments. _P_-values were calculated using Student's t-test.
Ioc4 localizes to ORFs in a Set2-dependent manner
Since Ioc4 interacts with H3K36me3-containing nucleosomes in vitro, we wanted to investigate whether this interaction also occurred in vivo. We used chromatin immunoprecipitation coupled with microarrays (ChIP-chip) to determine the genome-wide localization of Flag-tagged Ioc3 and Ioc4 in vivo for both wildtype and set2Δ yeast strains. Using average gene analysis (Supplementary Fig. 2a,b) we determined that Ioc4 localized primarily to the mid- and 3′ coding regions of genes (Fig. 3a), which are also generally associated with H3K36me3 (refs.8,30). By contrast, Ioc3 localized mostly to the regions surrounding the transcription start and end sites (Fig. 3b). Deletion of SET2 resulted in an almost complete abrogation of Ioc4 occupancy over ORFs (Fig. 3a). We also observed reductions in the association of Flag-tagged Ioc2 and Isw1 over ORFs in a set2Δ background, as determined by ChIP-qPCR and ChIP-chip respectively (Supplementary Fig. 2c–f). Taken together these data suggest that the H3K36me3 mark is involved in mediating Isw1b occupancy over coding regions. Ioc3 localization at intergenic regions was not affected by deletion of SET2, although Ioc3 occupancy over ORFs increased slightly in a set2Δ mutant background (Fig. 3b), perhaps as a consequence of Ioc4 delocalization. Based on the reduction of Ioc4, Ioc2 and Isw1 occupancy over ORFs in the set2Δ mutant we conclude that Isw1b occupancy depends on Set2 and H3K36 methylation.
Figure 3. Deletion of SET2 abrogates localization of Ioc4 to coding regions.

(a,b) ChIP-chip experiments were performed using yeast genomic tiling arrays. The log2 ratios of IP over input were subjected to average gene analysis (Supplementary Fig. 2a). Whole-genome average data were calculated and plotted as mean ± SEM. SE of mean are shown in grey and represent three independent experiments. The _T_ranscription _S_tart _S_ite (TSS) and _TE_rmination _S_ite (TES) are indicated. Flag-tagged Ioc4 (a) and Ioc3 (b) were immunoprecipitated from wildtype and set2Δ yeast strains.
Isw1 and Chd1 prevent intragenic transcription
Previous experiments have shown that Set2-dependent H3K36 methylation is vital for the catalytic activity of the Rpd3S histone deacetylase complex in order to maintain chromatin in a hypoacetylated state following transcription7. In the absence of H3K36 methylation nucleosomes in ORFs become hyperacetylated, resulting in aberrant transcription initiation from cryptic promoters inside ORFs3. Association of the Isw1b remodeling complex with H3K36 methylated nucleosomes (Fig. 1a) suggested its potential involvement in maintaining ordered chromatin structure following transcription elongation. Thus, we wanted to assess the involvement of Isw1 in suppressing cryptic transcription, as well as its involvement in the Set2 pathway. Indeed, previous studies using a yeast reporter strain for cryptic transcription showed that deletion of ISW1 did result in a selectable phenotype28. Deletion of ISW1 alone did cause the production of low to moderate amounts of cryptic transcripts at most genes tested when assessed by Northern blotting (Fig. 4 and Supplementary Fig. 3). The same phenotype is observed for a strain containing the ISW1K227R catalytic mutant (Fig. 4e–h), suggesting that Isw1 remodeling activity is required for the prevention of cryptic initiation in wildtype cells. Previous reports have indicated that Isw1, Isw2 and Chd1 can have overlapping functions12. While we have not observed cryptic transcription in an isw2Δ strain (Supplementary Fig. 3, data not shown), deletion of CHD1 alone caused low levels of cryptic transcripts to accumulate in some instances (Supplementary Fig. 3a,c)28. However, the isw1Δ chd1Δ double deletion strain did exhibit a substantially stronger cryptic transcript phenotype that was completely unaffected by the additional deletion of ISW2 (Fig. 4a–d) in agreement with previously published data17. These results suggest that Isw1 and Chd1 but not Isw2 affect chromatin structure during transcription. Deletion of SET2 in an isw1Δ chd1Δ background further exacerbates the cryptic transcript phenotype of this strain. However, the total levels of cryptic transcripts produced when SET2 is deleted in either the single deletion (Supplementary Fig. 3e) or double deletion backgrounds (Fig. 4) are similar to those obtained for set2Δ alone. In all instances the ISW1K227R catalytic mutant phenocopies an isw1Δ mutant (Fig. 4e–h). These results implicate ISW1, CHD1 and SET2 as being involved in the same pathway.
Figure 4. ISW1 and CHD1 have overlapping functions during transcription within the Set2 pathway.

(a,c,e,g) Total RNA for each strain was isolated and used for Northern blot analysis. The probes used were directed against the 3′ ends of the STE11 and PCA1 genes. ACT1 and rRNA were used as loading controls. The full-length (←) and cryptic transcripts (*) are indicated. (b,d,f,h) For each lane all cryptic transcript bands were quantitated and normalized against the ACT1 loading control. Total levels of cryptic transcription were expressed relative to set2Δ. Data were plotted as mean ± SEM for three independent experiments.
Isw1 and Chd1 prevent wide-spread cryptic transcription
It has been shown recently that deletion of ISW1 and CHD1 results in large-scale changes in chromatin structure24. Thus we wanted to investigate further whether deletion of both remodelers resulted in the appearance of cryptic transcripts genome-wide. For this purpose we purified mRNA from both wildtype and isw1Δ chd1Δ yeast cells, directly labeled the mRNA using fluorescent compounds and carried out competitive hybridizations to strand-specific yeast genome tiling arrays. These experiments allowed us to obtain strand-specific gene expression data as described previously8. While regularly transcribed genes show uniform signals all across open reading frames, cryptic transcripts transcribed from the sense strand cause a signal increase towards the 3′ end of genes. The reverse is true for cryptic transcripts initiated from the antisense strand. Previously, our laboratory has used K-means clustering to analyze this type of data8. While we found that this approach worked well for the antisense transcription data, regular transcriptional background proved problematic in the analysis of the sense data set. Instead, we directly compared the probe signals from the 3′ to the 5′ ends of open reading frames and selected genes that exhibited at least a 1.5-fold signal increase at the 3′ end for the sense data set. In order to validate our methodology we reanalyzed data for sense cryptic transcripts in a set2Δ mutant. This approach proved to be more stringent than K-means cluster analysis and underestimates the number of cryptic transcript genes for set2Δ, as only 327 out of 621 genes were identified. However, these genes retain the same characteristics, being predominantly long (89% > 1 kb) and infrequently transcribed (95% < 5 mRNA per hour), and include known cryptic transcript genes for set2Δ, such as STE11 and PCA1. We used the same approach for the data analysis of gene expression in the isw1Δ chd1Δ mutant, which revealed wide-spread cryptic initiation. A large number of genes with sense (646 genes) and/or antisense (962 genes) cryptic transcripts were identified (Fig. 5a). Thus, even though the approach used to identify sense cryptic transcript genes likely led to an underestimate, a substantial number of genes were identified indicating that both sense and antisense cryptic transcription is widespread in the isw1Δ chd1Δ mutant. Again, both STE11 and PCA1 were identified, in agreement with our Northern analysis (Fig. 4).
Figure 5. Deletion of ISW1 and CHD1 causes wide-spread intragenic transcription.
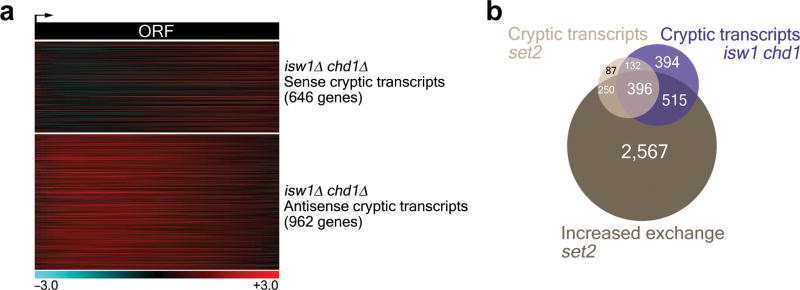
(a) Heatmap of cryptic transcript genes in an isw1Δ chd1Δ mutant. Probe intensities for the 5′ and 3′ ends of all genes were determined. Resulting 3′/5′ ratios with a cutoff value of log2 > 0.5 were used to define sense cryptic transcripts (n=646). K-means clustering of gene expression profiles was used to identify antisense cryptic transcripts (n=962). Only cryptic transcript genes are shown. (b) Venn diagram showing the overlaps between genes displaying increased histone exchange over ORFs in set2Δ (mean log2 > 0; n=3,728) and genes with cryptic transcription in set2Δ (n=865) and/or isw1Δ chd1Δ (n=1,437). Cryptic transcript sets contain genes with sense and/or antisense cryptic transcripts, whereby 68 and 171 genes show intragenic initiation in both the sense and antisense direction for set2Δ and isw1Δ chd1Δ, respectively. For the purpose of direct comparison we selected cryptic transcript genes for set2Δ according to the same criteria applied to isw1Δ chd1Δ (a), rather than the previously published data8.
Recent experiments from our laboratory have shown that the presence of H3K36 methylated nucleosomes is important for the retention of the original histones at the ORFs of transcribed genes by preventing trans-histone exchange, ie. the replacement of existing histones with histones from the free cellular pool9. Histone exchange affects many more genes in a set2Δ mutant when compared to the number of cryptic transcript genes (Supplementary Fig. 4a,b), presumably because many genes possess no cryptic promoters. Hence, we wanted to compare the genes that exhibit cryptic initiation in either the set2Δ or isw1Δ chd1Δ mutants to the group of genes with altered histone exchange over ORFs in a set2Δ background. Figure 5b shows that ca. 60% of genes with cryptic transcripts in an isw1Δ chd1Δ mutant also exhibit increased histone exchange over open reading frames in a set2Δ strain. Similarly, ca. 60% of set2Δ cryptic transcript genes also display intragenic initiation in an isw1Δ chd1Δ mutant. However, an appreciable number of cryptic transcript genes in isw1Δ chd1Δ show no overlap with genes associated with either increased histone exchange or cryptic transcription in set2Δ and are presumably regulated by Isw1 and Chd1 independently of Set2. In summary, these data show that deletion of ISW1 and CHD1 causes wide-spread intragenic transcription initiation in the yeast genome.
Isw1 and Chd1 prevent histone exchange over ORFs
Given the substantial overlap of genes displaying cryptic initiation in an isw1Δ chd1Δ background and Set2-regulated genes, and since the Isw1b complex is a reader of the H3K36 methyl mark in particular, we sought to investigate whether these proteins were involved in trans-histone exchange (Fig.6). Histone exchange was studied as described before31, using a yeast strain containing constitutively expressed, Myc-tagged H3 as well as Flag-tagged H3 under the control of the GAL1 promoter. In order to study transcription-coupled exchange, yeast were arrested in G1 with α-factor and expression of Flag-H3 was induced briefly by growth in galactose-containing media. Using microarray and average gene analysis, transcription-coupled histone dynamics can be followed by comparing occupancy levels for Flag-H3 relative to Myc-H3. In agreement with previously published work, histone exchange in wildtype yeast is high at the promoters of genes and low over coding regions (Supplementary Fig. 5a,b)31,32. We focused our analysis primarily on genes known to exhibit increased exchange over ORFs in a set2Δ background (n=3,728) (Supplementary Fig. 4a)9, although the profiles for the whole-genome data are very similar (Fig. 6a and Supplementary Fig. 5). Comparing data for both isw1Δ and chd1Δ relative to the wildtype, both mutants showed increased levels of histone exchange from mid-ORF to the 3′ ends of genes (Fig. 6a), areas particularly associated with H3K36me3 (Fig. 7a)8,30. Furthermore, the effect of each gene deletion was clearly additive (Fig. 6a), in agreement with the results obtained for cryptic transcription by Northern blotting (Fig. 4 and Supplementary Fig. 3a–d). We also assessed the effect of deletion of IOC4 on histone exchange and saw an increase in exchange over coding regions, similar to that observed for isw1Δ (Fig. 6b). Slight differences in the exchange profiles between isw1Δ and ioc4Δ presumably result from the fact that both the Isw1a and Isw1b remodelers are affected in the isw1Δ background. Genes that show increased exchange over ORFs in either an ioc4Δ, isw1Δ or chd1Δ background overlap to large extents with those known to be affected in a set2Δ mutant (Fig. 6c,d). The proportion of genes affected in isw1Δ and chd1Δ further increases when assessing increases in histone exchange specifically for the 3′ halfs of ORFs (Supplementary Fig. 4c). Also, genes exhibiting cryptic transcription in an isw1Δ chd1Δ background largely overlap with those genes that are affected by increased histone exchange over ORFs in both isw1Δ chd1Δ as well as set2Δ (Fig. 6g).
Figure 6. Deletion of ISW1 and CHD1 increases histone exchange over 3′ ends of ORFs.

(a–b,e–f) ChIP-chip experiments were performed using yeast genomic tiling arrays. Only genes known to display increased exchange over ORFs in a set2Δ mutant (n=3,728) (Supplementary Fig. 4a) were used for average gene analysis and plotted as mean ± SEM (grey) for three independent experiments. The _T_ranscription _S_tart _S_ite (TSS) and _TE_rmination _S_ite (TES) are indicated. (a–b) Histone exchange (Flag-H3/Myc-H3) was determined for wildtype, isw1Δ, chd1Δ, isw1Δ chd1Δ (a) and ioc4Δ (b) strains and presented as difference profiles for mutant (mut) over wildtype. (c–d) Genes were clustered into two groups based on their average histone exchange signals for mutant relative to wildtype profiles over ORFs (mean log2 < 0, mean log2 > 0). Venn diagrams show overlaps between genes displaying increased histone exchange over ORFs (mean log2 > 0) for the mutants indicated. (e–f) Average gene analysis for acetylated K56 (H3K56ac) immunoprecipitated from wildtype, isw1Δ, chd1Δ, isw1Δ chd1Δ (e) and the ISW1K227R catalytic mutant (f) strains. H3K56ac occupancy was normalized to histone H3 levels. Difference profiles are shown for mutant over wildtype. (g) Venn diagram showing the overlap between genes that exhibit cryptic transcription in an isw1Δ chd1Δ mutant (Fig. 5a,b) and genes that exhibit increased histone exchange over ORFs in an isw1Δ chd1Δ and set2Δ background. (h) Pearson correlation coefficients and _P_-values were calculated for histone exchange (a) and H3K56ac/H3 (e) profiles for each mutant background using R45. (i–l) For whole-genome analysis genes (n=4,894) were clustered into two groups (< 10 mRNA per hour (4,250 genes), ≥ 10 mRNA per hour (644 genes)) based on transcription rates published by Holstege et al.46. Average gene analysis for histone exchange (i–j) or H3K56ac/H3 (k–l) difference profiles clustered according to transcription rates are shown. Data were plotted for isw1Δ (i,k) or chd1Δ (j,l) relative to the wildtype.
Figure 7. Deletion of ISW1 and CHD1 increases histone acetylation over coding regions.

(a–d) ChIP-chip experiments were performed as described in Fig. 6. Only genes known to display increased exchange over ORFs in a set2Δ mutant (n=3,728) (Supplementary Fig. 4a) were used for average gene analysis and plotted as mean ± SEM (grey) for three independent experiments. The _T_ranscription _S_tart _S_ite (TSS) and _TE_rmination _S_ite (TES) are indicated. (a) Average gene analysis for H3K36me3 immunoprecipitated from wildtype, isw1Δ and chd1Δ yeast strains. (b) Average gene analysis for histone H3 immunoprecipitated from wildtype, isw1Δ and chd1Δ yeast strains. (c–d) Average gene analysis for acetylated H4 (AcH4) immunoprecipitated from wildtype, isw1Δ, chd1Δ, isw1Δ chd1Δ (c) as well as set2Δ, isw1Δ set2Δ and chd1Δ set2Δ (d) yeast strains. AcH4 occupancy was normalized to histone H3 levels. Difference profiles are shown for mutant over wildtype.
In order to confirm that deletion of ISW1 and CHD1 increases histone exchange over ORFs we also investigated incorporation of histone H3 acetylated on Lys56 (H3K56ac). Acetylation of H3K56 is mediated specifically by the Rtt109 acetyltransferase (KAT11), which acts on soluble histones, but not on chromatin33. Distribution of H3K56ac has been shown previously to correlate with both replication-dependent and -independent histone exchange32,34 and thus can be used as a marker for genomic regions undergoing trans-histone exchange. Using ChIP-chip we determined the distribution of H3K56ac and histone H3 in G1-arrested strains in a BY4741 background to confirm that increases in histone exchange in the mutant strains was not due to overexpression of histone H3. We observed increased levels of H3K56ac from mid-ORF to the 3′ ends of genes (Fig. 6e) for isw1Δ and chd1Δ as seen also for the histone exchange strains (Fig. 6a). Similarly, the effect of each gene deletion was additive (Fig. 6a,e). The increases in histone exchange for isw1Δ, chd1Δ and isw1Δ chd1Δ, whether represented as Flag/Myc in the histone exchange strains or by H3K56ac/H3, are very highly correlated (Fig. 6h). The ISW1K227R catalytic mutant strain also showed increased levels of H3K56ac over coding regions (Fig. 6f), similar to exchange in an ioc4Δ background (Fig. 6b). This indicates that the catalytic activity of Isw1 is required to suppress histone exchange and maintain ordered chromatin structure in wildtype yeast, in agreement with results on cryptic transcription obtained by Northern blotting (Fig. 4e–h).
Clustering all yeast genes according to transcription rates, we determined that exchange increased over the ORFs of lowly transcribed genes in an isw1Δ background relative to the wildtype, but did not affect exchange in highly transcribed genes (Fig. 6i). These results conform with expectations as genes known to rely on Set2 for accurate transcription are generally long and transcribed at low levels8. Interestingly, deletion of CHD1 increased exchange over both lowly and highly transcribed genes when compared to the wildtype (Fig. 6j). Similar analysis of H3K56ac levels over ORFs for both isw1Δ and chd1Δ show the same distributions (Fig. 6k,l), hinting at the complementary action of these two chromatin remodelers (see Discussion). From these experiments we conclude that the Isw1b and Chd1 chromatin remodeling factors both prevent co-transcriptional trans-histone exchange at largely overlapping, yet somewhat distinct groups of genes.
Deletion of ISW1 and CHD1 increases AcH4 over ORFs
The appearance of cryptic transcripts in a set2Δ background is associated with a lack of H3K36 methylation and hyperacetylation of ORF nucleosomes3. Also, we have recently shown that increased trans-histone exchange in a set2Δ mutant results in greater levels of histone acetylation over ORFs9. Therefore, we sought to determine whether deletion of ISW1 or CHD1 affected either of these histone marks. For purposes of direct comparison, we focused our analysis on the same group of genes that show increased histone exchange in set2Δ. However, the trends seen for each deletion are the same genome-wide (Supplementary Fig. 6).
Microarray analysis of H3K36me3 ChIPs clearly showed that the distribution and occupancy levels of H3K36me3 in an isw1Δ strain are highly similar compared to the wildtype (Fig. 7a). In contrast, deletion of CHD1 clearly affected the overall distribution but not the absolute levels of H3K36me3. Using histone H3 ChIP-chip data we determined the overall distribution of nucleosomes for both the isw1Δ and chd1Δ mutants. Histone H3 occupancy was slightly reduced in an isw1Δ background relative to the wildtype. However, deletion of CHD1 caused an overall redistribution of nucleosomes towards the 5′ end of genes (Fig. 7b), suggesting that Chd1 prevents loss of nucleosomes from the 3′ end of genes. Since deletion of ISW1 or CHD1 did not seem to affect absolute H3K36me3 levels in vivo, we asked whether deletion of either gene could cause an increase in co-transcriptional ORF acetylation, thought to result in sustained decompaction of chromatin. In wildtype cells histone H4 acetylation (AcH4) is high at promoters and low over the bodies of genes (Supplementary Fig. 6a). Promoter acetylation was unaffected in the isw1Δ mutant, but reduced in the chd1Δ background, indicating a secondary role for Chd1 at promoters. Importantly, deletion of either ISW1 or CHD1 resulted in a rise of ORF histone H4 acetylation with small additive effects in the isw1Δ chd1Δ mutant (Fig. 7c). We also assessed the effects of simultaneous deletion of ISW1 or CHD1 in a set2Δ background on histone H4 acetylation levels. As expected for proteins that act within the same pathway, AcH4 levels and distribution of either isw1Δ set2Δ or chd1Δ set2Δ mimic those of set2Δ alone (Fig. 7d), which also agrees with our results on cryptic transcription for those mutants (Supplementary Fig. 3e). Taken together, our results show that in the wildtype Isw1 and Chd1 function to suppress trans-histone exchange, thereby preventing the incorporation of soluble, highly acetylated histones over ORFs.
Discussion
We have shown here that the Isw1b chromatin remodeler is recruited to coding sequences by association of its Ioc4 subunit with H3K36 methylated nucleosomes, as interaction of Ioc4 with chromatin was reduced both in vivo and in vitro in the absence of H3K36 methylation (Figs. 2,3). By contrast, Ioc3, representative of the Isw1a complex was found preferentially at intergenic regions. Interestingly, its association with coding sequences increased in a set2Δ background, perhaps as a consequence of Ioc4 delocalization. Increased Ioc3 occupancy in a set2Δ mutant, taken together with the overall genome-wide distributions of Ioc3 and Ioc4 in the wildtype (Fig. 3), point towards the existence of a dynamic equilibrium between the Isw1a and Isw1b complexes in the cell. This is further supported by the observation that estimates on protein abundance imply that Isw1 is the least abundant of all ISWI subunits35.
In an earlier study, Isw1 was reported to preferentially pull down histone H3 Lys4 trimethylated (H3K4me3) nucleosomes36. Based on our ChIP-chip data for Ioc3 (Fig. 3b), we speculate that Isw1a may preferentially bind to H3K4me3-containing nucleosomes that are generally associated with promoters. In our hands TAP purifications of Isw1 result in near equal amounts of Ioc3 and Ioc2–Ioc4 being pulled down. Furthermore, in vitro gel shift experiments on mononucleosome binding by Isw1a and Isw1b have shown that Isw1a displays a much higher affinity for mononucleosomes due to its high affinity for DNA when compared to Isw1b13,27. Taken together, in the absence of crosslinks, any nucleosomes isolated from Isw1 pull-down experiments are more likely to be associated with Isw1a rather than Isw1b. Previous ChIP-qPCR experiments at the inducible MET16 locus suggested that Isw1a is recruited to the promoter only when the gene is inactive16. However, we observed Ioc3 occupancy at a large number of genes known to be actively transcribed, implying that Isw1a may play a more wide-spread role than previously anticipated.
Chd1 is known to associate with elongating RNAPII through interaction with the PAF complex and/or other RNAPII-associated proteins such as Spt5 and FACT19–21. Previous experiments using ChIP-qPCR could not establish differential association of Chd1 with chromatin when comparing wildtype cells to a set2Δ background17. Neither did Chd1 preferentially bind to H3K36 methylated nucleosomes in vitro37. While it is possible that Chd1 preferentially remodels H3K36 methylated nucleosomes, it is likely that its association with RNAPII mediates localization of its activity at coding sequences. This is in agreement with the observation that both histone exchange and H3K56ac in a chd1Δ but not an isw1Δ background are increased over genes with higher transcription rates (Fig. 6i–l).
Recent ChIP-seq experiments by Gkikopoulos et al.24 on nucleosome positioning in strains bearing single deletions of ISW1 or CHD1, as well as the combination of isw1Δ chd1Δ or isw1Δ isw2Δ chd1Δ show that nucleosome positioning is greatly perturbed across the genome in the double and triple deletion strains. The coding regions were severely affected in an isw1Δ chd1Δ background, in agreement with our own results showing that deletion of ISW1 and CHD1 has additive effects with respect to both cryptic transcription (Figs. 4,5) and histone exchange (Fig. 6a,e) and leads to wide-spread intragenic transcription initiation. Examining each deletion individually, single deletion of CHD1 had more severe effects overall compared to deletion of ISW1 alone24,38, also in agreement with our own data (Fig. 6a,e). These results presumably reflect the fact that Chd1 can compensate for the loss of Isw1 better than vice-versa. Chd1 association with RNAPII may well account for this effect; conversely, even though highly transcribed genes contain high levels of H3K36me3, their lower nucleosome density overall may translate as a reduced “interaction surface” for Isw1b recruitment.
Previous studies have shown that histone exchange of H3/H4 tetramers generally only takes place over promoters, while it is limited to highly transcribed genes over ORFs31,32,39. In contrast, exchange of H2A/H2B dimers is generally prevalent over transcribed regions39,40. This observation together with in vitro experiments lead to the suggestion that at low to moderately transcribed genes, RNAPII can transcribe through a hexasomal template following the eviction of a single H2A/H2B dimer, thus resulting in the retention of the H3/H4 tetramer on the DNA41,42. In contrast, the passage of multiple RNAPII molecules reminiscent of the transcription of highly transcribed genes causes complete dissociation of histone octamers from the DNA, which are subsequently reassembled behind the polymerase. We hypothesize that Isw1b and Chd1 may exert their overlapping, yet distinct functions in this context. Studies on Drosophila Chd1 and the Iswi containing ACF complex have shown that both enzymes can catalyze the transfer of histones from the histone chaperone Nap1 onto DNA and generate regularly spaced nucleosomal arrays in vitro26. Further experiments in Drosophila established that Chd1 together with the Hira chaperone is important for the replication-independent deposition of histone variant H3.3 in male fly pronuclei43. The yeast Chd1 and Isw1 chromatin remodelers are similarly known to efficiently space nucleosomes13,27,44. This opens up the intriguing possibility that yeast Isw1 and Chd1 may differ functionally in preventing histone exchange and promoting chromatin integrity at lowly and highly transcribed genes respectively: Isw1 in the form of the Isw1b complex by remodeling nucleosomes that were retained on the DNA in spite of transcription, and Chd1 by reassembling nucleosomes in cis in the wake of RNAPII (Fig. 8). In agreement with this hypothesis we see that in the wildtype Isw1 catalytic activity is required to suppress both cryptic transcription (Fig. 4e–h) as well as histone exchange (Fig. 6f) and thus maintains chromatin integrity. Such different functional roles for Isw1 and Chd1 could also account for the reduced capacity of Isw1 or Ioc4 to compensate for the loss of Chd1 activity, assuming that Isw1b is less able to recapture histones displaced by transcription. A number of histone chaperones have been implicated in RNAPII transcription elongation and it will be interesting to determine which may function in concert with these remodelers in vivo.
Figure 8. Isw1b and Chd1 maintain chromatin integrity over coding regions.

Isw1b is recruited to ORFs by interaction of its Ioc4 subunit with H3K36me3, while Chd1 is thought to co-localize with RNAPII due to its interaction with known transcription elongation factors (grey). In the wildtype both remodelers cooperate to preserve chromatin structure by preventing histone exchange over ORFs. Deletion of ISW1, IOC4 or CHD1, or abrogation of Isw1 catalytic activity allows for trans-histone exchange to occur over coding sequences and results in increased levels of histone acetylation over ORFs. This perturbation of ORF chromatin structure exposes normally hidden cryptic promoters and results in the production of internal, cryptic transcripts.
Supplementary Material
1
1 update
Acknowledgments
We thank O. Rando (University of Massachusetts Medical School) and T. Tsukiyama (Fred Hutchinson Cancer Research Center) for providing yeast strains, members of the Workman Lab for helpful discussions, including Arnob Dutta for technical advice on EMSAs and Vikki Weake for critical reading of the manuscript. This work was supported by NIH grant R01GM047867 to J.L.W. and the Stowers Institute for Medical Research.
Footnotes
Author Contributions: M.S. designed and carried out experiments, analyzed the data and wrote the manuscript. M.S., M.M.G., S.V. and H.L. carried out bioinformatics analyses. Y.Z., L.F. and M.P.W. performed mass spectrometry and analyzed the results. J.L.W. supervised experiments and wrote the manuscript.
Accession codes: ChIP-chip and gene expression data sets have been deposited in NCBI's Gene Expression Omnibus under GEO accession number GSE32071.
References
- 1.Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem. 2002;277:49383–8. doi: 10.1074/jbc.M209294200. [DOI] [PubMed] [Google Scholar]
- 2.Kizer KO, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–16. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrozza MJ, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–92. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 4.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–8. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 5.Keogh MC, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 6.Drouin S, et al. DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet. 2010;6:e1001173. doi: 10.1371/journal.pgen.1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Govind CK, et al. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol Cell. 2010;39:234–46. doi: 10.1016/j.molcel.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, et al. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007;21:1422–30. doi: 10.1101/gad.1539307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkatesh S, et al. Co-transcriptional acetylation is a consequence of histone exchange. Nature. 2012 Under revision. [Google Scholar]
- 10.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 11.Elfring LK, Deuring R, McCallum CM, Peterson CL, Tamkun JW. Identification and characterization of Drosophila relatives of the yeast transcriptional activator SNF2/SWI2. Mol Cell Biol. 1994;14:2225–34. doi: 10.1128/mcb.14.4.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C. Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev. 1999;13:686–97. doi: 10.1101/gad.13.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vary JC, Jr, et al. Yeast Isw1p forms two separable complexes in vivo. Mol Cell Biol. 2003;23:80–91. doi: 10.1128/MCB.23.1.80-91.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada K, et al. Structure and mechanism of the chromatin remodelling factor ISW1a. Nature. 2011;472:448–53. doi: 10.1038/nature09947. [DOI] [PubMed] [Google Scholar]
- 15.Pinskaya M, Nair A, Clynes D, Morillon A, Mellor J. Nucleosome remodeling and transcriptional repression are distinct functions of Isw1 in Saccharomyces cerevisiae. Mol Cell Biol. 2009;29:2419–30. doi: 10.1128/MCB.01050-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morillon A, et al. Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell. 2003;115:425–35. doi: 10.1016/s0092-8674(03)00880-8. [DOI] [PubMed] [Google Scholar]
- 17.Quan TK, Hartzog GA. Histone H3K4 and K36 methylation, Chd1 and Rpd3S oppose the functions of Saccharomyces cerevisiae Spt4–Spt5 in transcription. Genetics. 2010;184:321–34. doi: 10.1534/genetics.109.111526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morettini S, et al. The chromodomains of CHD1 are critical for enzymatic activity but less important for chromatin localization. Nucleic Acids Res. 2011;39:3103–15. doi: 10.1093/nar/gkq1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simic R, et al. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003;22:1846–56. doi: 10.1093/emboj/cdg179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krogan NJ, et al. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol. 2002;22:6979–92. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warner MH, Roinick KL, Arndt KM. Rtf1 is a multifunctional component of the Paf1 complex that regulates gene expression by directing cotranscriptional histone modification. Mol Cell Biol. 2007;27:6103–15. doi: 10.1128/MCB.00772-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stokes DG, Tartof KD, Perry RP. CHD1 is concentrated in interbands and puffed regions of Drosophila polytene chromosomes. Proc Natl Acad Sci U S A. 1996;93:7137–42. doi: 10.1073/pnas.93.14.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley DE, Stokes DG, Perry RP. CHD1 interacts with SSRP1 and depends on both its chromodomain and its ATPase/helicase-like domain for proper association with chromatin. Chromosoma. 1999;108:10–25. doi: 10.1007/s004120050347. [DOI] [PubMed] [Google Scholar]
- 24.Gkikopoulos T, et al. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333:1758–60. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xella B, Goding C, Agricola E, Di Mauro E, Caserta M. The ISWI and CHD1 chromatin remodelling activities influence ADH2 expression and chromatin organization. Mol Microbiol. 2006;59:1531–41. doi: 10.1111/j.1365-2958.2005.05031.x. [DOI] [PubMed] [Google Scholar]
- 26.Lusser A, Urwin DL, Kadonaga JT. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat Struct Mol Biol. 2005;12:160–6. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- 27.Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem. 2006;281:16279–88. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung V, et al. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008;6:e277. doi: 10.1371/journal.pbio.0060277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–12. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pokholok DK, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–27. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Dion MF, et al. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–8. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- 32.Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Tsubota T, et al. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–12. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan T, et al. Cell cycle- and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet. 2008;4:e1000270. doi: 10.1371/journal.pgen.1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 36.Santos-Rosa H, et al. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell. 2003;12:1325–32. doi: 10.1016/s1097-2765(03)00438-6. [DOI] [PubMed] [Google Scholar]
- 37.Li B, et al. Histone H3 lysine 36 di-methylation (H3K36ME2) is sufficient to recruit the Rpd3S histone deacetylase complex and to repress spurious transcription. J Biol Chem. 2009 doi: 10.1074/jbc.M808220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tirosh I, Sigal N, Barkai N. Widespread remodeling of mid-coding sequence nucleosomes by Isw1. Genome Biol. 2010;11:R49. doi: 10.1186/gb-2010-11-5-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiriet C, Hayes JJ. Replication-independent core histone dynamics at transcriptionally active loci in vivo. Genes Dev. 2005;19:677–82. doi: 10.1101/gad.1265205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jamai A, Imoberdorf RM, Strubin M. Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell. 2007;25:345–55. doi: 10.1016/j.molcel.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 41.Kireeva ML, et al. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9:541–52. doi: 10.1016/s1097-2765(02)00472-0. [DOI] [PubMed] [Google Scholar]
- 42.Belotserkovskaya R, et al. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–3. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 43.Konev AY, et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317:1087–90. doi: 10.1126/science.1145339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson KM, Schultz MC. Replication-independent assembly of nucleosome arrays in a novel yeast chromatin reconstitution system involves antisilencing factor Asf1p and chromodomain protein Chd1p. Mol Cell Biol. 2003;23:7937–46. doi: 10.1128/MCB.23.22.7937-7946.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Team RDC. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna (Austria): 2008. [Google Scholar]
- 46.Holstege FC, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–28. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
1 update