Orthologous Gene Clusters and Taxon Signature Genes for Viruses of Prokaryotes (original) (raw)
Abstract
Viruses are the most abundant biological entities on earth and encompass a vast amount of genetic diversity. The recent rapid increase in the number of sequenced viral genomes has created unprecedented opportunities for gaining new insight into the structure and evolution of the virosphere. Here, we present an update of the phage orthologous groups (POGs), a collection of 4,542 clusters of orthologous genes from bacteriophages that now also includes viruses infecting archaea and encompasses more than 1,000 distinct virus genomes. Analysis of this expanded data set shows that the number of POGs keeps growing without saturation and that a substantial majority of the POGs remain specific to viruses, lacking homologues in prokaryotic cells, outside known proviruses. Thus, the great majority of virus genes apparently remains to be discovered. A complementary observation is that numerous viral genomes remain poorly, if at all, covered by POGs. The genome coverage by POGs is expected to increase as more genomes are sequenced. Taxon-specific, single-copy signature genes that are not observed in prokaryotic genomes outside detected proviruses were identified for two-thirds of the 57 taxa (those with genomes available from at least 3 distinct viruses), with half of these present in all members of the respective taxon. These signatures can be used to specifically identify the presence and quantify the abundance of viruses from particular taxa in metagenomic samples and thus gain new insights into the ecology and evolution of viruses in relation to their hosts.
INTRODUCTION
There are an estimated 1031 virus particles on Earth (1, 2), most of which are bacteriophages in the oceans, causing on the order of 1023 infections per second globally (3). Phages also far outnumber eukaryotic viruses in the human intestinal tract and have been found to exist in relatively stable populations that are distinctive to individuals, even between identical twins (4). Recent advances in sequencing technologies combined with virus isolation protocols (5, 6) have enabled massive acquisition of virus genome sequences, most of which are not similar to any known genes, suggesting that most of the genetic diversity of viruses remains to be discovered (7, 8). Many novel features, including unexpected genes and unusual genome architectures, continue to be discovered in phages that infect bacteria (9–12) as well as in archaeal viruses (13–17).
Although the vast majority of viruses remain to be discovered, genome sequencing has already generated a large collection of well over a thousand phage genomes that altogether encompass >105 genes. With the fast-paced, and still accelerating, accumulation of genome sequences in the databases, automated approaches for genome analysis are essential to keep up with the data and provide the foundation for subsequent detailed evolutionary and functional studies. A well-established approach in comparative genomics involves construction of clusters of orthologous genes from large sets of diverse organisms (18–20). Computational methods have been developed to delineate clusters of likely orthologs from diverse organisms, and several collections of ortholog clusters have been constructed and have become indispensable tools for genome annotation and phylogenomics (21–23).
In previous work, we constructed >2,000 phage orthologous groups (POGs), including genes from >500 phage genomes (24–26). We found that despite the ability of phages to acquire genes from their bacterial hosts, at least half of these POGs consist of genes that were never or only very rarely observed in bacteria outside recently integrated prophages. In addition, the fraction of phage-specific gene families among all phage genes has remained high and stable over the last decade (24) despite the advent of next-generation sequencing technologies, the burst of interest in sampling diverse habitats, and the proliferation of phage genes and genomes in public databases. Some of these phage-specific genes could represent hallmarks of families or even larger groups of phages and could serve as diagnostic probes of phage presence in a given environment.
Here, we expand the POG collection, previously limited to the genomes of double-stranded DNA (dsDNA) phages, to incorporate over 1,000 genomes, including genomes of single-stranded DNA (ssDNA) and ss- and dsRNA phages, as well as archaeal viruses (notwithstanding the inclusion of archaeal viruses, we keep the acronym POGs for continuity and convenience). Using this expanded set of POGs, we demonstrate that despite the genomic fluidity that is characteristic of the viral pangenome, many taxa contain one or more genes that can serve as diagnostic signatures of the presence of viruses from a given taxon.
MATERIALS AND METHODS
The data set: viruses and genomes.
The query used for retrieval from the NCBI Nucleotide database included the following: Viruses[Organism] NOT cellular organisms [ORGN] NOT srcdb_refseq[PROP] AND vhost bacteria[filter] AND “complete genome”[All Fields]. It was followed by curation to remove entries that matched the query but which were not actually complete genomes, including merely complete coding sequences, partial genomes, single genes, and mutants. The nucleotide database contains several genomes labeled as a “complete sequence” rather than a “complete genome,” most notably over 30 mycophages, that were added separately, again followed by manual curation.
As described previously, single-linkage clustering was used to join groups of very closely related (essentially the same) phages. The resulting group contains the union of all proteins from each member phage genome. For the larger genomes containing ≥20 genes, viruses sharing ≥90% of their genes were joined, whereas for the smaller genomes of <20 genes, viruses must share all of their genes to be joined. Shared genes were defined as symmetric best matches between a pair of phage genomes. However, no procedure using gene content could be found to work for the genus Microvirus, because the phage species ϕ_X174_, G4, and alpha3 share the same set of 11 proteins (44), so the viruses of the genus Microvirus were specifically exempted from this procedure in order to allow these species to remain distinct despite their identical gene content.
Orthologous groups.
The Edge-Search algorithm (26) was used to implement the standard approach (18, 19, 23) of collecting 3-way reciprocal best matches. Only matches with E values of <10 and covering at least 50% of the protein lengths were considered. The ability of the new clusters of orthologous group (COG)-building algorithm to report all POGs that a protein appears to belong to was left enabled. A protein belonging to multiple POGs is always an error, due to unresolved paralogy or unrecognized domain fusion, but this option flags such errors so that they can be resolved later. Less than 1% of proteins were affected by such errors.
Virus-specific genes.
POGs were represented as profiles (position-specific scoring matrices), and PSI-BLAST (45) was used to search for matches in virus genomes and in the major chromosomes of each of the 2,005 prokaryotic genomes available in NCBI as of August 2012 for which PhiSpy predictions could be obtained (including those for which no proviral insertions were found). PhiSpy was run with default parameters and using the generic training set (39). Matches to PhiSpy-identified proviral regions in prokaryotic genomes were ignored for the purposes of virus quotient (VQ) computation, and only matches to the nonviral regions were counted. Matches were defined as hits occurring within a single iteration of PSI-BLAST, below an E value cutoff of 0.001, with a bit score of at least 40, and with the region of homology extending over at least 40 amino acids. The VQ was measured as the quotient of the frequency of matches to viral genomes (v = number of viral genomes matched/total number of viral genomes) versus the sum of the frequency of matches to viral and cellular genomes (h = number of prokaryotic genomes matched/total number of prokaryotic genomes), i.e., VQ = v/(v + h). This procedure differs from that used previously to determine the Phageness Quotient in that it scales between 0 and 1 rather than negative infinity to positive infinity. In addition, a match length cutoff relative to the full protein size is no longer used because multidomain proteins were split in viruses but not in cells. As before, however, some POGs find no matches to virus genomes using these criteria. For instance, the largest POG finds zero matches even with no length or bit score criteria and an E value of up to 10, presumably due to its profile being too diverse to adequately represent the protein family. For these 69 POGs, VQ is undefined and thus they are not represented in Fig. 5.
Fig 5.
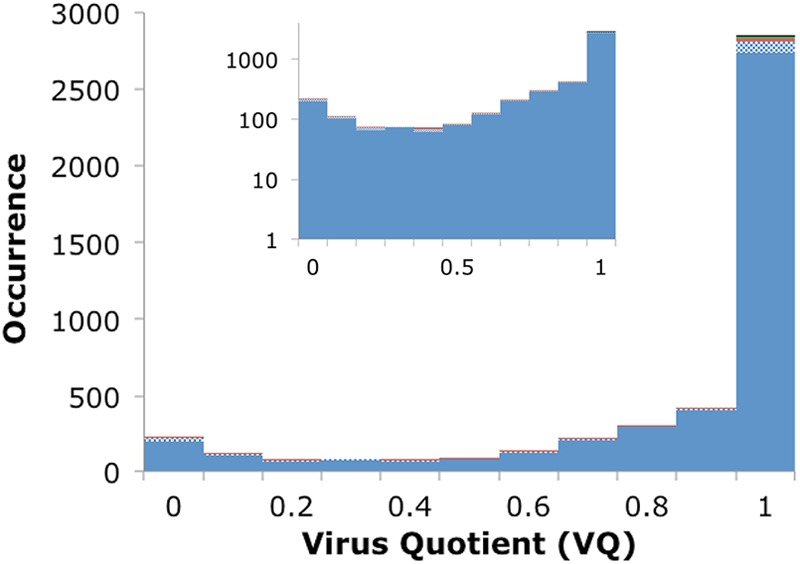
Distribution of the frequency of POGs with the indicated range of VQ. The inset shows the y axis on a log scale. The color scheme is the same as that for Fig. 1.
Identification of signature genes for viral taxa.
The complete genomes of viruses belong to 1,158 distinct taxon groups at various levels of their hierarchy (all the way from all viruses through orders, families, genera, and individual species). Individual virus species only represented once make up the majority of these groups, 991 (85%), and 1,072 (93%) are represented by fewer than 3 distinct viruses. To aim for higher-level clades, these groups were discarded, leaving 86 taxa represented by at least 3 distinct viruses. However, 25 of these represent temporary collections of unclassified or unassigned viruses or environmental samples, and since these do not represent bona fide clades, they were removed to be analyzed separately (although descendant groups below them in the hierarchy were still retained if they met the other criteria). Four more groups were also redundant (containing only a single descendant taxonomic node; these are ssRNA viruses/Leviviridae, dsRNA viruses/Cystoviridae, Rudiviridae/Rudivirus, and Tectiviridae/Tectivirus) and thus were collapsed, leaving a data set of 57 taxa for which we attempted to find signatures. These are listed in File S5 in the supplemental material and include 6 clades above the family level (the 4 genomic types dsDNA, ssDNA, dsRNA, and ssRNA, the order “Caudovirales; tailed phages,” and “all viruses”), 9 clades at the family level, 6 at the subfamily level, 28 at the genus level, and 8 groups of individual viruses. As an example for the latter, the Enterobacteria phage ϕ_X174_ species clade consists of the 19 genomes of the Enterobacteria phages S13, ID1, ID22, ID34, ID45, NC1, NC5, NC7, NC11, NC16, NC37, NC41, NC51, NC56, WA4, WA10, WA11, S13, and ϕ_X174 sensu lato_.
The POGs to be used as signatures for particular groups of viruses were chosen using a 3-tiered procedure. In each case, only individual POGs were considered, and the ability of compound signatures consisting of multiple genes to represent a taxa (e.g., gene A or gene B or genes A, B, and C) was not evaluated.
First, candidate signatures were chosen from the information contained only within the POGs themselves. Among the POGs appearing only within a given group of viruses and never outside that group, candidates were chosen that had the highest recall (found in the most genomes) and/or the highest VQ. Because taxonomy is hierarchical by nature, whenever a POG could serve as a signature for multiple taxonomic groups at different levels of the hierarchy, the group maximizing precision and recall, in that order, was chosen, with ties broken by assignment to the highest taxonomic level available. For instance, the RNA-dependent RNA polymerase (46) is found in all dsRNA viruses, and because in the present data set all dsRNA viruses are assigned to the family Cystoviridae and also to the genus Cystovirus, the precision and recall were both tied at 100% for all 3 of these clades, thus this POG was assigned to dsRNA viruses at the highest taxonomic level. In another example, the major coat protein present only within the genomes of members of the genus Inovirus could also serve as a signature for the higher-level clades of the family Inoviridae or for all ssDNA viruses. Doing so would yield 100% precision, because this protein is not observed outside Inoviridae or ssDNA viruses but would reduce recall because it is not present in other viruses within Inoviridae (plectroviruses) or in other ssDNA viruses (Microviridae).
Second, precision and recall was evaluated against the protein sequences of viral genomes. Once candidate signatures were chosen for each taxon, a sequence profile was constructed (multiple sequence alignment constructed by MUSCLE [47] and PSI-BLAST [45]) and used to search for matches among the protein sequences of the 1,027 viruses with completely sequenced genomes. An E value threshold of 1e−5 was used, as it was found to provide the highest recall and precision in small-scale tests with several signatures (data not shown). Matches were also required to have a bit score of at least 40 and a region of sequence similarity extending over at least 40 amino acids. In some cases, the use of a profile significantly enhanced the recall of the candidate signature compared to simply testing the POG membership. For instance, the maturation protein and RNA replicase beta protein are found in 10 of the 12 (recall of 83%) ssRNA viruses by the POG-making procedure, but matches to the respective profiles were identified in all 12 (recall of 100%). In other cases, the recall and/or precision were reduced by the use of profiles. For instance, although the RNA polymerase subunit of N4-like viruses never appears outside that clade in the POGs, the profile apparently raised the sensitivity enough to find matches in phages in other genera and even another family, thus lowering its precision to only 50%. However, this does not translate to a loss of a signature for the N4-like viruses, because 7 other signature candidates exist for it that each have 100% recall, 100% precision, and a VQ of 1.0.
Third, because the diversity of viruses is undersampled, in order to test for bias presented by the set of virus genomes, these profiles were also tested against all known virus proteins present in the NCBI nr nucleotide database. Occasionally this reduced the precision of a signature candidate; for instance, 2 of the 3 signatures in CBA120_-_like viruses (in the family Myoviridae) had precision lowered from 100 to 80% due to matches among virus proteins for which complete genomes were not yet available. In order to use a conservative estimate of precision, the lowest value among the two profile searches was reported.
Unclassified viruses represent a complication to the search for signatures, because only partial taxonomic information is available for these. Nearly 80% of the 1,027 genomes in the POG data set are listed as unclassified or unassigned at some level of the taxonomic hierarchy, with 78 (8%) even unclassified at the root (although all but one of these was found to be dsDNA by its genome size or manual literature search; see File S1 in the supplemental material). For all of these, matches were counted as far as the available partial information would allow. For instance, a virus unclassified at the root could match any signature gene for any clade without penalty to precision, because it is possible that the unclassified virus legitimately belongs to any clade. However, an unclassified ssDNA virus could only match (without penalty) signatures to clades below ssDNA viruses in the hierarchy, such as the family Inoviridae or the genera Inovirus or Plectrovirus, but could not legitimately match a clade within dsDNA, ssRNA, or dsRNA viruses, because the partial information would conflict in the latter cases. A further complication arises from taxonomic groups that appear as descendants of an internal taxonomic node that contains an unclassified label; for example, the 936 group of lactococcal phages, whose full taxonomic classification is the following: dsDNA viruses, no RNA stage→Caudovirales; tailed phages→Siphoviridae; phages with long noncontractile tails→unclassified _Siphoviridae_→936 group of lactococcal phages. By the procedure listed above, these viruses would be allowed to be matched without penalty by a potential signature for any other clade within the Siphoviridae family, such as lambda-like viruses, despite the fact that the 936 group forms a distinct clade on its own. To prevent this from occurring, all internal nodes containing an unclassified label were collapsed, effectively promoting groups such as this to become a distinct group within the Siphoviridae family, thereby penalizing any matches to it from another Siphoviridae virus, such as lambda-like virus. Finally, in addition to the 78 taxa that include at least 3 virus genomes present in the POG data set, an additional 26 unclassified or unassigned taxon nodes were found in the taxonomy information supplied in the GenBank entries of these genomes. Since these are not true taxa, they were not included in Fig. 6; however, occasionally signature genes could be found for them, and both the list of these taxa and those signatures are included in File S5 in the supplemental material.
Fig 6.
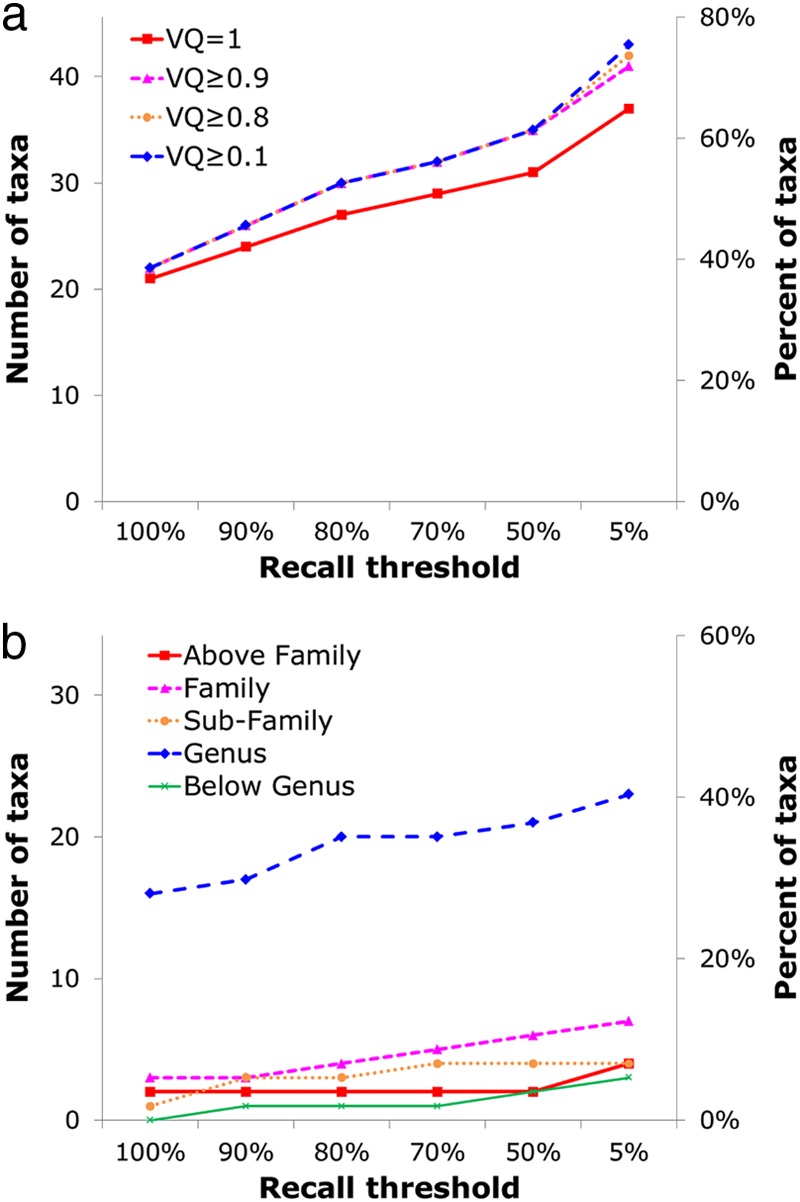
Number and percentage of taxa that can be represented by at least one signature gene, with precision fixed at 100% and recall (x axis) allowed to vary. (a) The dependence of signatures on VQ value. (b) Breakdown of signatures into taxonomic levels.
RESULTS AND DISCUSSION
The data set: viruses and genomes.
The POGs were constructed mostly by following the approach described previously (24; also see Materials and Methods). More than 1,700 completely sequenced prokaryotic virus genomes are deposited in the NCBI databases, but only about a third are represented by a manually curated RefSeq entry. The RefSeq genomes were supplemented by additional genomes from the NCBI Nucleotide database (including Bacillus phage G, the largest known phage genome). Because this database often contains multiple genome entries for some well-studied viruses (such as the Enterobacteria phages ϕ_X174_, f1, and P22, which are represented by 121, 10, and 4 genomes, respectively), to avoid redundancy and bias, genomes were only included from the Nucleotide database when a virus with an identical name was not already present in RefSeq. When an entry from RefSeq was not available, all genomes from Nucleotide were included, which led to 5 Enterobacteria phages being represented multiple times, with f1 represented 10 times and four others represented twice. Using this procedure, 385 genomes from the Nucleotide database were added to the 642 genomes from RefSeq, for a total of 1,027 genomes in the final data set.
In the final prokaryotic virus genome data set, 88% of the genomes are dsDNA viruses, whereas viruses with ssDNA genomes make up a further 10% and RNA phages (ssRNA and dsRNA together) amount to only 2% (Fig. 1). The great majority (93%) of dsDNA genomes for which taxonomic information was available belong to tailed phages of the order Caudovirales (including Siphoviridae, Myoviridae, and Podoviridae); among the other viruses, only three groups were represented by more than 3 genomes, namely, the phage family Tectiviridae and the archaeal virus families Fuselloviridae, Lipothrixviridae, and Rudiviridae. The ssDNA genomes come from Microviruses and Gokushovirinae of the family Microviridae, along with the Inovirus and Plectrovirus genera of the family Inoviridae. The RNA phages are sparsely represented, with 12 positive-strand ssRNA viruses of the family Leviviridae (including the genera Levivirus and Allolevivirus) and 5 dsRNA viruses of the family Cystoviridae. See File S1 in the supplemental material for more details.
Fig 1.
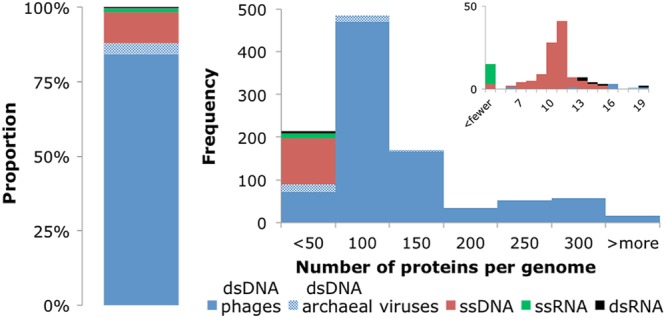
Proportions of prokaryotic virus types (dsDNA phages, dsDNA archaeal viruses, ssDNA, ssRNA, or dsRNA) in the data set and distribution of the number of protein-coding genes in virus genomes. The inset shows in more detail the part of the distribution that includes small virus genomes with <20 protein-coding genes.
Although the host ranges of even the best-studied viruses are not completely defined (10, 20), the host listings in GenBank indicate a substantial diversity of bacterial and archaeal hosts from 196 species that represent 100 genera. Nearly 200 phages are listed as infecting Escherichia coli (although removal of redundancy brings this figure down to 72 genomes, 13% of the viruses for which host information is available). Other bacterial host genera known to be infected by more than 30 viruses include Staphylococcus, Pseudomonas, Vibrio, Lactococcus, Mycobacterium, and Streptococcus (see File S2 in the supplemental material for more details).
Although redundancy was reduced by filtering multiple genome sequences from the same virus present in both RefSeq and Nucleotide, many of the genomes in this set are nearly identical at the level of amino acid sequences of the gene products, and some isolates of the same virus even share >99% sequence identity at the DNA level. To prevent the formation of POGs from only trivially related isolates and instead allow only those that include orthologs shared by 3 distinct viruses, groups of isolates were formed by merging all genomes that shared a large fraction of genes (see Materials and Methods for details). Altogether, the 1,027 genome isolates were classified into 790 groups, with 10% of the groups containing multiple members, mostly among the highly sampled Enterobacteria and Mycobacteria phages (a textbook example is the M13/f1/fd isolates of filamentous phages; see File S2 in the supplemental material for additional details).
The data set: proteins and domains.
More than 90,000 protein-coding genes were extracted from the GenBank entries of the virus genomes included in the data set. To identify genes that were missed in the original annotations, an automated gene prediction procedure was applied that found an additional 3,260 genes in 595 genomes, primarily in the large genomes of dsDNA phages. This procedure was previously tested on 28 well-annotated genomes from curated databases (including thoroughly studied phages T4, T7, and lambda) and was shown to provide high sensitivity and precision, missing only a small number of short or overlapping open reading frames (24). At least a third of the newly predicted genes are conserved in more than two phage genomes and belong to a POG, with this percentage being positively correlated with the protein length.
Each of these previously annotated or newly predicted gene products was examined by the automated method of domain identification described previously (HHpred-based matching to the known database of domains present in NCBI's CDD database [27], which includes entries from SMART, PFAM, LOAD, and CD) (24). Matches to a known domain were detected for 40% of the proteins, with 6% of the proteins containing multiple domains. The multidomain proteins were split into their component domains on the grounds that orthology in general is more properly measured at the level of individual domains than at the level of full-length proteins (19). Furthermore, splitting alleviates the need for manual curation of the resulting POGs to handle cases of artifactual POG fusion (24). This step yielded 97,707 protein-coding genes or domains from the 1,027 virus genomes. This is more than twice the number of genomes and proteins in the previous release of the POGs and three times the size of the latest manually curated set in the so-called annotated POGs 2007.
Orthologous genes in viruses and coverage of viral genomes by POGs.
Using the standard COG-building method (26), the 97,731 proteins or domains from the 1,027 virus genomes (790 distinct viruses) were clustered into 4,542 POGs. This procedure yielded nearly twice as many POGs as in the so-called extended POGs-2010 set and nearly three times the number in the annotated POGs 2007. Most of this increase is due to the additional sampling of the diverse phages discovered in recent years and also to the inclusion of ssDNA and RNA phages (∼1% of POGs) and archaeal viruses (3% of POGs). The representation of POGs in each of the major taxonomic groups of prokaryotic viruses is detailed in File S1 in the supplemental material (for details on POG coverage of individual virus genomes and representation of viruses in individual POGs, see File S2 and S3, respectively).
In most virus genomes, between 50 and 70% of the proteins are included in a POG, typically with low levels of in-paralogy. This coverage is substantially lower than the respective values for bacterial and archaeal COGs (28, 29), implying a vast, sparsely represented, and highly diverse virus pangenome. Moreover, these averages obscure extensive diversity, with some viruses being completely covered (100% of genes appearing in POGs, i.e., present in 3 or more distinct viruses), while others have no conserved genes at all (see File S2 in the supplemental material). Archaeal viruses are covered less extensively, at only 32% on average, and again with a great diversity. For example, the well-studied family Fuselloviridae, with 9 genomes, displays 67% coverage, and Lipothrixviridae, with 8 genomes, is covered at 52%; however, other less-well-studied families have much lower coverage (such as Globuloviridae, with only 2 genomes and 0% coverage; note that a gene must be present in at least 3 distinct viruses to form a POG). Examples of fully covered viruses include some Staphylococcus phages, Mycobacterium phages, phages of Lactobacillales, lambda-like E. coli phages, and _T4_-like phages of the Myoviridae family. Viruses with no representation in POGs include the tiny 2.4-kbp Leuconostoc phage L5 and, strikingly (but not unexpectedly for an archaeal virus), His1, a spindle-shaped halovirus, with none of its 36 protein products assigned to POGs.
Reflecting the diversity of the prokaryotic viruses, the characteristics of the POGs also widely differ. For instance, the size range spans 2 orders of magnitude, from a minimum of 3 proteins from 3 distinct viruses up to 673 proteins from 378 viruses (Fig. 2). However, most of the POGs are small, with a median size of 5 proteins from 5 viruses. The POGs specific to archaeal viruses comprise 60% of the 149 POGs represented in archaeal viruses (mostly genes with uncharacterized functions) and display an identical median size. The other 40%, namely, those shared with bacteriophages, display a median size of 59 proteins in 61 viruses and encompass virion components such as tail and capsid proteins, virulence-related proteins, and typical mobile elements, such as HNH endonucleases. The paralogy level within the POGs is uniformly low, with 95% containing no paralogs, and fewer than 1% contain multiple paralogs. However, a small number of POGs are (relatively) paralog rich, such as a POG that consists of HNH endonucleases that are present in multiple copies per genome in a third of its member viruses, up to a maximum of 8 copies in Salmonella phage PVP-SE1 and 8 and 7 copies of phytanoyl-coenzyme A (CoA)-dioxygenases in two cyanophages, Prochlorococcus phage P-SSM2 and Synechococcus phage S-SSM7, respectively (30). In these properties, the updated POGs are similar to the previous set (24), although a slight increase in genome coverage with the POGs was observed, as expected due to increases in the depth (number of genomes) and breadth (widely diverse viruses) of genomic sampling.
Fig 2.
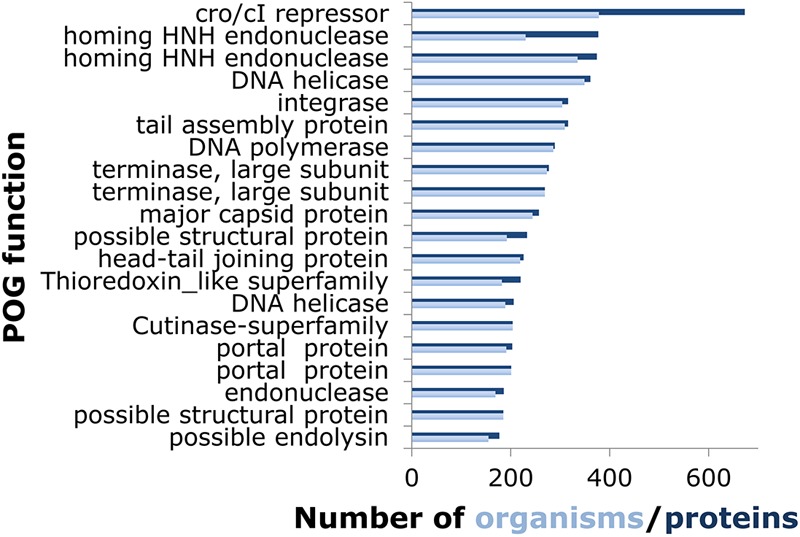
Functions and sizes of the 20 largest POGs. When the number of proteins (dark blue) is greater than the number of organisms (light blue), the excess is due to paralogy.
Despite the increased density of genomic sampling, no POG was found in more than 37% of the 1,027 virus genomes (Fig. 3). This is in contrast to the conserved gene families of the bacterial and archaeal orthologous clusters, where several proteins are found to be nearly universally present in 100% of all cellular organisms, and in general overall levels of coverage are ∼75 to 90% (for instance, 88% of archaeal COGs are found in 41 archaeal genomes) (28, 29). Furthermore, the gene frequency plot for prokaryotes shows a tripartite distribution, with a conserved core found in nearly all organisms under study, a smaller and nearly log-linearly growing shell of genes conserved in a varied number of organisms, and a numerically dominant cloud of genes that are present in only a small number of genomes (31). In the COGs shared between bacteria and archaea, the core consists mostly of the protein components of the translation apparatus, whereas within each prokaryotic domain the core incorporates additional genes, such as those encoding components of the transcription and replication machineries. Conversely, although prokaryotic viruses have a large number of gene families shared among a relatively small number of organisms, they conspicuously lack any semblance of a universal core or even much of a shell, with only 8 genes shared by ≥25% of the 1,027 virus genomes (Fig. 3). The lack of POGs shared among many diverse virus groups suggests that evolution of viruses is substantially distinct from the evolution of their cellular hosts. More specifically, evolution of both cellular life forms and viruses combines tree-like (vertical, from ancestor-to-descendant) and network-like (gene exchange) components (32). Although tree-like evolution might dominate for tight groups of viruses, such as _T4_-like phages (33), among more distant viruses the network component dominates, with no single underlying tree being detectable (15, 34).
Fig 3.
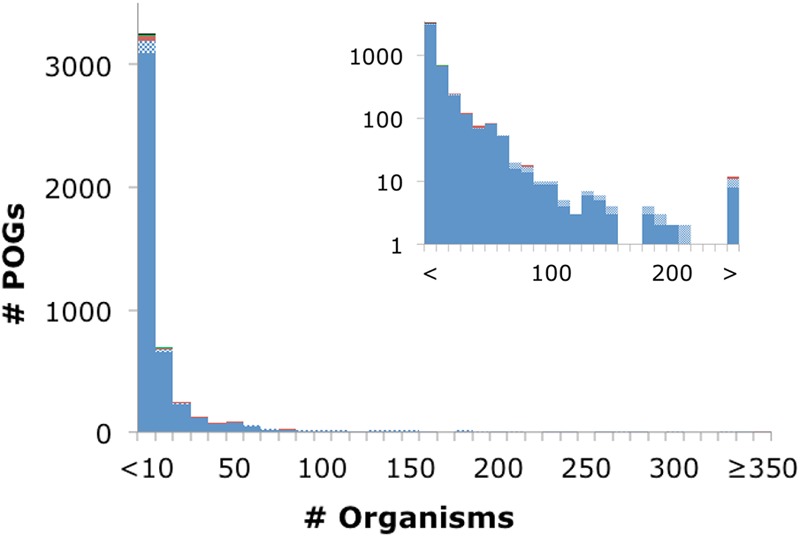
Distribution of the number of organisms in POGs, with the inset using a log scale on the y axis. The color scheme is the same as that for Fig. 1.
The network of evolutionary relationships between prokaryotic virus genomes.
Further evidence of the extensive network of gene exchange among viruses of prokaryotes is given in Fig. 4, where all analyzed genomes form a single giant connected network in which every virus is directly or indirectly connected to every other virus. The only two exceptions are Leviviridae and Cystoviridae, which form their own separate networks because they share no genes with the other viruses, at least not at the level that sequence similarity analysis can detect. The network includes some particularly dense modules, such as the well-characterized, highly connected subnetwork formed by the tailed phages in the order Caudovirales, including the families Myoviridae, Siphoviridae, and Podoviridae. This subnetwork also often acts as a bridge between groups that would otherwise be disjointed, such as Tectiviridae and Inoviridae. Many connections between closely related viruses are likely to stem from vertical rather than horizontal transmission of genetic information. However, given that no POG is shared by more than 37% of genomes and only 1% of the POGs are shared by more than a fifth of the genomes, most of the distant connections are made by different genes. This is in sharp contrast to cellular organisms, where most of the distant connections come from the core gene set that is inferred to have been present in the common ancestor (35). For instance, although no POGs are shared between the ssDNA bacteriophage families Microviridae and Inoviridae, each shares a single (different) POG with dsDNA viruses. Several members of the genus Microvirus share a DNA maturation protein with Pseudomonas phage ϕKZ of the family Myoviridae, and other members of the family Microviridae (Gokushovirinae and environmental samples) share a replication initiation protein with Clostridium phage ϕSM101, an unclassified dsDNA phage. Members of the family Inoviridae share several different POGs with diverse dsDNA viruses, such as a Cro/CI-like repressor and a recombinase/resolvase/invertase that is present in several families within the order Caudovirales, and a transposase also is found in Caudovirales as well as Bicaudaviridae and other archaeal viruses. Although differential gene loss from a common ancestor of these viruses in principle cannot be ruled out, gene exchange is by far the simplest explanation for the large number of POGs exhibiting such a widely scattered distribution among genomes.
Fig 4.
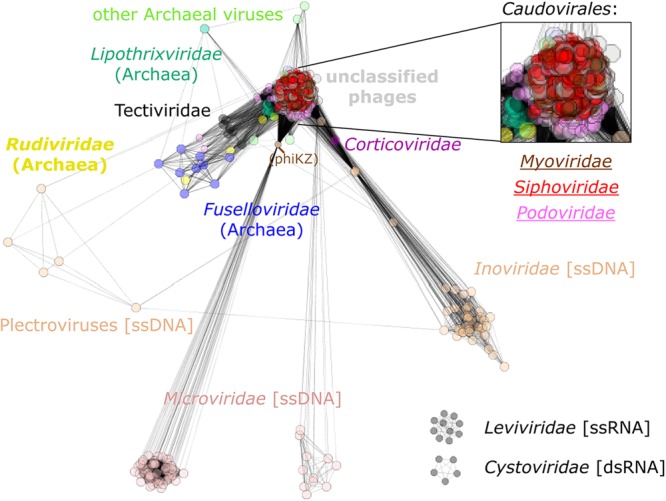
Network of phage genomes. The genomes of each phage are represented as boxes, which are colored according to the indicated taxonomic affiliation (type of dsDNA and with bacteria as their host except where specified otherwise), with connections drawn between genomes that share at least one POG. The distances between genomes are inversely proportional to the number of genes shared between neighbors. The inset is a zoomed-in region of the tightly connected subnetwork among the tailed phages.
Functional classification of the POGs.
The known and predicted functions of the large POGs were examined in greater detail (the information for the 20 largest POGs is shown in Fig. 2; see File S3 in the supplemental material for the 100 largest POGs, which also includes a list of predicted functions for all POGs). This group of widespread POGs is functionally diverse. The largest POG consists of helix-turn-helix DNA-binding proteins related to Cro/CI repressors, which are key regulators of the life cycle in diverse phages and are encoded by multiple paralogous genes in many of these. Among the 100 largest POGs, there are additional transcriptional regulators; proteins involved in phage DNA replication (DNA polymerases, nucleases, replication-initiating ATPases, ATP-dependent DNA ligases, recombinases, single-strand DNA-binding proteins, and terminases); nucleotide salvage enzymes (ribonucleoside reductases, dUTPases, thymidylate synthases, nucleoside kinases); virion structural components and maturation factors (this group appears to contain more head proteins than tail components, probably because of greater diversity of tail structures); and various lysins. A substantial fraction of POGs, including 10 of the top 100 largest POGs, are completely uncharacterized proteins. Some of these are present only in the extensively studied mycophages (16) and their close relatives infecting other coryneform bacteria, but others are widely distributed; these common but enigmatic phage proteins could be particularly attractive for experimental study.
Further analysis of the molecular functions represented in the Top 100 POG list identifies many pairs of isofunctional proteins. The examples include flavin-dependent and flavin-independent thymidylate synthases (POGs 1033 and 0092, ThyX and ThyA homologs, respectively); DnaG-like TOPRIM-domain DNA primase and archaeo-eukaryotic-like Primpol DNA primase (POGs 0084 and 0326); and head maturation proteases from distinct assemblin and ClpP families (POGs 0060 and 0304). These are pairs of nonhomologous proteins with essential functions that tend to displace each other in the individual phage genomes. However, in several cases apparently equivalent functions in different viruses are represented by multiple POGs that appear to include homologous proteins sharing limited sequence similarity. In particular, 13 POGs, including 2 of the 20 largest POGs, are annotated as terminase large subunits; all of these, however, are members of a distinct family of P-loop ATPases (36) but show extreme sequence divergence. The case of small terminase subunits is even more dramatic, with 30 POGs delineated for these highly diverged proteins (37). A similar situation was observed with amidases of the NlpC/P60 family (38), which are represented by at least 8 POGs. The failure of all of these proteins to resolve as a single POG each, despite the sensitive approach used (see Materials and Methods), suggests that our current approach has the tendency to oversplit POGs. Further sampling of the virus genome space is expected to help in establishing more robust evolutionary links between these POGs.
Despite these functional redundancies, it is notable that the repertoire of molecular functions represented in the Top 100 POG list appears diverse enough to allow for the assembly of a functionally coherent phage genome from these frequently occurring phage proteins, even though in actuality no such consensus virus genome has been identified.
Virus-specific POGs.
Despite the extensive gene transfer between viruses and their prokaryotic hosts (31), many of the POGs are virus specific. Specifically, 60 to 70% of the POGs are never or extremely rarely present in any prokaryotic genomes outside proviral regions that were identified using PhiSpy (39). This fraction of virus-specific POGs is slightly greater than that described previously (24), most likely due to the greater sensitivity of PhiSpy than ACLAME, which was used for the previous POG analysis (see Materials and Methods for details).
To quantify the likelihood of a POG appearing in virus versus prokaryotic genomes, the VQ of each POG was calculated (see Materials and Methods). There was no obvious correlation between the POG size and the VQ, and the main observed trend (Fig. 5) was that for a large fraction of the POGs VQ approached unity. Indeed, 62% of POGs have a VQ of 1.0, another 9% have a VQ between 0.9 and 1, and over three-fourths of the POGs have a VQ of ≥0.8. A weaker trend was also observed in the opposite direction, toward a VQ of 0, representing those genes that are more often observed in prokaryotes than in viruses; 4% of POGs have a VQ of ≤0.1, and nearly 7% have a VQ of ≤0.2. Apparently, these are bacterial genes that have been acquired by and transferred between viruses on a relatively small scale. It is well known that gene exchange also occurs in the reverse direction, from phages to hosts: for instance, many virulence factors in pathogenic bacteria come from integrated prophages (40, 41). However, due to the discounting of matches in the prophage regions for the calculation of VQ, most such genes are recognized as viral with a medium to high VQ; for example, Shiga toxins that are never observed in bacteria outside detected prophages have a VQ of 1.0. For each POG, the number of detected homologs in viral genomes, prokaryotic genomes, and the resulting VQ are given in File S4 in the supplemental material.
Taxon signature genes.
Given the high propensity of genes to be horizontally transferred between viruses as well as between viruses and their hosts, and despite the lack of a universal core of virus genes, it is notable that for many virus taxa, taxon-specific signature genes could be identified. For such a signature to serve as a useful diagnostic indicator of viral presence, it should satisfy several criteria. First, it should be a conserved gene present in all or at least most of the members of a particular taxon, i.e., it should provide high sensitivity (recall). Second, and more important, a signature should never or only very rarely be observed outside that taxon, i.e., it should provide high specificity (precision). Third, for analysis of samples containing a mixture of genomic sequences from both viruses and prokaryotes, a signature should never or very rarely be observed in prokaryotic genomes outside identifiable provirus regions (since this criterion can be difficult or virtually impossible to apply to metagenomic sequence samples). For example, photosystem components would serve as good signatures for cyanophages given a sample containing only phages (42), but in a mixed sample they might reflect the presence of cyanobacteria themselves to a greater extent than the presence of phages. Fourth, genes present in only a single copy per virus are desirable, because using such genes as signatures allows quantitative abundance measurements to be performed.
The POGs are well suited for defining signature genes, because collectively these conserved evolutionary families already encompass all of the information on the presence, absence, and frequencies of a gene in viral genomes. Sequence profiles built from POGs have the potential to provide both high sensitivity and high specificity in the search for a signature gene in a given sample. However, profile searches can introduce complications, such as matches to distant paralogs, so in practice we chose a hybrid approach whereby we used POGs to define several initial candidates that were then screened under an operational definition with parameters tuned to maximum precision.
For the 57 clades that include at least 3 distinct viruses present in the POG data set (see Materials and Methods), POGs were used to define several candidate signatures that were tested for precision and recall by profile searches against proteins in the completely sequenced virus genomes and also against all known virus sequences present in the NCBI nr database (see Materials and Methods). This procedure allowed us to select the most conservative (high-precision) signatures. Given that the diversity of viruses currently appears to be vastly undersampled, future genomic sequencing is likely to improve the performance of some, if not most, of these signatures. However, with POGs formed from a data set containing over 1,000 complete genomes of viruses, and having tested the precision of each signature against all known virus proteins both in complete genomes and in the nr database, these signatures should represent a reasonable first approximation reflecting the majority of what is currently known about virus proteins. The list of taxa tested and candidate signature POGs, with the results of their searches against proteins in complete viral genomes and against all known virus proteins, are given in File S5 in the supplemental material.
The functions of the majority of the candidate signature genes are uncharacterized, and no functional information could be extracted by searching the sequence databases with POG protein profiles using either PSI-BLAST or HHpred, apparently reflecting the current paucity of knowledge about the biological roles of virus-specific proteins. Those signatures that do have a discernible function tend to be structural proteins or enzymes involved in virion morphogenesis rather than proteins involved in viral genome replication and expression. This trend reflects the fact that the taxonomy of viruses is mostly defined by structural characteristics of the virion (with some supporting evidence from shared gene content [15]) and that the goal of the current study was to find virus-specific, single-copy signature genes for existing taxonomic groups.
For 37% of the tested viral taxa (21 of the 57 taxa), at least one signature was identified that met the strictest criteria of 100% recall, 100% precision, and a VQ of 1.0 and appeared in only a single copy per genome in the POGs (Table 1). As these criteria were relaxed, signatures were found for many more taxa. Figure 6a shows that at the precision threshold at 100% (i.e., allow zero false positives), the VQ threshold at 1.0 (allow no matches to cells), but using no recall threshold (allow false negatives), the number of identifiable signatures nearly doubles to 37 (65% of the 57 taxa). For instance, a few genes are present only in the genus Lambda-like viruses (precision of 100%) but are found in at most 14% of the members of that group. These genes remain somewhat useful, as highly specific signatures of the given taxon but multiple overlapping sets of signatures (not considered here) will likely be necessary to detect all lambda-like viruses.
Table 1.
Top-quality POG signatures for virus taxaa
| Virus clade | No. of genomes | Signature gene(s) | |
|---|---|---|---|
| POG no. | Function(s) | ||
| Order Caudovirales, family Siphoviridae | |||
| T1-like viruses | 5 | 2763, 2765, 2766, 2771, 2773, 2778, 2780, 2802 | Holin, transcriptional regulator, hypothetical proteins |
| L5-like viruses | 3 | 1603, 1605 | Hypothetical proteins |
| ϕC31-like viruses | 3 | 3419, 3420, 3421, 3422 | Major capsid, hypothetical proteins (possible RNA polymerase sigma factor) |
| Order Caudovirales, family Myoviridae | |||
| CBA120-like viruses | 4 | 3145 | Calcium-binding hemolysin protein |
| Hp1-like virus | 6 | 2211, 2212, 2213, 2216 | Tail sheath and tail completion proteins, hypothetical proteins |
| Bcep781-like virus | 5 | 3660, 3661, 3664 | Hypothetical proteins (possible terminase small subunit) |
| FelixO1-like virus | 3 | 1350, 1425 | Structural protein, hypothetical protein |
| ϕCD119-like virus | 3 | 4377, 4378, 4382, 4383 | Resolvase/integrase, hypothetical proteins |
| Spounavirinae | 12 | 0072 | Tail protein |
| ϕKZ-like viruses | 3 | 3252, 3254, 3245 | DNA-directed RNA polymerase beta subunit, structural protein, hypothetical protein |
| Order Caudovirales, family Podoviridae | |||
| T7-like viruses | 29 | 0036, 0041 | Internal virion protein, DNA packaging/maturation protein |
| SP6-like viruses | 6 | 1055, 1056, 1057, 1058, 1061, 1064, 1065, 1066 | DNA-endonuclease-like protein, internal virion protein, hypothetical proteins (possible tail assembly) |
| AHJD-like viruses | 4 | 3732 | Hypothetical protein (CHAP domain) |
| ϕ29-like viruses | 5 | 0875, 0876, 0878, 0879, 0885 | Terminal protein, transcriptional regulator, dsDNA-binding protein, scaffolding protein, early protein |
| N4-like viruses | 8 | 2377, 2380, 2383, 2384, 2387, 2389, 2390 | Major coat protein, ssDNA-binding protein, hypothetical proteins (several possibly structural) |
| Unassigned | |||
| Fuselloviridae | 6 | 3362 | Hypothetical protein |
| Fusellovirus | 6 | 3354, 3362 | Hypothetical proteins |
| Lipothrixviridae | 8 | 3538 | Hypothetical protein |
| Betalipothrixvirus | 6 | 3504, 3508, 3508 | RHH transcriptional regulator, structural protein, hypothetical protein (possibly structural) |
| ssRNA viruses/Leviviridae | 10 | 0166, 0167 | Maturation protein, RNA replicase beta |
| dsRNA viruses/Cystoviridae | 5 | 4542 | RNA-dependent RNA polymerase |
Allowing VQ to relax (i.e., allowing a few matches to cells) also increases the number of taxa for which signatures could be found. However, for a VQ of ≤0.95, not many additional signatures are found, because for most taxa, genes with high VQ were easily identified (with 83% of signature candidates having a VQ of 1). Relaxing the copy number has a similar effect (data not shown), with 90% of signature candidates being present in a single copy, thus relaxation usually is not needed in order to find a signature. Precision was not relaxed, as it was deemed the most important attribute for a signature to display. Removing the thresholds for the other 3 parameters, recall, VQ, and copy number, but keeping precision fixed at 100% allows at least one signature to be found for 47 of the 57 taxa (82%), whereas signatures for the remaining 18% could not be found without relaxing precision.
Figure 6b further shows the breakdown of matches to the identified taxon-specific signatures at different taxonomic levels. Most of the signatures identified with each recall threshold are at the genus level (56 to 73%), which constitutes 28 (49%) of the 57 taxa tested, followed by the family and subfamily levels, and above-family and below-genus levels make up <10% of the signatures.
The signature approach can help to identify taxonomic membership of currently unclassified viruses and to create markers for which fewer than 3 genomes exist. As an example of both tasks, the genome of Enterobacteria phage EPS7 (NC_010583) is currently described as an unclassified Siphoviridae phage, despite its obvious identification as a T5-like virus in the title of the publication describing this phage (43). Comparison of EPS7 to two other T5-like virus genomes represented in the database yielded 129 POGs. Of these, 55 (covering ∼30% of each of the three genomes) could be used as markers for T5-like viruses, with 100% recall, 100% precision, VQ of 1.0, and appearing in a single copy. In another example, virus N15 is currently the only member of the N15-like viruses recognized in the NCBI taxonomy database, but 4 of its 70 genes comprise markers with 100% recall, 100% precision, VQ of 1.0 and appear in a single copy. These markers are shared with 2 other linear plasmid prophages labeled only as unclassified Siphoviridae: Yersinia phage PY54 (accession no. NC_005069) and Klebsiella phage ϕKO2 (accession no. NC_005857). Thus, the signature approach has the potential to tentatively classify currently unclassified viruses.
Conclusions.
The current update of the POGs reinforces and generalizes trends noticed previously, above all the vastness of the virus pangenome and the dominance of the network trend in virus evolution. The majority of the POGs remain virus specific, and the number of POGs keeps growing without any sign of saturation, suggesting that numerous virus gene families remain to be discovered. A complementary observation is that many viral genomes remain sparsely, if at all, covered by POGs, apparently because the currently known viruses represent a small fraction of the vast virosphere. These limitations notwithstanding, it appears that POGs can be of immediate value as a tool for identification of viruses in metagenomic samples, given that virus-specific, single-copy signature genes were found with high precision and recall for the majority of the prokaryotic virus taxa tested (those with completely sequenced genomes of at least 3 distinct viruses). Further search for such signatures and refinement of the search strategies, in particular toward using multiple signatures with partially overlapping ranges for comprehensive coverage of virus taxa, is expected to help researchers obtain insights into the ecology of viruses and structure of the virosphere. All POG and marker data, including profile alignments and BLAST-formatted searchable databases (of both all proteins conserved in POGs and also those with a VQ of ≥0.9) are available for download at ftp.ncbi.nlm.nih.gov/pub/kristensen/thousandgenomespogs/.
Supplementary Material
Supplemental material
ACKNOWLEDGMENTS
D.M.K. and E.V.K. are supported by intramural funds of the U.S. Department of Health and Human Services (to the National Library of Medicine). A.R.M. was supported by the Stowers Institute for Medical Research.
We thank Yoo-Ah Kim for helpful discussions on network visualization.
D.M.K. collected the data; D.M.K., A.S.W., T.Y., P.B., A.R.M., and E.V.K. analyzed the data; D.M.K. and E.V.K. wrote the manuscript, which was edited and approved by all authors.
Footnotes
Published ahead of print 7 December 2012
REFERENCES
- 1.Breitbart M, Rohwer F. 2005. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13(6):278–284 [DOI] [PubMed] [Google Scholar]
- 2.Wommack KE, Colwell RR. 2000. Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64(1):69–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suttle CA. 2007. Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 5(10):801–812 [DOI] [PubMed] [Google Scholar]
- 4.Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. 2010. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466(7304):334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ansorge WJ. 2009. Next-generation DNA sequencing techniques. Nat. Biotechnol. 25(4):195–203 [DOI] [PubMed] [Google Scholar]
- 6.Reyes A, Semenkovich NP, Whiteson K, Rohwer F, Gordon JI. 2012. Going viral: next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 10(9):607–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurwitz BL, Deng L, Poulos BT, Sullivan MB. 2012. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. [Epub ahead of print.] doi:10.1111/j.1462-2920.2012.02836.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mokili JL, Rohwer F, Dutilh BE. 2012. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2(1):63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatfull GF. 2008. Bacteriophage genomics. Curr. Opin. Microbiol. 11(5):447–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. 2011. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 75(4):610–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharon I, Battchikova N, Aro EM, Giglione C, Meinnel T, Glaser F, Pinter RY, Breitbart M, Rohwer F, Beja O. 2011. Comparative metagenomics of microbial traits within oceanic viral communities. ISME J. 5(7):1178–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swanson MM, Reavy B, Makarova KS, Cock PJ, Hopkins DW, Torrance L, Koonin EV, Taliansky M. 2012. Novel bacteriophages containing a genome of another bacteriophage within their genomes. PLoS One 7(7):e40683 doi:10.1371/journal.pone.0040683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackermann HW, Prangishvili D. 2012. Prokaryote viruses studied by electron microscopy. Arch. Virol. 157(10):1843–1849 [DOI] [PubMed] [Google Scholar]
- 14.Bolduc B, Shaughnessy DP, Wolf YI, Koonin EV, Roberto FF, Young M. 2012. Identification of novel positive-strand RNA viruses by metagenomic analysis of archaea-dominated Yellowstone hot springs. J. Virol. 86(10):5562–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavigne R, Darius P, Summer EJ, Seto D, Mahadevan P, Nilsson AS, Ackermann HW, Kropinski AM. 2009. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 9:224 doi:10.1186/1471-2180-9-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mochizuki T, Krupovic M, Pehau-Arnaudet G, Sako Y, Forterre P, Prangishvili D. 2012. Archaeal virus with exceptional virion architecture and the largest single-stranded DNA genome. Proc. Natl. Acad. Sci. U. S. A. 109(33):13386–13391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sencilo A, Paulin L, Kellner S, Helm M, Roine E. 2012. Related haloarchaeal pleomorphic viruses contain different genome types. Nucleic Acids Res. 40(12):5523–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koonin EV. 2005. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet 39:309–338 [DOI] [PubMed] [Google Scholar]
- 19.Kristensen DM, Wolf YI, Mushegian AR, Koonin EV. 2011. Computational methods for Gene Orthology inference. Brief Bioinform. 12(5):379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trachana K, Larsson TA, Powell S, Chen WH, Doerks T, Muller J, Bork P. 2011. Orthology prediction methods: a quality assessment using curated protein families. Bioessays 33(10):769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Stoeckert CJ, Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13(9):2178–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, Jensen LJ, von Mering C, Bork P. 2012. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 40:D284–D289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tatusov RL, Koonin EV, Lipman DJ. 1997. A genomic perspective on protein families. Science 278(5338):631–637 [DOI] [PubMed] [Google Scholar]
- 24.Kristensen DM, Cai X, Mushegian A. 2011. Evolutionarily conserved orthologous families in phages are relatively rare in their prokaryotic hosts. J. Bacteriol. 193(8):1806–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Glazko G, Mushegian A. 2006. Protein repertoire of double-stranded DNA bacteriophages. Virus Res. 117(1):68–80 [DOI] [PubMed] [Google Scholar]
- 26.Kristensen DM, Kannan L, Coleman MK, Wolf YI, Sorokin A, Koonin EV, Mushegian A. 2010. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26(12):1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makarova KS, Sorokin AV, Novichkov PS, Wolf YI, Koonin EV. 2007. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol. Direct 2:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41 doi:10.1186/1471-2105-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, DeFrancesco AS, Kern SE, Thompson LR, Young S, Yandava C, Fu R, Krastins B, Chase M, Sarracino D, Osburne MS, Henn MR, Chisholm SW. 2010. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 12(11):3035–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koonin EV, Wolf YI. 2008. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36(21):6688–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puigbo P, Wolf YI, Koonin EV. 2010. The tree and net components of prokaryote evolution. Genome Biol. Evol. 2:745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krisch HM, Comeau AM. 2008. The immense journey of bacteriophage T4–from d'Herelle to Delbruck and then to Darwin and beyond. Res. Microbiol. 159(5):314–324 [DOI] [PubMed] [Google Scholar]
- 34.Glazko G, Makarenkov V, Liu J, Mushegian A. 2007. Evolutionary history of bacteriophages with double-stranded DNA genomes. Biol. Direct 2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puigbo P, Wolf YI, Koonin EV. 2009. Search for a “Tree of Life” in the thicket of the phylogenetic forest. J. Biol. 8(6):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell MS, Matsuzaki S, Imai S, Rao VB. 2002. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 30(18):4009–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun S, Gao S, Kondabagil K, Xiang Y, Rossmann MG, Rao VB. 2012. Structure and function of the small terminase component of the DNA packaging machine in T4-like bacteriophages. Proc. Natl. Acad. Sci. U. S. A. 109(3):817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anantharaman V, Aravind L. 2003. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol. 4(2):R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akhter S, Aziz RK, Edwards RA. 2012. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 40(16):e126 doi:10.1093/nar/gks406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busby B, Kristensen DM, Koonin EV. 2012. Contribution of phage-derived genomic islands to the virulence of facultative bacterial pathogens. Environ. Microbiol. [Epub ahead of print.] doi:10.1111/j.1462-2920.2012.02886.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hacker J, Kaper JB. 2000. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54:641–679 [DOI] [PubMed] [Google Scholar]
- 42.Chenard C, Suttle CA. 2008. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl. Environ. Microbiol. 74(17):5317–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong J, Kim KP, Heu S, Lee SJ, Adhya S, Ryu S. 2008. Identification of host receptor and receptor-binding module of a newly sequenced T5-like phage EPS7. FEMS Microbiol. Lett. 289(2):202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roux S, Krupovic M, Poulet A, Debroas D, Enault F. 2012. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS One 7(7):e40418 doi:10.1371/journal.pone.0040418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koonin EV. 1991. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 72(Pt 9):2197–2206 [DOI] [PubMed] [Google Scholar]
- 47.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material