Sorafenib and its derivative SC-49 sensitize hepatocellular carcinoma cells to CS-1008, a humanized anti-TNFRSF10B (DR5) antibody (original) (raw)
Abstract
Background and Purpose
Previously, we have shown that sorafenib sensitizes hepatocellular carcinoma (HCC) to apoptosis induced by TNF-related apoptosis-inducing ligand (TNFSF10; TRAIL). Here, we report that sorafenib and SC-49 sensitize HCC cells to CS-1008, a novel anti-human death receptor 5 (TNFRSF10B) antibody.
Experimental Approach
HCC cell lines (PLC5, Huh-7, and Hep3B) were treated with CS-1008 and/or sorafenib and analysed in terms of apoptosis and signal transductions.
Key Results
SC-49 is a sorafenib derivative, which is devoid of kinase inhibitory activity. Both sorafenib and SC-49 down-regulated the phosphorylation of STAT3 at Tyr705 and subsequently reduced the levels of STAT3-regulated proteins, Mcl-1, survivin and cylcin D1, in CS-1008-treated HCC cells. Knockdown of STAT3 by RNA interference overcame apoptotic resistance to CS-1008 in HCC cells, and ectopic expression of STAT3 in HCC cells abolished the sensitizing effects of sorafenib and SC-49 on CS-1008-induced apoptosis, indicating that inhibition of STAT3 mediates the enhancing effects of these compounds when combined with CS-1008. Importantly, inhibition of SHP-1 by adding a specific SHP-1 inhibitor reduced the effects of SC-49 and CS-1008 on p-STAT3 and apoptosis, whereas co-treatment of CS-1008 with SC-49 increased the activity of SHP-1. These data indicate that the combined effects of CS-1008 and SC-49 on HCC are mediated by SHP-1. Moreover, the combination of CS-1008 and SC-49 inhibited HCC xenograft tumour growth in vivo.
Conclusions and Implications
Sorafenib and its derivative SC-49 sensitize HCC cells to the antitumour effects of CS-1008 through SHP-1-dependent inactivation of STAT3.
Keywords: CS-1008, SC-49, sorafenib, STAT3, HCC
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world. It is a highly lethal malignancy that has a high recurrence rate despite surgical resection (Tanaka and Arii, 2009; Verslype et al., 2009). Advanced or recurrent HCC is frequently resistant to current chemotherapeutic agents and radiation; therefore, the development of targeted agents with tolerable toxicity is needed to advance anti-HCC therapy (Tanaka and Arii, 2009;). Sorafenib, a multi-kinase inhibitor, has been approved for clinical use in advance HCC after it was found to improve overall survival significantly in two prospective randomized phase III trials for patients with advanced HCC (Llovet et al., 2008; Cheng et al., 2009). Sorafenib inhibits multiple kinases, including the Ras/Raf/MAPK/ERK signalling pathway, the angiogenic pathways VEGFR2, VEGFR3, as well as PDGFRβ and other kinases such as FLT3 and FGFR1 (Adnane et al., 2006; Liu et al., 2006; Wilhelm et al., 2006; Auclair et al., 2007).
Among various targeted strategies for HCC treatment, TNF-related apoptosis inducing ligand (TNFSF10; TRAIL), which targets receptor-mediated apoptosis, represents an attractive option (Johnstone et al., 2008; Wang, 2008; Falschlehner et al., 2009). As a member of the TNF superfamily, TNFSF10 initiates apoptosis by binding to two important death domain-containing death receptors (DRs), TNFRSF10A (DR4) and TNFRSF10B (DR5) (Wang and El-Deiry, 2003; Rowinsky, 2005; Wiezorek et al., 2010). TNFSF10 or TNFSF10 agonists bind to TNFRSF10A or TNFRSF10B and form death-inducing signalling complex (DISC), which is a multi-protein complex consisting of an adaptor molecule, FADD and the initiator of extrinsic pathway caspase-8. (Wang and El-Deiry, 2003; Johnstone et al., 2008) Activated caspase-8 is capable of both initiating an extrinsic apoptotic pathway in type I cells (through activation of caspase-3, -6, and -7) and triggering the intrinsic pathway in type II cells (through activation of Bid) (Li et al., 1998; Johnstone et al., 2008).
CS-1008 is a novel TNFRSF10B agonist that exerts TNFSF10-like activity. It is a humanized anti-human TNFRSF10B antibody manufactured from a murine anti-human TNFRSF10B monoclonal antibody, TRA-8 (Yada et al., 2008). So far, CS-1008 has shown selective cytotoxicity towards tumour cells expressing TNFRSF10B (Yada et al., 2008) and an excellent safety profile in humans (Saleh et al., 2008). CS-1008 monotherapy induces apoptosis in various cancer cells and CS-1008 in combination with some chemotherapeutic agents (such as gemcitabine or docetaxel) has also been found to have enhanced antitumour activity (Yada et al., 2008). In a phase I trial, no dose-limiting toxicity was reported for CS-1008 at doses up to 8 mg·kg−1 weekly (Saleh et al., 2008).
Although TNFSF10 may be applied as anti-HCC strategy, more and more studies have reported that the efficacy of TNFSF10-induced apoptosis in HCC cells is insufficient, often due to resistance to TNFSF10 or its agonists (Shin et al., 2002; Pathil et al., 2006; Chen et al., 2009). Resistance to TNFSF10 may be induced at any step in the apoptosis signalling cascade, from the receptor level (mutations or overexpression of TNFRSF10A or TNFRSF10B) (Zhang and Fang, 2005), or defects in DISC assembly, (Eggert et al., 2001; Okano et al., 2003) through to dysfunctions of the anti-apoptotic Bcl-2 family proteins (Bcl-2, Bcl-xl, Mcl-1, etc) (Fulda et al., 2002; Kim et al., 2008) and pro-apoptotic proteins (Bax or Bak) or defects in mitochondria-derived activator of caspases (Smac/Diablo) (Zhang et al., 2001; Zhang and Fang, 2005). Of particular note, Mcl-1, an anti-apoptotic Bcl-2 family protein, plays a critical role in conferring TNFSF10 resistance (Kim et al., 2008). Data have shown that overexpression of Mcl-1 can neutralize TNFSF10-induced signalling (Meng et al., 2007; Ricci et al., 2007). Moreover, directly or indirectly destabilizing or disabling Mcl-1 can restore TNFSF10 sensitivity (Taniai et al., 2004; Hall and Cleveland, 2007). Interestingly, Mcl-1 is a highly regulated cell death and survival controller that responds to various cytokines and growth factors (Yang-Yen, 2006), and can be regulated by a number of transcription factors, including NF-κB targeting the cAMP response element (CRE-2) motif, and STAT3 targeting the sis-inducible element (SIE) motif of mcl-1 promoter region (Wang et al., 2003; Yang-Yen, 2006; Kim et al., 2008).
STAT3 is considered a potential anti-cancer therapeutic target because of its crucial role in transcriptional regulation of genes involved in cell proliferation and survival and it is constitutively activated in common human cancers, including HCC (Li et al., 2006; Germain and Frank, 2007; Kusaba et al., 2007). In response to the stimulation of cytokines, growth factors and hormones, STAT3 is phosphorylated (activated) and homodimerizes or heterodimerizes with STAT1 in the cytoplasm; it then translocates to the nucleus to regulate a number of genes, including genes that encode apoptosis-related proteins and cell cycle regulators (i.e. Bcl-2, Bcl-xl, Mcl-1, survivin and cyclin D1). In cancer cells, constitutively activated STAT3 directly contributes to tumourigenesis, invasion and metastasis (Germain and Frank, 2007). Targeting STAT3 using antisense oligonucleotide reduces the growth and metastasis of HCC cells in vitro and in vivo (Li et al., 2006). Importantly, reducing constitutive STAT3 activity has been shown to sensitize human hepatoma cells to TNFSF10-mediated apoptosis (Kusaba et al., 2007). Moreover, a number of protein tyrosine phosphatases have been shown to negatively regulate STAT3 signalling through direct dephosphorylation of p-STAT3 (Tyr705); these include members of the SH2-domain containing tyrosine phosphatase family (SHP-1 and SHP-2) and protein tyrosine phosphatase 1B (PTP-1B) (Ke et al., 2007; Chen et al., 2008; Kunnumakkara et al., 2009; Pandey et al., 2009). Therefore, activity of protein tyrosine phosphatases may be critical for the regulation of STAT3 phosphorylation in cancer cells.
Recently, we have reported that sorafenib sensitizes HCC cells to TNFSF10 (Chen et al., 2010), and STAT3 is a major kinase-independent target of sorafenib in HCC (Tai et al., 2011). We have discovered that several sorafenib derivatives are novel STAT3 inhibitors (Chen et al., 2011). In this study, we demonstrated that SC-49, a novel sorafenib analogue, is able to sensitize HCC cells to the antitumour effects of CS-1008.
Methods
The receptor nomenclature used in this paper conforms to Br J Pharmacol's Guide to Receptors and Channels (Alexander et al., 2011).
Reagents and antibodies
CS-1008 and sorafenib (Nexavar®) were kindly provided by Daiichi Sankyo Co., Ltd. (Tokyo, Japan) and Bayer Pharmaceuticals (West Haven, CT, USA) respectively. For in vitro studies, sorafenib at various concentrations was dissolved in DMSO and then added to the cells in 5% FBS-containing DMEM. Antibodies for immunoblotting such as Akt1, Mcl-1 and PARP were purchased from Santa Cruz Biotechnology (San Diego, CA, USA). Other antibodies such as anti-pERK (1/2), ERK2, survivin, cylcin D1, Bcl-xL, Bid, caspase-3, caspase-8, caspase-9, phospho-STAT3 (Tyr705), STAT3 and phosphor-Akt (Ser473) were from Cell Signaling (Danvers, MA, USA).
Cell culture and Western blot analysis
The Huh-7 HCC cell line was obtained from the Health Science Research Resources Bank (HSRRB; Osaka, Japan; JCRB0403). The PLC/PRF/5 (PLC5), Sk-Hep-1, Hep3B and U937 cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). All cells obtained from HSRRB or ATCC were immediately expanded and frozen such that all cell lines could be restarted every 3 months from a frozen vial of the same batch of cells. No further authentication was done in our lab. Cells were maintained in DMEM supplemented with 10% FBS, 100 U·mL−1 penicillin G, 100 μg·mL−1 streptomycin sulfate and 25 μg·mL−1 amphotericin B in a humidified incubator at 37°C in an atmosphere of 5% CO2 in air. Lysates of HCC cells treated with drugs at the indicated concentrations for various periods of time were prepared for immunoblotting of caspase-3, PARP, p-STAT3, STAT3, etc. Western blot analysis was performed as previously reported (Chen et al., 2008).
Apoptosis analysis
The following three methods were used to assess drug-induced apoptotic cell death: detection of DNA fragmentation with the Cell Death Detection ELISA kit (Roche Diagnostics, Indianapolis, IN, USA), Western blot analysis of caspase activation and PARP cleavage, and measurement of apoptotic cells by flow cytometry (sub-G1). The elisa was conducted according to the manufacturer's instructions.
Gene knockdown using siRNA
Smart pool siRNA reagents, including a control (D-001810-10), and mcl-1, STAT3, SHP-1, SHP-2, and PTP-1B were all purchased from Dharmacon Inc. (Chicago, IL, USA). The procedure has been described previously (Chen et al., 2008).
PLC5 with ectopic expression of STAT3
STAT3 cDNA (KIAA1524) was purchased from Addgene plasmid repository (http://www.addgene.org/). PLC5 cells with stable expression of STAT3 were then treated with drugs, harvested, and processed for Western blot analysis as described previously (Chen et al., 2008).
Activity of Raf-1 and SHP-1
A tyrosine phosphatase assay kit (R-22067) was used for assessing SHP-1 activity (Molecular Probes, Invitrogen, Grand Island, NY, USA). The Raf-1 kinase cascade assay kit (Upstate-Millipore, Billerica, MA) was used to examine Raf-1 kinase activity. The VEGFR1 kinase activity kit was purchased from Reaction Biology Corp. (Malvern, PA, USA).
Xenograft tumour growth
Male NCr athymic nude mice (5–7 weeks of age) were obtained from the National Laboratory Animal Center (Taipei, Taiwan). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). We used a total of 30 mice for the therapeutic evaluations and each separate experimental group consisted of 6-8 mice. In addition, all nude mice were housed in a light-controlled room with a 12 h day/night cycle and were given free access to water and food. The temperature of the animal room was kept at 25 °C.
All experimental procedures using these mice were performed in accordance with protocols approved by the Institutional Laboratory Animal Care and Use Committee of National Taiwan University. Each mouse was inoculated s.c. in the dorsal flank with 1 × 106 PLC5 cells suspended in 0.1 mL of serum-free medium containing 50% Matrigel (BD Biosciences, Bedford, MA, USA). When tumours reached 200–300 mm3, mice received an i.v. injection of CS-1008 (200 μg) three times a week, SC-49 (5 mg·kg−1) p.o. once daily or in combination. Controls received vehicle. Tumours were measured weekly using calipers, and their volumes calculated using the following standard formula: width2 × length × 0.52.
Immunohistochemistry
Immunohistochemical studies were performed on formalin-fixed, paraffin-embedded tissue. The primary monoclonal antibodies used were p-STAT3 (1:50 dilution; Cell Signaling), STAT3 (1:200; Cell Signaling), CD31 (1:40; BD Pharmingen, San Jose CA, USA). Antigen retrieval was performed at pH 9.0 using Epitope Retrieval 2 solution (Leica Microsystems, Wetzlar, Germany) for 30 min at 100°C. Slides were then stained using the Leica Microsystems BONDMAX autostainer according to the manufacturer's protocol.
Statistical analysis
Comparisons of mean values were performed using the independent samples _t_-test in SPSS for Windows 11.5 software (SPSS, Inc., Chicago, IL, USA).
Results
Sorafenib sensitizes resistant HCC cells to CS-1008-induced apoptosis
To investigate the effects of sorafenib and CS-1008 on HCC cells, we first examined the apoptotic effects of both drugs on a panel of three human HCC cell lines Hep3B, PLC5 and Huh-7 at clinically relevant concentrations (≤1000 ng·mL−1); however, the combination of sorafenib and CS-1008 overcame the resistance and induced apoptosis in all cell lines tested, in a dose-dependent manner (Figure 1A). Next, we examined the effect of sorafenib on CS-1008-induced apoptosis, as assessed by DNA fragmentation in all HCC cell lines. DNA fragmentation was determined by cell death elisa after 48 h of treatment. As shown in Figure 1B, combining sorafenib at 7.5 μM with CS-1008 reversed the resistance in all three cell lines and induced significant apoptosis. Moreover, we further examined the apoptotic pathway by Western blot analysis. Our data indicated that co-treatment with CS-1008 and sorafenib activated caspase-8 then induced cleavage of Bid and subsequently activated caspase-9 and caspase-3 and PARP cleavage (Figure 1C). These data suggest that this intrinsic pathway played a role in mediating the combined effect of sorafenib and CS-1008 on apoptosis in HCC cells.
Figure 1.

Sorafenib enhances CS-1008-induced apoptosis in resistant HCC cells. (A) Dose escalation effects of a combination of CS-1008 and sorafenib (5 or 7.5 μM) on apoptosis in 3 TNFSF10-resistant HCC cells. Cells were exposed to CS-1008 and/or sorafenib at the indicated doses for 48 h. Apoptotic cells were analysed by flow cytometry (sub-G1). (B) Effects of CS-1008 and sorafenib on DNA fragmentation in three HCC cell lines. Cells were treated with CS-1008 (1000 ng·mL−1) and/or sorafenib (7.5 μM) for 48 h and DNA fragmentation was analysed by using a cell death elisa kit. (C) Effects of sorafenib on CS-1008-induced apoptosis in PLC5 cells. CF, cleaved form (activated form).
Sorafenib and CS-1008 co-treatment down-regulates p-STAT3 in HCC cells
Previous studies have suggested that Mcl-1, an anti-apoptotic Bcl-2 family protein, may play a role in mediating the sensitizing effect of sorafenib to TNFSF10 in cancer cells (Meng et al., 2007; Ricci et al., 2007; Kim et al., 2008). As STAT3 regulates the expression of Mcl-1, we next examined its related proteins including phospho-STAT3 (p-STAT3), STAT3 and STAT3-regulated proteins, which include Mcl-1, survivin and cyclin D1. As shown in Figure 2A, co-treatment with sorafenib and CS-1008 down-regulated p-STAT3 (Tyr705) and related proteins, Mcl-1, survivin and cyclin D1, in all the cells tested without altering of total protein levels of STAT3. In addition, down-regulation of p-STAT3 was associated with the cleavage of PARP as shown by the analysis of apoptosis induced in cells exposed to sorafenib and CS-1008 for 48 h (Figure 2A). Furthermore, we found that the combination of sorafenib and CS-1008 down-regulated p-STAT3 in PLC5 cells in both a dose- and time-dependent manner (Figure 2B). Interestingly, co-treatment with sorafenib and CS-1008 did not affect phospho-Erk, suggesting that the sensitizing effect of sorafenib on CS-1008 is not associated with its Raf-1 activity (Figure 2C). Notably, the combination of sorafenib and CS-1008 did not alter the expression of Bax and Bcl-xl (Figure 2C).
Figure 2.

STAT3 mediates the sensitizing effect of sorafenib on CS-1008. (A) Effects of sorafenib (7.5 μM) and/or CS-1008 (1000 ng·mL−1) on STAT3-related proteins. Cells were exposed to the drugs for 48 h. (B) Effects of sorafenib on phospho-STAT3 in PLC5 cells. (C) Effects of sorafenib and/or CS-1008 on p-Erk, Bax and Bcl-xl. Cells were treated with 1000 ng·mL−1 CS-1008 and/or sorafenib at 7.5 μM for 48 h. (D) Left, down-regulation of mcl-1 by siRNA overcame the resistance to CS-1008 in PLC5 cells. Middle, down-regulation of STAT3 by siRNA overcomes resistance to CS-1008 in PLC5 cells. Right, ectopic expression of STAT3 reduced apoptosis induced by the combination of CS-1008 and sorafenib in PLC5 cells. Cells (wild-type PLC5 or PLC5 with STAT3 overexpression) were exposed to drugs at the indicated doses for 48 h. Apoptotic cells were analysed by flow cytometry (sub-G1). Columns show means with bars representing SD (n = 3). *P < 0.05.
Validation of STAT3
Several approaches were used to validate the finding that inhibition of STAT3 signals is responsible for the sensitizing effect of sorafenib on CS-1008-induced apoptosis in HCC cells. Firstly, we knocked down the protein expression of Mcl-1 and STAT3 by use of small interference RNA (siRNA). PLC5 cells were transfected with either control, survivin siRNA or STAT3 siRNA for 48 h then exposed to DMSO or CS-1008 at the indicated doses for another 48 h. Silencing Mcl-1 and STAT3 significantly sensitized PLC5 cells to CS-1008-induced apoptosis (P < 0.05) (Figure 2D, left and middle), suggesting that inhibition of the STAT3 signalling pathway is important for the sensitivity of HCC cells towards CS-1008. Next, we examined the effects of sorafenib in combination with CS-1008 in both wild-type PLC5 cells and PLC5 cells with ectopic expression (overexpression) of STAT3. Over-expression of STAT3 significantly reduced the combined effects of sorafenib plus CS-1008 on p-STAT3 and apoptosis (P < 0.05) (Figure 2D, right). Together, these results confirm the importance of STAT3 inhibition in mediating the combined effect of CS-1008 and sorafenib.
SHP-1 plays a role in mediating the effects of apoptosis induced by sorafenib and CS-1008
To elucidate the mechanism by which sorafenib plus CS-1008 down-regulated p-STAT3 in HCC cells, we investigated the roles of several protein phosphatases on the effect of sorafenib plus CS-1008 on p-STAT3 and apoptosis. Firstly, we altered the expression of SHP-1, by using siRNA, in PLC5 cells and showed that silencing SHP-1 significantly reduced the effects of sorafenib plus CS-1008 on p-STAT3 and apoptosis (Figure 3A, left). This suggests that SHP-1 mediates the effects of these drugs on p-STAT3 and apoptosis. Notably, co-treatment with sorafenib and CS-1008 did not affect the expression level of SHP-1 in HCC cells. Therefore, we measured SHP-1 phosphatase activity in PLC5 cells that were treated with sorafenib plus CS-1008. As shown in Figure 3A (right), sorafenib plus CS-1008 significantly increased the activity of SHP-1 (P < 0.05). Moreover, as sorafenib is a kinase inhibitor, we examined whether sorafenib plus CS-1008 enhanced SHP-1 activity by affecting the phosphorylation of SHP-1. According to previous reports, phosphorylation of SHP-1 at Tyr536 may enhance its activity and phosphorylation at Ser591 may down-regulate its activity. However, our data showed that neither sorafenib alone nor co-treatment with CS-1008 altered phospho-SHP-1 at either site (Figure 3B, left). In addition, we examined whether the combination of drugs affected the protein-protein interactions between SHP-1 and STAT3. Our data show that the amount of STAT-SHP1 complex did not alter significantly after co-treatment with the two drugs, suggesting that this combination treatment did not affect the interaction between SHP-1 and STAT3 protein (Figure 3B right). Finally we also examined other protein tyrosine phosphatases such as SHP-2 and PTP-1B that could also regulate the STAT3 signalling pathway. However, neither knockdown of SHP-2 nor silencing of PTP-1B affected the effect of sorafenib plus CS-1008 on p-STAT3 signalling and apoptosis (Figure 3C and D). These data indicate that SHP-2 and PTP-1B are not involved in mediating the effects of sorafenib on p-STAT3 signalling and apoptosis induced by CS-1008.
Figure 3.
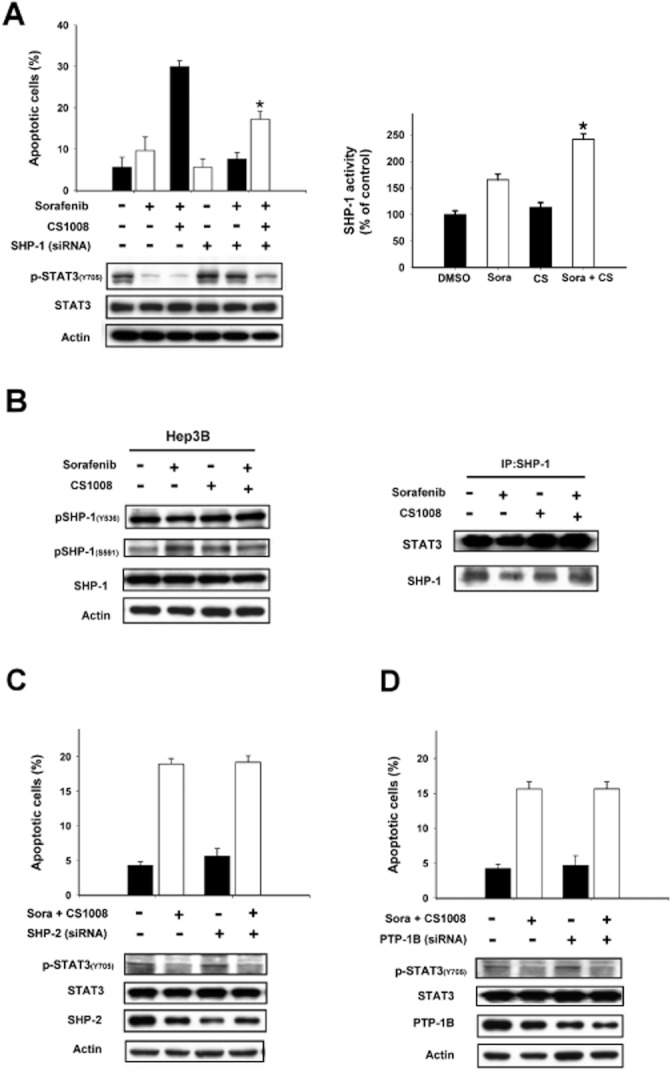
SHP-1 plays a role in mediating the effects of the combination of CS-1008 plus sorafenib on p-STAT3 and apoptosis. (A) Left, silencing SHP-1 by siRNA reduced the effects of sorafenib on p-STAT3 in HCC cells. Columns are means and bars represent SD (n = 3). *P < 0.05. Right, co-treatment of CS-1008 and sorafenib enhanced the activity of SHP-1 in PLC5 cells. (B) Left, effects of CS-1008 and/or sorafenib on phospho-SHP-1 in Hep3B cells. Columns show means with bars representing SD (n = 3). *P < 0.05. Right, effects of CS-1008 and/or sorafenib on SHP-1 and STAT3 protein interactions. (C) Knock down of SHP-2 did not affect effects of co-treatment of sorafenib and CS-1008 on p-STAT3 and apoptosis. (D) Knock down of PTP-1B did not affect effects of sorafenib on p-STAT3 and apoptosis. Columns show means with bars representing SD (n = 6). *P < 0.05.
SC-49, a sorafenib derivative, sensitizes HCC cells to CS-1008
Previously, we showed that STAT3 mediates the anti-tumour effect of sorafenib on HCC, and this effect was not related to inhibition of kinase activity (Tai et al., 2011). We then modified sorafenib and synthesized several new sorafenib analogues, which, by enhancing the activity of SHP-1, are potent inhibitors of STAT3. Here, we investigated the ability of SC-49 to sensitize HCC cells to CS-1008. As shown in Figure 4A, SC-49 is similar to sorafenib structurally but without Raf-1 inhibitory activity. Like sorafenib, the combination of SC-49 and CS-1008 also induced significant apoptosis in TNFSF10-resistant HCC cells. Combining sorafenib at 5 μM with CS-1008 reversed the resistance in all three cell lines and induced significant apoptosis within 24 h (Figure 4B and C). As shown in Figure 4D, co-treatment with SC-49 and CS-1008 activated caspase-8 then induced cleavage of Bid and subsequently activated caspase-9 and caspase-3 and PARP cleavage. It is noteworthy that in comparison with sorafenib, SC-49 sensitized HCC cells to CS-1008 at lower concentrations (5 μM vs. 7.5 μM) and in a shorter period of time (24 h vs. 48 h). These data suggest that SC-49 is more potent than sorafenib in sensitizing HCC cells to CS-1008.
Figure 4.
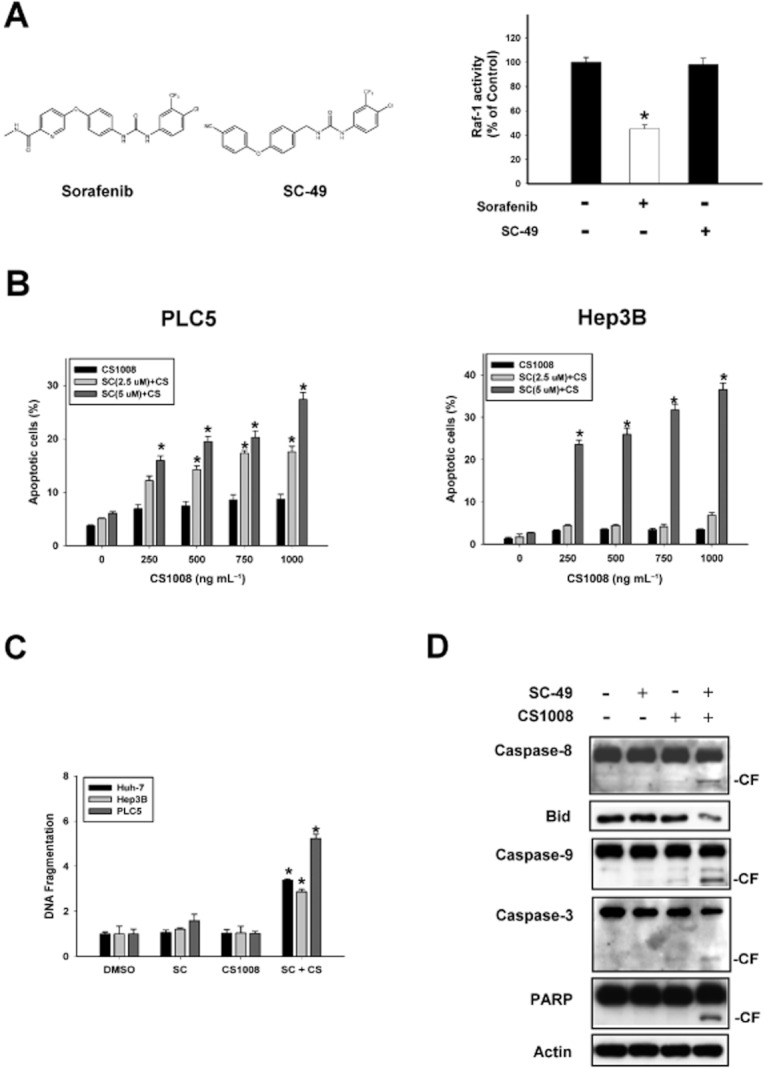
SC-49 sensitized HCC cells to CS-1008. (A) Left, chemical structures of sorafenib and SC-49. Right, effect of sorafenib and SC-49 on Raf-1 activity. PLC5 cells were exposed to sorafenib or SC-49 at 10 μM for 24 h. Points show means with bars representing SD (n = 6). (B) Dose escalation effects of a combination of CS-1008 and SC-49 (2.5 μM or 5 μM) on apoptosis in HCC cells. Cells were exposed to CS-1008 and/or SC-49 at the indicated doses for 24 h. (C) Effects of CS-1008 and SC-49 on DNA fragmentation in three HCC cell lines. Cells were treated with CS-1008 (1000 ng·mL−1) and/or sorafenib (5 μM) for 24 h and DNA fragmentation was analysed by using a cell death elisa kit. (D) Effects of SC-49 on CS-1008-induced apoptosis in PLC5 cells. CF, cleaved form (activated form).
SC-49 showed better apoptotic effects than sorafenib in HCC
To further investigate the effect of SC-49 on angiogenesis, we tested the effect of SC-49 on the activity of VEGFR1 in HUVEC cells. As shown in Figure 5A (left), sorafenib as a kinase inhibitor significantly inhibited the activity of VEGFR1 in HUVEC cells. However, unlike sorafenib, SC-49 did not affect the activity of VEGFR1 in HUVEC cells. Next, we examined the effect of SC-49 on p-STAT1 and p-STAT5; SC-49 down-regulated both p-STAT1 and p-STAT5 in a dose-dependent manner (Figure 5A, right). Furthermore, in HCC cell lines, Huh-7 and Hep3B, we found that SC-49 induced more apoptotic cell death than sorafenib (Figure 5B). In addition, SC-49 was more effective at down-regulating p-STAT3 than sorafenib in HCC cells (Figure 5C). These data suggest that SC-49, a sorafenib derivative without kinase inhibitory activity, is a more potent anti-tumour agent than sorafenib and that its effect is induced by targeting the STAT3 signalling pathway. Notably, a recent study has shown that depletion of tumour-associated macrophages may be associated with the effect of sorafenib on a liver metastasis model (Zhang et al., 2010). In this regard, we tested the effect of SC-49 on U937, a human macrophage cell line (Passmore et al., 2001) and again found that SC-49 down-regulated p-STAT3 in a dose-dependent manner in these cells (Figure 5D).
Figure 5.

SC-49 showed better apoptotic effects than sorafenib in HCC. (A) Left, effect of sorafenib and SC-49 on VEGFR1 activity. Right, effect of sorafenib and SC-49 on p-STAT1 and p-STAT5. Cells were exposed to sorafenib or SC-49 at the indicated doses for 24 h. Points show means with bars representing SD (n = 6). (B) Dose escalation effects of sorafenib and SC-49 on apoptosis in HCC cells. Cells were exposed to sorafenib or SC-49 at the indicated doses for 24 h. Apoptotic cells were analysed by flow cytometry (sub-G1). (C) Effects of SC-49 on p-STAT3. Cells were treated with SC-49 or sorafenib at the indicated doses for 24 h. (D) Effects of SC-49 on p-STAT3 in U937 cells. Cells were treated with SC-49 at the indicated doses for 24 h.
SHP-1-dependent inhibition of STAT3 mediates the effect of SC-49
We next examined the mechanism by which SC-49 sensitized HCC cells to CS-1008. Our data showed that the combination of SC-49 and CS-1008 decreased p-STAT3 in three TNFSF10-resistant cell lines. Co-treatment with SC-49 plus CS-1008 also down-regulated STAT3-driven proteins, Mcl-1, survivin and cylcin D1, in all three HCC cell lines (PLC5, Huh-7 and Hep3B) (Figure 6A). As shown in Figure 6B, overexpression of STAT3 reversed the sensitizing effects of SC-49 on CS-1008, suggesting that STAT3 plays a role in mediating the effect of SC-49. We next employed a specific SHP-1 inhibitor to test whether SHP-1 mediates the effect of SC-49 on p-STAT3, and found that the SHP-1 inhibitor abolished the sensitizing effect of SC-49 on the action of CS-1008 significantly, indicating that SHP-1 is a mediator of this effect of SC-49 (Figure 6C). As shown in Figure 6D (left), SC-49 at 5 μM enhanced the activity of SHP-1. Notably, SC-49 did not affect the phosphorylation of SHP-1 (Figure 6D, right). To further explore the mechanism by which SC-49 affected the activity of SHP-1, we tested the effect of SC-49 on SHP-1-containing cell lysates to determine whether SC-49 enhances the activity of SHP-1 by affecting its interactions with other proteins. Briefly, PLC5 cells were immunoprecipitated with anti-SHP-1 antibody. Protein extract which included SHP-1 complex was further incubated with SC-49 at 7.5 μM and/or CS-1008 at 1000 ng·mL−1 for 30 min and then SHP-1 phosphatase activity assay was performed. Our data showed that SC-49 alone or in combination with CS-1008 increased the phosphatase activity of SHP-1-containg lysates (Figure 6E). These data suggest that SC-49 enhances the activity of SHP-1 by directly interacting with it. However, further work is needed to elucidate details of the interactions between SC-49 and SHP-1.
Figure 6.

SHP-1 mediates the effect of SC-49 plus CS-1008 on p-STAT3 and apoptosis. (A) Effects of SC-49 (5 μM) and/or CS-1008 (1000 ng·mL−1) on STAT3-related proteins. Cells were exposed to the drugs for 24 h. (B) Ectopic expression of STAT3 reduced apoptosis induced by SC-49 plus CS-1008 in PLC5 cells. (C) Inhibition of SHP-1 by adding a SHP-1 inhibitor reduced the effects of sorafenib on p-STAT3 in Hep3B cells. Columns show means with bars representing SD (n = 3). *P < 0.05. (D) Left, co-treatment of SC-49 (5 μM) and CS-1008 (1000 ng·mL−1) enhanced the activity of SHP-1 in PLC5 cells. Right, effects of SC-49 (5 μM) and/or CS-1008 (1000 ng·mL−1) on phospho-SHP-1 in Hep3B cells. (E) Effects of dovitinib on phosphatase activity in SHP-1-containing lysates. PLC5 cells were immunoprecipitated with anti-SHP-1 antibody. The lysates were incubated with dovitinib (10 nM) for 30 min then analysed by SHP-1 phosphatase activity.
In vivo effects of SC-49 in Huh-7 xenograft tumours
To further examine the effect of SC-49, we next tested the effect of SC-49 on Huh-7 xenograft tumours in vivo. As shown in Figure 7A (left), treatment of mice with SC-49 at a dose of 10 mg·kg−1·day−1 p.o. significantly reduced the growth of the Huh-7 tumour and this anti-tumour effect was better than that of sorafenib in vivo. As shown in Figure 7A (right), animals had stable body weights throughout the course of study. In addition, SC-49 down-regulated p-STAT3 in Huh-7 tumours (Figure 7B). SC-49 and sorafenib enhanced the activity of SHP-1 in Huh-7 tumours (Figure 7C). Immunohistochemical staining for STAT3 showed no obvious significantly different cytoplasmic expression in all groups (Figure 7D). The treatment of both sorafenib and SC-49 decreased the nuclear expression of P-STAT3 (Figure 7D). From the immunohistochemical stain for CD-31, all the groups showed a similar vascular density in the tumour areas (Figure 7D).
Figure 7.

In vivo effects of SC-49 in Huh-7 xenograft tumour. (A) Left, SC-49 showed significant anti-tumour effect on Huh-7 tumours. Right, body weight. Points show means with bars representing SEM (n = 6). *, P < 0.05; **, P < 0.01. (B) Western blot analysis of p-STAT3, STAT3, and SHP-1 in Huh7 tumours. (C) Analysis of SHP-1 activity. Columns show means with bars representing SD (n = 6). *P < 0.05 versus vehicle group. (D) Immunohistochemical staining for tumours. Slides were then stained using the Leica Microsystems BONDMAX autostainer according to the manufacturer's protocol (400 folds).
These data indicate that SC-49 exhibited better in vivo effects than sorafenib through an SHP-1-dependent inhibitory effect on STAT3.
The effect of the combination of SC-49 and CS-1008 in vivo
To confirm whether the sensitizing effect of SC-49 in resistant cell lines has potentially relevant clinical implications, we assessed the effect of the combination of CS-1008 plus SC-49 on the growth of PLC5 tumours in vivo. Tumour-bearing mice were treated with vehicle or CS-1008 i.v. at a dose of 200 μg three times a week or SC-49 p.o. at a dose of 5 mg·kg−1·day−1, or a combination of the two, for the duration of the study. All animals tolerated the treatments well without an observable signs of toxicity and had stable body weights throughout the course of study. No gross pathological abnormalities were noted at necropsy.
Tumour growth was significantly inhibited by co-treatment with CS-1008 and SC-49 for 2 weeks (vs. control, P < 0.05), and tumour size in the co-treatment group was only one third of that of the control group at the end of the study (Figure 8A). Treatment with CS-1008 had no significant effect on PLC5 tumour growth. SC-49 alone showed modest effects on tumour growth. In addition, co-treatment of SC-49 and CS1008 significantly down-regulated p-STAT3 in PLC5 tumours (Figure 8B). Moreover, as shown in Figure 8C, co-treatment with SC-49 and CS-1008 enhanced SHP-1 activity significantly, indicating that SHP-1 plays a role in mediating the effects of the combination of drugs on PLC5 tumours. Together, these data indicate that a combination of CS-1008 and SC-49 exhibits good anti-tumour activity in vivo. Further clinical investigations are warranted.
Figure 8.

The effect of the combination of SC-49 and CS-1008 in vivo. (A) The combination of SC-49 and CS-1008 showed significant anti-tumour effect on PLC5 tumours. Columns show means with bars representing SEM (n = 6). *, P < 0.05; **, P < 0.01. (B) Western blot analysis of p-STAT3, STAT3, and SHP-1 in PLC5 tumours. (C) Analysis of SHP-1 activity. Columns show means with bars representing SD (n = 6). *P < 0.05 versus vehicle group.
Discussion
Previous literature has consistently shown that sorafenib is capable of sensitizing various cancer cells, including HCC, to TNFSF10-induced apoptosis (Hall and Cleveland, 2007; Ricci et al., 2007; Koehler et al., 2009; Huang and Sinicrope, 2010; Llobet et al., 2010). For that reason, CS-1008 is currently undergoing phase II trials for the treatment of advanced HCC (NCI clinical trial: NCT01033240). The results should provide informative in vivo evidence of the activity of this combination strategy.
Although the mechanisms employed by various tumours to evade TNFSF10-induced apoptosis are heterogeneous (Zhang and Fang, 2005), it has been suggested that Mcl-1 is the gateway to the sensitizing effect of sorafenib in cells (including HCC cells) that harbour defects in apoptosis mediated by the intrinsic pathway (Meng et al., 2007; Kim et al., 2008). In addition, aberrant activation of anti-apoptotic pathways, such as PI3K/Akt signalling, MAPK pathway and the NF-κB pathway, may also contribute to the development of TNFSF10 resistance in HCC cells (Bortul et al., 2003; Ehrhardt et al., 2003; Zhang and Fang, 2005). In particular, TNFSF10 treatment in resistant cells has been shown to induce Mcl-1 expression through the Raf and NF-κB-dependent pathway. Sorafenib, as a Raf kinase inhibitor, could, therefore, potentially block this TNFSF10-induced NF-κB–mediated transcriptional activation of Mcl-1, and NF-κB binding to the Mcl-1 promoter region (Kim et al., 2008). In the present study, we added to previous data by further showing that sorafenib suppresses other STAT3-regulated proteins (i.e. survivin and cyclin D1) in HCC. Our data also confirmed that the inhibition of STAT3 is the major mechanism by which sorafenib sensitizes TNFSF10 in HCC. It has previously been demonstrated that sorafenib inhibits STAT3 activity and enhances TNFSF10-mediated apoptosis in other cancer cells, including pancreatic cancer cells (Huang and Sinicrope, 2010), medulloblastomas cells (Yang et al., 2008) and cholangiocarcinoma cells (Blechacz et al., 2009). Taken together, these data indicate that STAT3 represents a novel anti-cancer target of sorafenib.
Another important finding in the current study is that sorafenib inhibits STAT3 by increasing SHP-1 activity (Figure 3). Our results showed that sorafenib increased SHP-1 activity but did not alter SHP-1 protein expression level and, despite being a kinase inhibitor, did not alter the phosphorylation of SHP-1 at either the Y-536 or S-591 sites, both known to change SHP-1 activity upon phosphorylation. Moreover, sorafenib did not influence the SHP-1 and STAT3 protein–protein interactions. In contrast, several chemical compounds such as acety-11-keto-β-boswellic acid and butein (3,4,2′,4′-tetrahydroxychalcone), are thought to inhibit STAT3 by the induction of SHP-1 expression (Kunnumakkara et al., 2009; Pandey et al., 2009). Nevertheless, the mechanism by which sorafenib influences SHP-1 activity remains to be elucidated and further studies are needed to address this issue. Interestingly, Blechacz et al. (2009) suggested that sorafenib inhibits STAT3 in cholangiocarcinoma cells by influencing SHP-2 activity through the down-regulation of phospho-SHP-2. However,Blechacz et al. (2009) did not show whether sorafenib also affects SHP-1 in cholangiocarcinoma cells. In contrast, in the present work we have shown that knockdown of SHP-2 did not alter the sensitizing effect of sorafenib on apoptosis and STAT3 phosphorylation in HCC cells (Figure 3C). However, it is possible that sorafenib inhibits STAT3 by affecting different protein tyrosine phosphatases in various cancer cells. More effort is needed to fully understand why sorafenib affects different protein tyrosine phosphatases in HCC and cholangiocarcinoma, and perhaps other cancer cells.
In conclusion, our results revealed that sorafenib as well as SC-49 have a synergistic effect with CS-1008 on HCC through SHP-1-dependent inhibition of STAT3 and indicate that the STAT3 signalling pathway may be a suitable target for the development of anti-HCC targeted agents. Sorafenib may serve as a lead compound for the development of more potent STAT3 inhibitors.
Acknowledgments
This study is supported by grants, NTUH 100P04 from National Taiwan University Hospital (K-F Chen), NSC99-2314-B-002-017-MY2 (K-F Chen), NSC 100-3112-B-002 -013 (A-L. Cheng), NSC 100-2325-B-002 -036 (K-F Chen), and NSC 100-2325-B-010 -007 (C-W Shiau) from National Science Council, Taiwan, Taiwan Clinical Oncology Research Foundation (C-Y Liu).
Glossary
c-FLIP
cellular FLICE-inhibitory protein
FADD
Fas-associated protein with death domain
HCC
hepatocellular carcinoma
TNFSF10 (TRAIL)
TNF-related apoptosis-inducing ligand
Disclosure of potential conflict of interest
A-LC is a consultant for and a member of the speaker's bureau of Bayer-Schering and a consultant of Daiichi Sankyo. K I is an employee of Daiichi Sankyo. Other authors have nothing relevant to this manuscript to disclose.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612. doi: 10.1016/S0076-6879(05)07047-3. [DOI] [PubMed] [Google Scholar]
- Auclair D, Miller D, Yatsula V, Pickett W, Carter C, Chang Y, et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia. 2007;21:439–445. doi: 10.1038/sj.leu.2404508. [DOI] [PubMed] [Google Scholar]
- Blechacz BR, Smoot RL, Bronk SF, Werneburg NW, Sirica AE, Gores GJ. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology. 2009;50:1861–1870. doi: 10.1002/hep.23214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortul R, Tazzari PL, Cappellini A, Tabellini G, Billi AM, Bareggi R, et al. Constitutively active Akt1 protects HL60 leukemia cells from TRAIL-induced apoptosis through a mechanism involving NF-kappaB activation and cFLIP(L) up-regulation. Leukemia. 2003;17:379–389. doi: 10.1038/sj.leu.2402793. [DOI] [PubMed] [Google Scholar]
- Chen KF, Yeh PY, Yeh KH, Lu YS, Huang SY, Cheng AL. Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2008;68:6698–6707. doi: 10.1158/0008-5472.CAN-08-0257. [DOI] [PubMed] [Google Scholar]
- Chen KF, Yeh PY, Hsu C, Hsu CH, Lu YS, Hsieh HP, et al. Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem. 2009;284:11121–11133. doi: 10.1074/jbc.M806268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, et al. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res. 2010;16:5189–5199. doi: 10.1158/1078-0432.CCR-09-3389. [DOI] [PubMed] [Google Scholar]
- Chen KF, Tai WT, Huang JW, Hsu CY, Chen WL, Cheng AL, et al. Sorafenib derivatives induce apoptosis through inhibition of STAT3 independent of Raf. Eur J Med Chem. 2011;46:2845–2851. doi: 10.1016/j.ejmech.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, et al. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–1319. [PubMed] [Google Scholar]
- Ehrhardt H, Fulda S, Schmid I, Hiscott J, Debatin KM, Jeremias I. TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene. 2003;22:3842–3852. doi: 10.1038/sj.onc.1206520. [DOI] [PubMed] [Google Scholar]
- Falschlehner C, Ganten TM, Koschny R, Schaefer U, Walczak H. TRAIL and other TRAIL receptor agonists as novel cancer therapeutics. Adv Exp Med Biol. 2009;647:195–206. doi: 10.1007/978-0-387-89520-8_14. [DOI] [PubMed] [Google Scholar]
- Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002;21:2283–2294. doi: 10.1038/sj.onc.1205258. [DOI] [PubMed] [Google Scholar]
- Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13:5665–5669. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- Hall MA, Cleveland JL. Clearing the TRAIL for cancer therapy. Cancer Cell. 2007;12:4–6. doi: 10.1016/j.ccr.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Huang S, Sinicrope FA. Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Mol Cancer Ther. 2010;9:742–750. doi: 10.1158/1535-7163.MCT-09-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- Ke Y, Zhang EE, Hagihara K, Wu D, Pang Y, Klein R, et al. Deletion of Shp2 in the brain leads to defective proliferation and differentiation in neural stem cells and early postnatal lethality. Mol Cell Biol. 2007;27:6706–6717. doi: 10.1128/MCB.01225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ricci MS, El-Deiry WS. Mcl-1: a gateway to TRAIL sensitization. Cancer Res. 2008;68:2062–2064. doi: 10.1158/0008-5472.CAN-07-6278. [DOI] [PubMed] [Google Scholar]
- Koehler BC, Urbanik T, Vick B, Boger RJ, Heeger S, Galle PR, et al. TRAIL-induced apoptosis of hepatocellular carcinoma cells is augmented by targeted therapies. World J Gastroenterol. 2009;15:5924–5935. doi: 10.3748/wjg.15.5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunnumakkara AB, Nair AS, Sung B, Pandey MK, Aggarwal BB. Boswellic acid blocks signal transducers and activators of transcription 3 signaling, proliferation, and survival of multiple myeloma via the protein tyrosine phosphatase SHP-1. Mol Cancer Res. 2009;7:118–128. doi: 10.1158/1541-7786.MCR-08-0154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kusaba M, Nakao K, Goto T, Nishimura D, Kawashimo H, Shibata H, et al. Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL-mediated apoptosis. J Hepatol. 2007;47:546–555. doi: 10.1016/j.jhep.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li WC, Ye SL, Sun RX, Liu YK, Tang ZY, Kim Y, et al. Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clin Cancer Res. 2006;12:7140–7148. doi: 10.1158/1078-0432.CCR-06-0484. [DOI] [PubMed] [Google Scholar]
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- Llobet D, Eritja N, Yeramian A, Pallares J, Sorolla A, Domingo M, et al. The multikinase inhibitor Sorafenib induces apoptosis and sensitises endometrial cancer cells to TRAIL by different mechanisms. Eur J Cancer. 2010;46:836–850. doi: 10.1016/j.ejca.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K, et al. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem. 2007;282:29831–29846. doi: 10.1074/jbc.M706110200. [DOI] [PubMed] [Google Scholar]
- Okano H, Shiraki K, Inoue H, Kawakita T, Yamanaka T, Deguchi M, et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest. 2003;83:1033–1043. doi: 10.1097/01.lab.0000079328.76631.28. [DOI] [PubMed] [Google Scholar]
- Pandey MK, Sung B, Ahn KS, Aggarwal BB. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol Pharmacol. 2009;75:525–533. doi: 10.1124/mol.108.052548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore JS, Lukey PT, Ress SR. The human macrophage cell line U937 as an in vitro model for selective evaluation of mycobacterial antigen-specific cytotoxic T-cell function. Immunology. 2001;102:146–156. doi: 10.1046/j.1365-2567.2001.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M, et al. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43:425–434. doi: 10.1002/hep.21054. [DOI] [PubMed] [Google Scholar]
- Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell. 2007;12:66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Rowinsky EK. Targeted induction of apoptosis in cancer management: the emerging role of tumor necrosis factor-related apoptosis-inducing ligand receptor activating agents. J Clin Oncol. 2005;23:9394–9407. doi: 10.1200/JCO.2005.02.2889. [DOI] [PubMed] [Google Scholar]
- Saleh MN, Percent I, Wood TE, Posey J, III, Shah J, Carlisle R, et al. A phase I study of CS-1008 (humanized monoclonal antibody targeting death receptor 5 or DR5), administered weekly to patients with advanced solid tumors or lymphomas. J Clin Oncol. 2008;26:3537. [Google Scholar]
- Shin EC, Seong YR, Kim CH, Kim H, Ahn YS, Kim K, et al. Human hepatocellular carcinoma cells resist to TRAIL-induced apoptosis, and the resistance is abolished by cisplatin. Exp Mol Med. 2002;34:114–122. doi: 10.1038/emm.2002.17. [DOI] [PubMed] [Google Scholar]
- Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ, et al. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol. 2011;55:1041–1048. doi: 10.1016/j.jhep.2011.01.047. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Arii S. Molecularly targeted therapy for hepatocellular carcinoma. Cancer Sci. 2009;100:1–8. doi: 10.1111/j.1349-7006.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64:3517–3524. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- Verslype C, Van Cutsem E, Dicato M, Arber N, Berlin JD, Cunningham D, et al. The management of hepatocellular carcinoma. Current expert opinion and recommendations derived from the 10th World Congress on Gastrointestinal Cancer, Barcelona, 2008. Ann Oncol. 2009;20(Suppl 7):vii1–vii6. doi: 10.1093/annonc/mdp281. [DOI] [PubMed] [Google Scholar]
- Wang JM, Lai MZ, Yang-Yen HF. Interleukin-3 stimulation of mcl-1 gene transcription involves activation of the PU.1 transcription factor through a p38 mitogen-activated protein kinase-dependent pathway. Mol Cell Biol. 2003;23:1896–1909. doi: 10.1128/MCB.23.6.1896-1909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene. 2008;27:6207–6215. doi: 10.1038/onc.2008.298. [DOI] [PubMed] [Google Scholar]
- Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res. 2010;16:1701–1708. doi: 10.1158/1078-0432.CCR-09-1692. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Yada A, Yazawa M, Ishida S, Yoshida H, Ichikawa K, Kurakata S, et al. A novel humanized anti-human death receptor 5 antibody CS-1008 induces apoptosis in tumor cells without toxicity in hepatocytes. Ann Oncol. 2008;19:1060–1067. doi: 10.1093/annonc/mdn015. [DOI] [PubMed] [Google Scholar]
- Yang F, Van Meter TE, Buettner R, Hedvat M, Liang W, Kowolik CM, et al. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol Cancer Ther. 2008;7:3519–3526. doi: 10.1158/1535-7163.MCT-08-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang-Yen HF. Mcl-1: a highly regulated cell death and survival controller. J Biomed Sci. 2006;13:201–204. doi: 10.1007/s11373-005-9064-4. [DOI] [PubMed] [Google Scholar]
- Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–237. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16:3420–3430. doi: 10.1158/1078-0432.CCR-09-2904. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Zhang XY, Gray CP, Nguyen T, Hersey P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res. 2001;61:7339–7348. [PubMed] [Google Scholar]