Evidence of Missense Mutations on the Neuregulin 1 Gene Affecting Function of Prepulse Inhibition (original) (raw)
. Author manuscript; available in PMC: 2013 Feb 12.
Abstract
Background
Neuregulin 1 (NRG1) is one of the leading candidate genes in schizophrenia. Rodents with NRG1 knock-out showed significantly impaired prepulse inhibition (PPI) in the original report linking NRG1 to schizophrenia (Stefansson et al 2002). PPI is a widely used surrogate measure of psychosis in animal models and is considered a schizophrenia endophenotype. We hypothesized that if NRG1 influences PPI in rodents, then it should have a similar effect on PPI in humans.
Methods
We examined the potential neurophysiological effects of two nonsynonymous single nucleotide polymorphisms (SNPs) located on NRG1 (rs3924999 and rs10503929) on PPI. Genotyping were completed in 430 unrelated individuals, including 244 schizophrenia cases and 186 controls. PPI was available in a subgroup of 113 cases and 63 controls.
Results
Rs3924999 genotype was significantly associated with PPI (p=0.003): PPI was lowest in the subjects who were homozygous for the minor allele A/A carriers, intermediate in A/G carriers, and highest in homozygous major alleles G/G carriers. The associations persisted within cases (p=0.02) and controls (p=0.02) analyzed separately. An additive model suggested that rs3924999 alone contributes to 7.9% of the PPI variance. In contrast, rs10503929 genotype was not associated with PPI (p=0.85). Schizophrenia patients had reduced PPI compared to control subjects (p=0.04). Neither SNP was associated with schizophrenia (all p>0.37). However, schizophrenia patients with abnormal PPI may be associated with rs3924999 (p=0.05).
Conclusions
A missense mutation on rs3924999 of the neuregulin 1 gene may have a functional effect on prepulse inhibition in both schizophrenia and healthy control populations.
Keywords: schizophrenia, glutamatergic, psychosis, endophenotype, nonsynonymous, polymorphism, PPI, NRG, NMDA, SNP
Introduction
Neuregulin 1 (NRG1) is a leading candidate gene in schizophrenia. In Stefansson and colleagues' original report linking NRG1 to schizophrenia, mice with hypomorphic NRG1 had reduced prepulse inhibition (PPI) (1). PPI is a measure of inhibitory sensorimotor gating of the startle response, thought to be under genetic control, and shown to be abnormal in some schizophrenia patients and in their family members (2–5). PPI is increasingly being adopted as a surrogate measure of psychosis in animal studies for investigating neural mechanisms and antipsychotic treatments related to schizophrenia (6;7). We hypothesized that if NRG1 is involved in the function of PPI as shown in the rodent model, mutations in the gene might similarly affect PPI in humans, and may explain part of the PPI abnormality seen in schizophrenia patients.
NRG1 is a signaling protein that plays an important role in neural development and synaptic functions (8;9). Increasing interest in NRG1 as a schizophrenia candidate gene is motivated by a number of positive linkage and association findings (10–16). NRG1 is involved in neural development, glutamatergic function, and synaptic plasticity, all of which are plausibly relevant to the pathophysiology of schizophrenia (9;17;18). Furthermore, the gene is located at chromosomal 8p12–p21, a “hot spot” linked to schizophrenia (1;19–26). However, there are also notable failed replications in Caucasian (27–29) and Asian samples (30–33), and even in those studies reporting positive replications, the same SNPs or haplotypes did not always replicate [for reviews see (27) and (34)]. Difficulties in identifying susceptibility genes in schizophrenia are in part due to the likely clinical and genetic heterogeneity of this disorder (35–37). Schizophrenia is likely caused by multiple genes, perhaps acting in combination, which interact with environmental factors, leading to clinical manifestation of the overt disorder when the genetic loading and/or environmental insults cross a certain threshold of liability (35;38–41). Under these circumstances, identifying physiological measures that are stable, heritable and associated with the disease risk may facilitate identification of disease susceptibility genes. Such intermediate measures that index specific physiological deficits occurring along the causal pathway from a gene(s) to the clinical symptoms are termed endophenotypes (38). Endophenotypes are likely to be more homogenous than the clinical syndrome, with the effects of the associated gene being more robust than observed on the clinical syndrome. The current study is based on this paradigm, by testing the association of polymorphisms in NRG1 with both PPI and schizophrenia.
Since previous studies have not identified individual SNPs in NRG1 that consistently replicate across studies, and the functions of those replicated SNPs have as yet been determined, we focused our study on identifying variants of the NRG1 gene that were likely to be functional. Thus, we searched publicly available databases for polymorphisms on NRG1 that are known to change amino acid, or affect splicing or transcription, and have a minor allele frequency of at least 5%. These criteria led to only two nonsynonymous SNPs at the time of genotyping. Rs3924999 is located in the 2nd exon, a G>A polymorphism at the 2nd codon, which exchanges arginine to glutamine. Rs10503929 is in exon 12, a T>C polymorphism at the 2nd codon exchanging methionine to threonine. The functional effects of these amino acid exchanges are currently unknown. In this study, we examined the potential neurophysiological effects of these two missense mutations on PPI function in schizophrenia patients and healthy controls.
Methods and Materials
Subjects
Genotyping was carried out in 430 unrelated individuals, including 244 schizophrenia patients and 186 non-schizophrenia controls without family history of schizophrenia. Their age ranged from 16 to 70 years. Among them PPI was tested in 113 patients and 63 controls, who were enrolled in the later stage of the study when the PPI phenotype was added. Patients were recruited from the outpatient clinics of the Maryland Psychiatric Research Center and the neighboring community clinics. The Structured Clinical Interview for DSM-IV (SCID) was administered to all subjects to obtain DSM-IV diagnoses. Patients were individuals with DSM-IV schizophrenia who were medicated and clinically stable. The 113 patients tested for PPI were on the following medications: 9 patients were on first generation antipsychotic medications (mean±s.d.: 828±671 mg in CPZ equivalent), 83 patients were on second generation antipsychotic medications, including 21 on clozapine (442±165 mg; 25 on olanzapine (19±9 mg), 25 on risperidone (5±2 mg), 2 on quetiapine (750±212 mg), 6 on aripiprazole (19±6 mg), and 4 on ziprasidone (150±20 mg). Three patients were not on antipsychotic medications. The remaining 18 patients were on 2 antipsychotic medications. Symptoms in patients were measured with the 20-item Brief Psychiatric Rating Scale (BPRS) using 1 – 7 scores on each item. Non-schizophrenia controls were recruited using local community newspaper advertisements. The controls had no DSM IV Axis I diagnosis, no Axis II cluster A personality disorder, and no family history of psychosis in 3 generations. The University of Maryland IRB approved written informed consent was obtained from each subject.
Genotyping
Genotyping was conducted on DNA isolated from whole blood using the QIAamp DNA Blood Maxi Kit (Qiagen). The 2 SNPs (rs3924999 and rs10503929) were ordered from the Applied Biosystems (AB) assay-on-demand service (http://www.appliedbiosystems.com/) and genotyped using AB TaqMan technology. Genotyping was successful in all but 3 subjects for rs3924999 and 2 subjects for rs10503929.
PPI laboratory procedures
A hearing screen was carried out. All subjects had hearing threshold of 40 db or better at 500 Hz. The orbicularis oculi electromyographic (EMG) activity was recorded from the right eye, filtered (1–1000Hz, 60-Hz notch filter) and digitized at a 1-kHz rate. The acoustic stimuli were generated by Psylab Stand Alone Monitor and a tone generator (Contact Precision Instruments, Cambridge, MA) and delivered with headphones. EMG was directed through a Grass A.C. Amplifier (model 1CP511, Astro-Med., Inc.) and was acquired using commercially available hardware and software (BioPac, Gloeta, CA) (42). The sound intensity was measured by a Sound Level Meter (Model 2700, Quest Technologies, Oconomowoc, WI) on the A scale and a headphone coupler (Model EC-9A) with a standard 1-pound weight applied to the headphone during measurement. A standard 100 micro-voltage pulse was applied to the beginning of each record allowing off-line calibration. The EMG recording was processed off-line with a 100 Hz high pass filter, which was used for response amplitude measurement. To identify a response, baseline EMG was obtained by smoothing (binomial filter) a 100 ms artifact-free recording segment prior to stimulus onset. Response onset was defined by the first crossing from baseline within a 20–120-msec window after pulse onset; and was automatically identified using an in-house algorithm in an interactive software environment (Igor Pro, Wavemetrics, Inc, Lake Oswego, OR). The computer also presented the scoring trial-by-trial to the scorer. If the scorer concurred for a response, the algorithm calculated the response amplitude, defined as the difference of the most positive peak and most negative trough in a 20- to 150- msec window using the 100 Hz filtered recording. The trial was marked as no response if there was a lack of response onset. Scorers were trained to be in high agreement in defining response or non-response trials. The scorers were concurred on 95.8% of the trials (kappa=0.92) before scoring began. Non-responders to startle stimuli were defined as subjects who responded to less than 50% of the first 8 pulse alone trials. A session started with a 3-minute acclimation period with 70-dB white noise. Startle pulse alone trials contained 116 dB white noise lasting 40ms, and the prepulse-pulse trials contained a 20ms, 80 dB white noise prepulse, or 10 dB above the background noise. The prepulse and pulse was 120ms apart at stimulus onset. The first three responses were discarded. After the 3 trials, 18 pulse alone trials and 12 prepulse-pulse trials were administered in a pseudorandom order. Inter-trial intervals varied from 12 to 20s. Subjects were told to relax and keep their eyes open. PPI was calculated as percent suppression in response amplitudes [% PPI = (startle alone trial – prepulse trial)/startle alone trial × 100]. Responses were scored blind to subject identity and group membership.
Analysis
PPI data distributions were assessed for normality by Kolmogorov-Smirnov goodness-of-fit test. ANOVA was used to assess patient-control differences in PPI. Chi-square tests were used to compare observed genotype frequencies with those expected under Hardy-Weinberg equilibrium. Linkage disequilibrium between the two SNPs was evaluated using the D' statistic, as computed by Haploview software version 3.3 (43). Differences in allele frequencies between schizophrenia cases and controls were assessed by Chi-square test, as implemented in Haploview. The effect of genotype on PPI levels was tested by linear regression based on an additive model in which PPI was the dependent variable and the genotypes were predictors (coded as 0, 1, and 2 for homozygous minor alleles, heterozygous, and homozygous major alleles, respectively). Subsequent regression models tested the joint effects of age, schizophrenia diagnosis, and age by diagnosis interaction on PPI. Significant interactions and main effects were followed up by individual group post hoc tests with Tukey adjustment. For the calculation of relative risk and attributable risk, PPI was defined as abnormal if the value was below the mean minus 1 s.d. of the PPI in the control sample. All tests were two-tailed.
Results
1. PPI and schizophrenia
The distribution of PPI was consistent with normal distribution (Kolmogov-Smirnov Z statistic =0.71, p=0.69). Demographic information was presented at Table 1. Twenty-two (19.5%) of the patients and 19 (30.2%) of the controls were non-responders to startle sound (χ2 =2.59, p=0.14), and were excluded from PPI analysis. There were no significant differences between responders vs. non-responders in age (41.1± 11.0 vs. 37.6 ± 12.1, p=0.10), gender (female 16.4% vs. 83.6%; male 27.5% vs. 72.5%, p=0.10), or ethnic groups (Caucasians 16.5% vs. 83.5%; African Americans 32.9% vs. 67.1%; Asians 20.0% vs. 80.0%; p=0.15). There was no significant difference in mean eyeblink response amplitudes, but there was a significant difference in %PPI between schizophrenia patients and controls (Table 1). Age was modestly, albeit significantly, correlated with PPI (n=135, Pearson's r=−0.23, p=0.01). PPI did not differ significantly between those on first and second generation psychotic medications (36.4±20.3 vs. 41.1±25.5, p=0.66). Nor was medication dose correlated with PPI in any category of antipsychotic medications (all p>0.05). PPI was not significantly correlated with BPRS total score (r=−0.20, p=0.07, ns) or any subscales (all p≥0.09, ns).
Table 1.
Demographics and PPI group comparisons
| Schizophrenia probands (n=45) | Healthy Controls (n=42) | Chi-Square or F value | P value | |
|---|---|---|---|---|
| Age | 39.1 ± 10.8b | 37.1 ± 13.7 | 1.21 | 0.27 |
| Ethnicity (% cauc vs. afri vs. others) | 63:47:3 | 34:23:6 | 4.60a | 0.33 |
| Gender (%male) | 72.6 | 42.8 | 15.0a | 0.001c |
| Years of education | 12.8 ± 8.4b | 14.1 ± 2.5 | 1.35 | 0.25 |
| Eyeblink amplitude (mV) | 47.0 ± 31.3b | 47.1 ± 33.2 | 0.00 | 0.98 |
| %PPI | 40.8 ± 24.8b | 49.0 ± 15.8 | 4.00 | 0.04c |
2. NRG1 SNPs and schizophrenia phenotype
The distribution of genotypes was consistent with those predicted under Hardy Weinberg equilibrium in both the control and patient groups (all p>0.05). The two SNPs were in linkage equilibrium (D'=0.01) and therefore only single SNP analyses were performed. Genotypes at neither SNP were associated with presence of schizophrenia: the minor allele frequencies in cases (n=241) and controls (n=186) were 27.6% and 30.4% for rs3924999 (χ2 =0.79, p=0.37) and 9.5% and 10.2% for rs10503929 (χ2=0.11, p=0.74). The subjects with PPI phenotype were enrolled in the later stage of the study when the PPI measurement was implemented to the protocol. There was no change in enrollment criteria. There were no differences in key clinical characteristics, including age, gender, or ethnicity, between individuals phenotyped for PPI and those not phenotyped (all p>0.05). The subsequent results involve only subjects with PPI measurements unless specified otherwise.
3. NRG1 SNPs and PPI phenotype
The effect of neither SNPs on PPI differed significantly between schizophrenia cases and controls (all p ≥ 0.17 for the interaction term). Rs3924999 was significantly associated with PPI (F(2, 131) =5.95, p=0.003). This association persisted even with adjustment for age and gender (p=0.003). Percent PPI was lowest in homozygous minor alleles A/A or glutamine/glutamine carriers (mean ± SD: 33.9±26.0), intermediate in heterozygous A/G (38.4±25.2), and highest in homozygous major alleles G/G or arginine/arginine carriers (50.0±17.6) (Figure 1). Post hoc test showed that A/A and A/G carriers had significantly reduced PPI compared to G/G (both p=0.02). The genotypic effects on PPI were apparent also in stratified analyses of schizophrenia patients (34.8±29.7, 33.1±28.3, and 48.5±18.2, for A/A, A/G, and G/G, respectively, F(2, 87) =4.07, p=0.02) and healthy controls (31.6±14.9, 49.6±11.0, and 52.2±16.6, F(2, 41) =4.10, p=0.02) separately. A regression analysis based on the additive model suggested that rs3924999 alone contributed to 7.9% (R2 change) of the PPI variance (p=0.001). In contrast rs10503929 was not associated with PPI (p=0.85). A regression analysis suggested that this SNP contributed 0.0% to the PPI variance.
Figure 1.
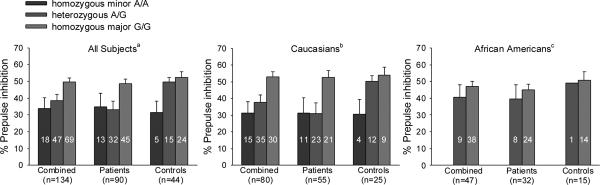
The figure shows the effects of rs3924999 on mean %PPI (error bars are SEM) in the combined sample, in Caucasians alone, and in African Americans alone. Numbers in bars indicate the number of subjects for each genotype.
aStatistically significant in the combined group (p=0.003): A/A and A/G carriers had significantly reduced PPI compared to G/G (both p=0.02). Similar effects were also found in schizophrenia patients (p=0.02) and in healthy controls (p=0.02).
bStatistically significant in the combined Caucasians group (p=0.01). Similar effects were also observed in Caucasian patients alone (p=0.02) and in Caucasian controls alone (p=0.03).
cStatistically not significant in African Americans. No homozygous minor allele genotype A/A in African Americans.
Endophenotypes can be used to reduce heterogeneity and identify subgroups of schizophrenia patients with similar biological deficits. With this in mind, we compared the allele frequencies in the subgroup of schizophrenia patients with abnormal PPI (defined as mean minus 1 s.d. of the controls) with the control group. The frequency of the rs3924999 mutation was significantly higher in patients with abnormal PPI (n=30) than the controls (n=44) (minor allele frequencies 45.0% and 28.4%, respectively, χ2 = 4.3, p=0.05, Fisher's exact test, two tailed). When comparing patients with abnormal PPI vs. controls with abnormal PPI (n=5, minor allele frequency 20.0%), there was no significant difference between the two groups (χ2 = 2.21, p=0.14). Therefore, the data did not support that this mutation had significantly more impact on the patients' PPI; even though the insignificant finding may be due to the small number of controls with abnormal PPI.
4. NRG1 rs3924999 and ethnic groups
Our sample included 239 Caucasians (132 patients and 107 controls), 169 African-Americans (106 patients and 63 controls), and 22 from other ethnic groups. Allele frequencies for rs3924999 in our sample (39.4% and 10.1%, in European Caucasians and African Americans) correspond closely to those reported in NCBI dbSNP (43.8% in North American Caucasians and 10.9% in African Americans; p=0.86, ns). Similarly, allele frequencies for rs10503929 in our sample (14.9% and 2.4%, in European Caucasians and African Americans) also correspond closely to those reported in NCBI dbSNP (12.5% and 0.0%, p=0.43, ns). Because of these ethnic differences in allele frequencies, we reanalyzed the rs3924999 data in Caucasians and African-Americans separately.
In Caucasians, there was a significant rs3924999 genotype effect on PPI (31.3±26.6, 37.5±27.0, and 52.9±17.4 for A/A, A/G, and G/G, respectively, p=0.01) (Figure 1). Similar effects were observed in Caucasian patients alone (31.4±30.0, 31.0±30.4, and 52.5±18.8, p=0.02) and in Caucasian controls alone (30.7±17.0, 50.1±12.2, and 53.9±14.5, p=0.03). In Caucasians, 8.0% of the controls and 39.3% of the patients had abnormal PPI. The relative risk (RR) of abnormal PPI for having A allele was 2.08 (95% confidence interval: 1.23 – 2.51). Based on a 39.4% population frequency of the A allele in Caucasians, the attributable risk (AR) of the A allele for abnormal PPI was 29.8%. The additive genotype effect of rs3924999 contributed to 11.3% of the variance of PPI in Caucasian subjects (p=0.002).
In African Americans, the genotypic effect on PPI was not significant (no A/A carrier, 40.6±22.8 and 47.2±17.8 for A/G and G/G, respectively, p=0.35, ns), indicating that a much larger sample of African Americans would be needed to allow meaningful interpretation of the effect of the minor allele on PPI in this population. In African Americans, the RR and AR of abnormal PPI for having A allele were 1.34 (95% CI: 0.40 – 5.38) and 3.2%, respectively. The additive genotype effect of rs3924999 contributed to 2.0% of the variance in PPI (p=0.35, ns).
Discussion
Prepulse inhibition has been studied as a neurophysiological endophenotype in schizophrenia, thought to be heritable and associated with the genetic risk of schizophrenia (2–5;44). The finding of association between reduced PPI in the carriers of a missense mutation on rs3924999 is the first reported association between a potentially functional variant in the NRG1 gene and the schizophrenia-related endophenotype in humans. Other findings included that schizophrenia patients with abnormal PPI had over-representation of the missense mutation, while there was no significant association of the mutation when all schizophrenia patients were considered.
The finding of association between PPI and a NRG1 missense mutation is predicated on the a priori hypothesis of the involvement of NRG1 on PPI, which was based on a rodent NRG1 knockout model, described in Stefansson et al's original report linking this gene to schizophrenia (1). However, this mutation was not significantly over-represented in the schizophrenia phenotype. Rather, it was associated with the PPI phenotype regardless of schizophrenia presence. Three reported studies have genotyped rs3924999 in schizophrenia patients. The first report found a significant association between rs3924999 and schizophrenia in a Chinese Han sample (12). Two subsequent studies did not replicate the association (27, 30). The lack of significant association of rs3924999 and schizophrenia in this study is thus another failed replication. In comparison, a narrower phenotype, i.e., schizophrenia patients with abnormal PPI, had over-representation of the missense mutation. This finding is preliminary given the small sample size.
There were two additional studies that found associations between psychosis-related alternative phenotypes and rs3924999 (45;46); one study included patients with Alzheimer's Disease with Psychosis (45), while the other included individuals with schizotypal personality features (46). Similar to the effects of the genotype on PPI impairment in the current study, the A/A carriers had the highest score on the schizotypal scale, while the A/G carriers scored in-between the A/A and G/G carriers. Schizotypal personality traits are thought as a sub-clinical representation of aspects of schizophrenia and are under significant genetic control (47–49). PPI has been found to be correlated with the severity of schizotypal personality (50;51).
How variation in rs3924999 or the NRG1 gene affects the risk for schizophrenia and PPI abnormality is not clear. Further elaboration on the NRG1 gene and its protein products is needed to address this question. In the original study by Steffanson and colleagues using heterozygote NRG1 knockout model, most of the mouse exon 11, which encodes the transmembrane domain, was replaced (1). The animals showed normal startle response but reduced PPI compared to the wild types. We observed similar findings with the rs3924999 polymorphism, which is located at human NRG1 exon 2. The NRG1 gene consists of about 15 exons; transcripts from these exons produce at least 14 isoforms, from which three main types of NRG1 proteins are assembled (9); but also see (52). All three main types have the transmembrane domain, which anchors the proteins on the membrane with different extracellular and intracellular domains. A transmembrane domain knockout would have affected all 3 NRG1 protein types. Therefore, the transmembrane domain knockout model does not exclusively implicate exon 11 in the PPI deficit. Exon 2 codes the N-terminal sequence of type 1 NRG1 protein. Type I and II NRG1 proteins have an extracellular immunoglobulin-like (lg) domain. The NRG1 lg domain knockout mice showed no impairment in PPI (53) but rather showed impaired latent inhibition (54). Since the N-terminal type 1 sequence needs to attach to the lg domain to form the type 1 NRG1 protein (9), one may assume that this mouse model would have altered type 1 NRG1. However, it is not clear whether the type 1 N-terminal sequence isoform was still translated and available to serve other functions. A recent report examining different types of NRG1 transcripts showed that postmortem hippocampal tissue of schizophrenia patients had deregulated type I mRNA, suggesting that type 1 NRG1 may be particularly relevant to the pathology of schizophrenia (55). A mouse model with targeted transgenic alternation at the exon 2 may be needed to provide precise understanding between the genotype/phenotype relations in mice and in humans. Adding to the already complicated picture, mice with hypomorphic type III NRG1 (its type-specific isoform is coded by exon 8 in humans) seemed to have impaired PPI (56). Finally, a majority of the positive association studies identified positive markers around the 5' end of the gene, even though the positively associated SNPs or haplotypes were not always the same (1;10–15). The only exon located in this area is exon 1, which encodes the N-terminal sequence of the type 2 NRG1 protein (9;57), but no known functional mutation within this exon has been found segregated with schizophrenia. Overall, while accumulating evidence supports the link between the NRG1 gene and schizophrenia, there is insufficient information to reconcile these very different aspects of the genotype/phenotype observations available thus far. The evidence seems to suggest that multiple alleles of NRG1 may be involved and each may variably increase the risk of schizophrenia and/or the associated endophenotypes.
NRG1 and its ErbB receptors play important roles in glutamatergic neurotransmission. ErbB receptors colocalize with NMDA receptors at neuronal synaptic sites and together they may regulate synaptic plasticity (58;59). NRG1 changes the composition of glutamatergic neurotransmitter receptors in prefrontal cortex and cerebellum (17;18;60) and regulates cellular NMDA receptor currents in the prefrontal cortex (17). Mice with hypomorphic NRG1 had reduced NMDA receptors in the forebrain (1). Studies show that noncompetitive N-methyl-D-aspartate (NMDA) antagonist phencyclidine induces schizophrenia-like symptoms in humans and reduce PPI in monkeys (61). However, another NIMA antagonist ketamine has been shown not to affect PPI or even increase PPI in humans (62–64) (see (44) for a review). It is plausible that variation in the NRG1 gene affects PPI through alterations in the glutamatergic signaling, but further studies to delineate the genetic and neurochemical pathways leading to PPI change are needed.
Many genetic mutations have been associated with deficient PPI, mostly in animal models. Mutations in 2 genes at the 22q11 locus, Tbx1 and Gnb1l, were shown to cause reduced PPI in mouse (65). Transgenic mouse carrying Huntington's disease (HD) gene showed significant impairment in PPI (66). Tbx1 gene has been associated with Asperger's syndrome. Both Asperger's syndrome and HD patients have shown to have deficient PPI (65;67). Mutations in the Prodh gene at 22q, the PACAP gene at 18p, glutermatergic receptor genes mGluR1 and mGluR5 (68;69), beta3 nicotinic receptor subunit (70), alpha3 subunit of the GABA(A) gene (71), GAD65 (72), Homer1 (73), ZDHHC8 (74) and a number of other genes have shown to caused deficient PPI in animals; and many of these genes have been associated with schizophrenia, a disease associated with deficient PPI. A more comprehensive review of mouse genetic models for PPI can be found in Geyer et al (75). However, the current study appeared to be one of the first direct associations between a genetic mutation and PPI in humans.
The study is limited by its modest sample size. In addition, multiple comparisons were made when data were analyzed by several stratifications, which may also introduce Type I error. Replication studies will be critical for increasing the confidence for the association findings. Certainly, even if our results are replicated, there remains the possibility that this missense mutation is not functional at the protein level, and is rather in linkage disequalibrium with another etiological locus that affects the PPI function. We have attempted to address population stratification of allele frequencies between major ethnic groups. However, our results can potentially still be affected by sub-population stratification, which is an inherent concern in population-based association studies and may lead to Type I error (76). Future replications of the findings using a different study design that include appropriate controls for stratification such as family-based association and genomic control would be critical. The minor allele frequency of rs3924999 was much smaller in the African American population, a larger sample would be needed to examine whether this mutation would also affect PPI in this population.
There was a marginally insignificant correlation between symptoms and PPI (p=0.07). Acute symptoms as a “state” factor may affect the genotype-phenotype results. To address this issue, data were re-analyzed using the BPRS score as a covariate. We found that rs3924999 remained significantly associated with PPI in all the patients (p=0.02) and in Caucasian patients alone (p=0.05). This as well as findings in healthy control subjects suggested that symptoms did not substantially affect the association between rs3924999 and PPI.
Since the time of genotyping on our sample, several other potentially functional nonsynonymous SNPs and polymorphisms at the promotor regions and splicing sites of the NRG1 gene were identified (77;78). Future replication efforts on NRG1 and PPI should consider including these and perhaps other potentially functional loci.
In conclusion, the results of this study suggested that a missense mutation on the SNP rs3924999 located in exon 2 of the neuregulin 1 gene may have a functional effect on prepulse inhibition in both schizophrenia and non-schizophrenia populations.
Acknowledgements
We thank Robert P. McMahon, Ph.D. for statistical consultations. Support was received from NIMH grants MH 68282, 49826, 67014, 70644, 68580, and from General Clinical Research Center grant # M01-RR16500
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures None.
Reference List
- 1.Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch.Gen.Psychiatry. 1990;47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- 3.Cadenhead KS, Light GA, Geyer MA, Braff DL. Sensory gating deficits assessed by the P50 event-related potential in subjects with schizotypal personality disorder. Am.J.Psychiatry. 2000;157:55–59. doi: 10.1176/ajp.157.1.55. [DOI] [PubMed] [Google Scholar]
- 4.Anokhin AP, Heath AC, Myers E, Ralano A, Wood S. Genetic influences on prepulse inhibition of startle reflex in humans. Neurosci Lett. 2003;353:45–48. doi: 10.1016/j.neulet.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Kumari V, Das M, Zachariah E, Ettinger U, Sharma T. Reduced prepulse inhibition in unaffected siblings of schizophrenia patients. Psychophysiology. 2005;42:588–594. doi: 10.1111/j.1469-8986.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- 6.Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. 1994;51:139–154. doi: 10.1001/archpsyc.1994.03950020063007. [DOI] [PubMed] [Google Scholar]
- 7.Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 8.Buonanno A, Fischbach GD. Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr.Opin.Neurobiol. 2001;11:287–296. doi: 10.1016/s0959-4388(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 9.Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- 10.Stefansson H, Thorgeirsson TE, Gulcher JR, Stefansson K. Neuregulin 1 in schizophrenia: out of Iceland. Mol.Psychiatry. 2003;8:639–640. doi: 10.1038/sj.mp.4001384. [DOI] [PubMed] [Google Scholar]
- 11.Williams NM, Preece A, Spurlock G, Norton N, Williams HJ, Zammit S, et al. Support for genetic variation in neuregulin 1 and susceptibility to schizophrenia. Mol.Psychiatry. 2003;8:485–487. doi: 10.1038/sj.mp.4001348. [DOI] [PubMed] [Google Scholar]
- 12.Yang JZ, Si TM, Ruan Y, Ling YS, Han YH, Wang XL, et al. Association study of neuregulin 1 gene with schizophrenia. Mol.Psychiatry. 2003;8:706–709. doi: 10.1038/sj.mp.4001377. [DOI] [PubMed] [Google Scholar]
- 13.Petryshen TL, Middleton FA, Kirby A, Aldinger KA, Purcell S, Tahl AR, et al. Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol.Psychiatry. 2005;10:366–74. 328. doi: 10.1038/sj.mp.4001608. [DOI] [PubMed] [Google Scholar]
- 14.Tang JX, Chen WY, He G, Zhou J, Gu NF, Feng GY, et al. Polymorphisms within 5' end of the Neuregulin 1 gene are genetically associated with schizophrenia in the Chinese population. Mol.Psychiatry. 2004;9:11–12. doi: 10.1038/sj.mp.4001436. [DOI] [PubMed] [Google Scholar]
- 15.Norton N, Moskvina V, Morris DW, Bray NJ, Zammit S, Williams NM, et al. Evidence that interaction between neuregulin 1 and its receptor erbB4 increases susceptibility to schizophrenia. Am J Med Genet B Neuropsychiatr.Genet. 2006;141:96–101. doi: 10.1002/ajmg.b.30236. [DOI] [PubMed] [Google Scholar]
- 16.Thomson PA, Christoforou A, Morris SW, Adie E, Pickard BS, Porteous DJ, et al. Association of Neuregulin 1 with schizophrenia and bipolar disorder in a second cohort from the Scottish population. Mol.Psychiatry. 2007;12:94–104. doi: 10.1038/sj.mp.4001889. [DOI] [PubMed] [Google Scholar]
- 17.Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci. 2005;25:4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat.Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- 19.Kendler KS, MacLean CJ, O'Neill FA, Burke J, Murphy B, Duke F, et al. Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry. 1996;153:1534–1540. doi: 10.1176/ajp.153.12.1534. [DOI] [PubMed] [Google Scholar]
- 20.Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, et al. Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat.Genet. 1998;20:70–73. doi: 10.1038/1734. [DOI] [PubMed] [Google Scholar]
- 21.Kaufmann CA, Suarez B, Malaspina D, Pepple J, Svrakic D, Markel PD, et al. NIMH Genetics Initiative Millenium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med.Genet. 1998;81:282–289. [PubMed] [Google Scholar]
- 22.Brzustowicz LM, Honer WG, Chow EW, Little D, Hogan J, Hodgkinson K, et al. Linkage of familial schizophrenia to chromosome 13q32. Am J Hum Genet. 1999;65:1096–1103. doi: 10.1086/302579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, et al. Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21–22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3–24 and 20q12.1–11.23. Am J Hum Genet. 2001;68:661–673. doi: 10.1086/318788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suarez BK, Duan J, Sanders AR, Hinrichs AL, Jin CH, Hou C, et al. Genomewide Linkage Scan of 409 European-Ancestry and African American Families with Schizophrenia: Suggestive Evidence of Linkage at 8p23.3–p21.2 and 11p13.1–q14.1 in the Combined Sample. Am J Hum Genet. 2006;78:315–333. doi: 10.1086/500272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badner JA, Gershon ES. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol.Psychiatry. 2002;7:405–411. doi: 10.1038/sj.mp.4001012. [DOI] [PubMed] [Google Scholar]
- 26.Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan J, Martinez M, Sanders AR, Hou C, Krasner AJ, Schwartz DB, et al. Neuregulin 1 (NRG1 ) and schizophrenia: analysis of a US family sample and the evidence in the balance. Psychol Med. 2005;35:1599–1610. doi: 10.1017/S0033291705005428. [DOI] [PubMed] [Google Scholar]
- 28.Thiselton DL, Webb BT, Neale BM, Ribble RC, O'Neill FA, Walsh D, et al. No evidence for linkage or association of neuregulin-1 (NRG1) with disease in the Irish study of high-density schizophrenia families (ISHDSF) Mol.Psychiatry. 2004;9:777–783. doi: 10.1038/sj.mp.4001530. [DOI] [PubMed] [Google Scholar]
- 29.Ingason A, Soeby K, Timm S, Wang AG, Jakobsen KD, Fink-Jensen A, et al. No significant association of the 5' end of neuregulin 1 and schizophrenia in a large Danish sample. Schizophr Res. 2006;83:1–5. doi: 10.1016/j.schres.2005.12.850. [DOI] [PubMed] [Google Scholar]
- 30.Hong CJ, Huo SJ, Liao DL, Lee K, Wu JY, Tsai SJ. Case-control and family-based association studies between the neuregulin 1 (Arg38Gln) polymorphism and schizophrenia. Neurosci Lett. 2004;366:158–161. doi: 10.1016/j.neulet.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 31.Li T, Stefansson H, Gudfinnsson E, Cai G, Liu X, Murray RM, et al. Identification of a novel neuregulin 1 at-risk haplotype in Han schizophrenia Chinese patients, but no association with the Icelandic/Scottish risk haplotype. Mol.Psychiatry. 2004;9:698–704. doi: 10.1038/sj.mp.4001485. [DOI] [PubMed] [Google Scholar]
- 32.Zhao X, Shi Y, Tang J, Tang R, Yu L, Gu N, et al. A case control and family based association study of the neuregulin1 gene and schizophrenia. J Med Genet. 2004;41:31–34. doi: 10.1136/jmg.2003.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwata N, Suzuki T, Ikeda M, Kitajima T, Yamanouchi Y, Inada T, et al. No association with the neuregulin 1 haplotype to Japanese schizophrenia. Mol.Psychiatry. 2004;9:126–127. doi: 10.1038/sj.mp.4001456. [DOI] [PubMed] [Google Scholar]
- 34.Tosato S, Dazzan P, Collier D. Association between the neuregulin 1 gene and schizophrenia: a systematic review. Schizophr Bull. 2005;31:613–617. doi: 10.1093/schbul/sbi043. [DOI] [PubMed] [Google Scholar]
- 35.McGue M, Gottesman II, Rao DC. The transmission of schizophrenia under a multifactorial threshold model. Am.J.Hum.Genet. 1983;35:1161–1178. [PMC free article] [PubMed] [Google Scholar]
- 36.Kendler KS, Myers JM, O'Neill FA, Martin R, Murphy B, MacLean CJ, et al. Clinical features of schizophrenia and linkage to chromosomes 5q, 6p, 8p, and 10p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry. 2000;157:402–408. doi: 10.1176/appi.ajp.157.3.402. [DOI] [PubMed] [Google Scholar]
- 37.Thaker GK. Current progress in schizophrenia research. Search for genes of schizophrenia: back to defining valid phenes. J Nerv Ment Dis. 2002;190:411–412. doi: 10.1097/00005053-200206000-00012. [DOI] [PubMed] [Google Scholar]
- 38.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am.J.Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 39.Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 40.Tsuang MT. Defining alternative phenotypes for genetic studies: what can we learn from studies of schizophrenia? Am.J.Med.Genet. 2001;105:8–10. [PubMed] [Google Scholar]
- 41.Cannon TD, Gasperoni TL, van Erp TG, Rosso IM. Quantitative neural indicators of liability to schizophrenia: implications for molecular genetic studies. Am.J.Med.Genet. 2001;105:16–19. [PubMed] [Google Scholar]
- 42.Hong LE, Summerfelt A, Wonodi I, Adami H, Buchanan RW, Thaker GK. Independent domains of inhibitory gating in schizophrenia and the effect of stimulus interval. Am J Psychiatry. 2007;164:61–65. doi: 10.1176/ajp.2007.164.1.61. [DOI] [PubMed] [Google Scholar]
- 43.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 44.Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- 45.Go RC, Perry RT, Wiener H, Bassett SS, Blacker D, Devlin B, et al. Neuregulin-1 polymorphism in late onset Alzheimer's disease families with psychoses. Am J Med Genet B Neuropsychiatr.Genet. 2005;139:28–32. doi: 10.1002/ajmg.b.30219. [DOI] [PubMed] [Google Scholar]
- 46.Lin HF, Liu YL, Liu CM, Hung SI, Hwu HG, Chen WJ. Neuregulin 1 gene and variations in perceptual aberration of schizotypal personality in adolescents. Psychol Med. 2005;35:1589–1598. doi: 10.1017/S0033291705005957. [DOI] [PubMed] [Google Scholar]
- 47.Meehl PE. Schizotaxia, schizotypy and schizophrenia. American Psychologist. 1962;17:827–838. [Google Scholar]
- 48.Lyons MJ, Toomey R, Faraone SV, Kremen WS, Yeung AS, Tsuang MT. Correlates of psychosis proneness in relatives of schizophrenic patients. Journal of Abnormal Psychology. 1995;104:390–394. doi: 10.1037//0021-843x.104.2.390. [DOI] [PubMed] [Google Scholar]
- 49.Kendler KS, Czajkowski N, Tambs K, Torgersen S, Aggen SH, Neale MC, et al. Dimensional representations of DSM-IV cluster A personality disorders in a population-based sample of Norwegian twins: a multivariate study. Psychol Med. 2006;36:1583–1591. doi: 10.1017/S0033291706008609. [DOI] [PubMed] [Google Scholar]
- 50.Cadenhead KS, Geyer MA, Braff DL. Impaired startle prepulse inhibition and habituation in patients with schizotypal personality disorder. Am J Psychiatry. 1993;150:1862–1867. doi: 10.1176/ajp.150.12.1862. [DOI] [PubMed] [Google Scholar]
- 51.Evans LH, Gray NS, Snowden RJ. Prepulse inhibition of startle and its moderation by schizotypy and smoking. Psychophysiology. 2005;42:223–231. doi: 10.1111/j.1469-8986.2005.00280.x. [DOI] [PubMed] [Google Scholar]
- 52.Steinthorsdottir V, Stefansson H, Ghosh S, Birgisdottir B, Bjornsdottir S, Fasquel AC, et al. Multiple novel transcription initiation sites for NRG1. Gene. 2004;342:97–105. doi: 10.1016/j.gene.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 53.Rao ST, Zhou MM, Merker RJ, Mann MA, Fischbach GD, Gingrich JA. Behavioral alteration in mice with a reduce neuregulin-1 ig domain isoform. Behavioral Genetics Association. 2004 [Program No 35] [Google Scholar]
- 54.Rimer M, Barrett DW, Maldonado MA, Vock VM, Gonzalez-Lima F. Neuregulin-1 immunoglobulin-like domain mutant mice: clozapine sensitivity and impaired latent inhibition. NeuroReport. 2005;16:271–275. doi: 10.1097/00001756-200502280-00014. [DOI] [PubMed] [Google Scholar]
- 55.Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, et al. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5' SNPs associated with the disease. Proc.Natl.Acad.Sci.U.S.A. 2006;103:6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen YJ, Talmage DA, Role LW. Mice haplo-insufficient for cysteine-rich domain containing neuregulin 1 have impaired prepulse inhibition, an endophenotype related to schizophrenia. Soc.Neurosci. 2004 [Program No 38.2.2] [Google Scholar]
- 57.Chen MS, Bermingham-McDonogh O, Danehy FT, Jr., Nolan C, Scherer SS, Lucas J, et al. Expression of multiple neuregulin transcripts in postnatal rat brains. J Comp Neurol. 1994;349:389–400. doi: 10.1002/cne.903490306. [DOI] [PubMed] [Google Scholar]
- 58.Garcia RA, Vasudevan K, Buonanno A. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc.Natl.Acad.Sci.U.S.A. 2000;97:3596–3601. doi: 10.1073/pnas.070042497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol.Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 60.Ozaki M, Sasner M, Yano R, Lu HS, Buonanno A. Neuregulin-beta induces expression of an NMDA-receptor subunit. Nature. 1997;390:691–694. doi: 10.1038/37795. [DOI] [PubMed] [Google Scholar]
- 61.Javitt DC, Lindsley RW. Effects of phencyclidine on prepulse inhibition of acoustic startle response in the macaque. Psychopharmacology (Berl) 2001;156:165–168. doi: 10.1007/s002130100758. [DOI] [PubMed] [Google Scholar]
- 62.van Berckel BN, Oranje B, van Ree JM, Verbaten MN, Kahn RS. The effects of low dose ketamine on sensory gating, neuroendocrine secretion and behavior in healthy human subjects. Psychopharmacology (Berl) 1998;137:271–281. doi: 10.1007/s002130050620. [DOI] [PubMed] [Google Scholar]
- 63.Duncan EJ, Madonick SH, Parwani A, Angrist B, Rajan R, Chakravorty S, et al. Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology. 2001;25:72–83. doi: 10.1016/S0893-133X(00)00240-2. [DOI] [PubMed] [Google Scholar]
- 64.Abel KM, Allin MP, Hemsley DR, Geyer MA. Low dose ketamine increases prepulse inhibition in healthy men. Neuropharmacology. 2003;44:729–737. doi: 10.1016/s0028-3908(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 65.Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc.Natl.Acad.Sci.U.S.A. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington's disease. J Neurol.Neurosurg.Psychiatry. 1995;58:192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brody SA, Conquet F, Geyer MA. Disruption of prepulse inhibition in mice lacking mGluR1. Eur.J Neurosci. 2003;18:3361–3366. doi: 10.1111/j.1460-9568.2003.03073.x. [DOI] [PubMed] [Google Scholar]
- 69.Brody SA, Dulawa SC, Conquet F, Geyer MA. Assessment of a prepulse inhibition deficit in a mutant mouse lacking mGlu5 receptors. Mol.Psychiatry. 2004;9:35–41. doi: 10.1038/sj.mp.4001404. [DOI] [PubMed] [Google Scholar]
- 70.Cui C, Booker TK, Allen RS, Grady SR, Whiteaker P, Marks MJ, et al. The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci. 2003;23:11045–11053. doi: 10.1523/JNEUROSCI.23-35-11045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yee BK, Keist R, von Boehmer L, Studer R, Benke D, Hagenbuch N, et al. A schizophrenia-related sensorimotor deficit links alpha 3-containing GABAA receptors to a dopamine hyperfunction. Proc.Natl.Acad.Sci.U.S.A. 2005;102:17154–17159. doi: 10.1073/pnas.0508752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heldt SA, Green A, Ressler KJ. Prepulse inhibition deficits in GAD65 knockout mice and the effect of antipsychotic treatment. Neuropsychopharmacology. 2004;29:1610–1619. doi: 10.1038/sj.npp.1300468. [DOI] [PubMed] [Google Scholar]
- 73.Szumlinski KK, Lominac KD, Kleschen MJ, Oleson EB, Dehoff MH, Schwarz MK, et al. Behavioral and neurochemical phenotyping of Homer1 mutant mice: possible relevance to schizophrenia. Genes Brain Behav. 2005;4:273–288. doi: 10.1111/j.1601-183X.2005.00120.x. [DOI] [PubMed] [Google Scholar]
- 74.Mukai J, Liu H, Burt RA, Swor DE, Lai WS, Karayiorgou M, et al. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat.Genet. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- 75.Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol.Psychiatry. 2002;7:1039–1053. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- 76.Cardon LR, Palmer LJ. Population stratification and spurious allelic association. Lancet. 2003;361:598–604. doi: 10.1016/S0140-6736(03)12520-2. [DOI] [PubMed] [Google Scholar]
- 77.Walss-Bass C, Liu W, Lew DF, Villegas R, Montero P, Dassori A, et al. A novel missense mutation in the transmembrane domain of neuregulin 1 is associated with schizophrenia. Biol.Psychiatry. 2006;60:548–553. doi: 10.1016/j.biopsych.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 78.Hall J, Whalley HC, Job DE, Baig BJ, McIntosh AM, Evans KL, et al. A neuregulin 1 variant associated with abnormal cortical function and psychotic symptoms. Nat.Neurosci. 2006 doi: 10.1038/nn1795. [DOI] [PubMed] [Google Scholar]