Molecular imaging of microglia/macrophages in the brain (original) (raw)
. Author manuscript; available in PMC: 2013 Feb 24.
Published in final edited form as: Glia. 2012 May 21;61(1):10–23. doi: 10.1002/glia.22357
Abstract
Neuroinflammation perpetuates neuronal damage in many neurological disorders. Activation of resident microglia and infiltration of monocytes/macrophages contributes to neuronal injury and synaptic damage. Non-invasive imaging of these cells in vivo provides a means to monitor progression of disease as well as assess efficacies of potential therapeutics. This review provides an overview of positron emission tomography (PET) and magnetic resonance (MR) imaging of microglia/macrophages in the brain. We describe the rationale behind PET imaging of microglia/macrophages with ligands that bind to translocator protein-18 kDa (TSPO). We discuss the prototype TSPO radioligand [11C]PK11195, its limitations, and the development of newer TSPO ligands as PET imaging agents. PET imaging agents for targets other than TSPO are emerging, and we outline the potential of these agents for imaging brain microglia/macrophage activity in vivo. Finally, we briefly summarize advances in MR imaging of microglia/macrophages using iron oxide nanoparticles and ultra-small super paramagnetic particles that are phagocytosed. Despite many technical advances, more sensitive agents are required to be useful indicators of neuroinflammation in brain.
Keywords: PET, microglia, macrophages, neuroinflammation, MRI
Introduction
All organ systems contain tissue-based macrophages referred to as histiocytes. These cells arise from yolk sac and bone marrow stem cells and populate systemic organs during development (Chan et al. 2007; Ransohoff and Perry 2009). Long considered to be quiescent cells, our understanding of the physiological role of histiocytes has changed dramatically. Today, histiocytes are considered local sentinels performing important physiological responses and molecularly primed to detect injury and initiate an innate immune response (Kofler and Wiley 2011).
The nature and origin of the central nervous system (CNS) histiocytes called microglia has been debated literally for centuries. While unquestionably of hematopoietic origin and not derived from the neuroectoderm, their renewal and physiological turnover during the life of the host remains controversial (Ransohoff and Perry 2009; Soulet and Rivest 2008). Perhaps this controversy is best summarized as: microglia have a significant capacity to locally renew and respond, but ingress of additional mononuclear elements can occur depending upon the experimental system under study. To avoid a Lilliputian controversy regarding distinguishing microglia from macrophages, it is important to clearly define what is meant by the term microglia where possible and necessary distinguish it from macrophages, and finally combine the terms when such a distinction is not possible. For this review, we will use the term “microglia” (μ) only when we are unambiguously referring to the myeloid elements located in or derived from those CNS histiocytes present at birth. We will use the general term “macrophages” (M) when referring to monocytes from the bone marrow that have trafficked into the CNS after birth or in response to injury. Since once the elements are in the brain it is difficult to distinguish them, especially from an in vivo imaging perspective, most of the time we will use the term “M/μ” to be clear of the ambiguity. When it is important to distinguish between the two origins, rigorous experimental manipulation is necessary. Fortunately for the purpose of this review, the semantic distinction is not crucial and it is more important to be unambiguous about intended meaning.
Non-invasive in vivo imaging of cells has a long history that has recently undergone revolutionary advances. With the advent of the first proto-microscope approximately 350 years ago, emerged the concept of a cell. First discovered by Robert Hooke in plant tissue, technical advances in the microscope soon allowed Anton Van Leeuwenhoek to describe single cell living organisms and then individual cells within animals. The capacity to resolve cells at the theoretical limit of visible light wavelengths led to an explosion of new knowledge and formed the basis of our understanding of tissue structure.
Several centuries later we are experiencing a new revolution utilizing novel technology that extends our abilities well beyond the simple configuration of ground glass lenses. As initially configured the microscope was severely limited by the need to project light through a specimen or gather light that was reflected from the specimen surface, this necessarily limited detection to thinly sliced preparations stained in various arcane manners to create contrast in structures. The advent of confocal microscopy reduced some of the need to physically slice a tissue and instead to optically section thicker material using confocal principles. Today multi-photon confocal microscopy provides a moderately non-invasive means (e.g. after creating a window through the overlying skin and bone) of imaging the CNS to a depth of ~200 microns. Sophisticated design of experimental animals and systems permits the introduction of molecular labels that can then be viewed in real time in the living animal.
Scientific and technical advances outside of the field of microscopy have unshackled investigators from dependence on the microscope to monitor cellular elements.. Bioluminescent imaging is founded on the capacity of visible light photons to penetrate many millimeters of soft tissue and be detected by sensitive photo-multipliers. Experiments utilizing bioluminescence have permitted real-time assessment of physiological processes and the tracking of cells in living animals. From metastasis of malignant cells to migration of stem cells, bioluminescence has dramatically expanded our understanding of cell movement within a living organism. As currently implemented, this technology does not permit tracking of individual cells and the capacity of these photons to penetrate tissues limits application to small rodents. The skull or vertebral column presents a formidable barrier to expanding this technology to studying the primate CNS. As discussed extensively below, high-energy photons, such as gamma radiation emitted during the decay of unstable nuclei, can substantially circumvent these tissue barriers. High-energy (511 keV) gamma photons arising from the interaction and mutual annihilation of an electron and a positron, the electron’s anti-particle, forms the basis for positron emission tomography (PET). Neutron-deficient nuclei are often unstable and typically decay by positron emission. It is possible to artificially produce some of these unstable radionuclides in small quantities using a proton accelerator called a cyclotron, which provides the source of positrons for PET imaging. Importantly, many of the positron-emitting nuclides that can be produced by a relatively low energy cyclotron (~11 MeV/proton) are species of biologically occurring elements such as carbon, nitrogen, oxygen, and fluorine. PET imaging is non-invasive and provides several advantages including direct applicability to human subjects and the capacity to extract quantitative information in vivo regarding specific biological phenomena relevant to a disease process.
Why image M/μ?
Microglia formally described by del Rio Hortega (Del Rio Hortega 1932) constitute approximately 5% of the CNS cell population, yet represent a significant component of the CNS microenvironment. Previously thought to be quiescent and non-motile (in the “resting state”) in the normal brain, microglia are active and mobile cells constantly sampling their surrounding milieu by extending processes to neurons, astrocytes and blood vessels suggesting that microglia contribute to maintaining homeostasis of the CNS microenvironments (Davalos et al. 2005; Nimmerjahn et al. 2005). Macrophages (M) derived from peripheral monocytes infiltrate the CNS in many disease states. Both microglia (μ) and macrophages (M) undergo “activation” in response to synaptic and neuronal injury in a variety of neurological disorders. This phenomenon of activation is complex and can occur along multiple pathways with varying functional outcomes (Kofler and Wiley 2011). Chronically activated M/μ exacerbate neuronal injury by producing a myriad of neurotoxins such as interleukins (Giulian et al. 1986; Righi et al. 1989), tumor necrosis factor α (Chao et al. 1995b), free radicals (Chao et al. 1995a), nitric oxide (Chao et al. 1992), proteinases (Colton et al. 1993) and eicosanoids (Heyes et al. 1996). These toxins trigger various cellular processes including synaptic damage, changes in extracellular matrix and finally neuronal death and are discussed more extensively in other articles accompanying this review. Since chronically activated microglia exacerbate neurological injury, imaging these cells in vivo can provide a means of non-invasively monitoring neuroinflammation and disease progression. Further, decreasing activation of M/μ is a viable therapeutic target that can be leveraged to diminish neuronal damage. Imaging M/μ in this context will provide a means to assess the efficacy of therapeutics targeted to diminish neuroinflammation. This review focuses on non-invasive means of imaging the primate brain using PET and MR.
PET imaging of M/μ
PET imaging has come to the forefront as a means of non-invasively assessing biological processes by targeting specific molecular biomarkers in disease. PET imaging of M/μ in the CNS takes advantage of expression of the translocator protein 18 kDa (TSPO, formerly termed the peripheral benzodiazepine receptor) in M/μ (Banati 2002) (Venneti et al. 2006). In the past decade radioligands that bind specifically to TSPO have been extensively used to image neuroinflammation non-invasively using PET. The most well characterized of these ligands is the isoquinoline carboximide derivative _R_-[_N-methyl_-11C]PK-11195, although, recent years have yielded several other putative TSPO-specific ligands with improved properties for use as in vivo imaging agents, We examine the rationale behind these studies and detail some of the findings. Beyond using radioligands that bind to TSPO, recent reports examine other potentially viable molecular targets in M/μ for in vivo PET imaging. We briefly discuss these data in the latter part of this review.
Principles of PET imaging
Positron Emission Tomography (PET) is a nuclear medicine imaging technique used to non-invasively assess various biological functions. The technique relies upon the injection of chemical compounds of biological significance that are labeled with one of several short-lived radionuclides such as 11C (t½ = 20.4 min) and 18F (t½ = 109.8 min), which are artificially produced by bombarding stable target nuclei with protons accelerated to relatively high energies (~11 MeV) by a cyclotron. These and other radionuclides useful for PET imaging decay by the emission of a positron, the anti-particle of the electron, from the decaying nucleus with a characteristic kinetic energy spectrum. The positron quickly loses its kinetic energy through a series of elastic electrostatic (Coulomb) interactions with electrons in matter until reaching thermal energies, which results in large angular deviations of the positron’s trajectory (figure 1). The distance the positron travels from the point of emission is termed the positron range, which is a function of the effective atomic number (Z), density of the attenuating medium and the positron energy (Levin and Hoffman 1999). The thermalized positron combines with a free electron, briefly forming a bound state called positronium before a matter-antimatter interaction results in the mutual annihilation of both particles. In accordance with Einstein’s concept of mass-energy equivalence, the rest masses of the electron and positron are converted to energy in the form of electromagnetic radiation. The emitted radiation consists of two 511 keV gamma ray photons that travel along a line in opposite directions. It is the collinear angular correlation of these gamma ray pairs that forms the basis for PET imagingn (figure 1). A PET scanner consists of an array of radiation detectors and signal processing electronics that are tuned to detect these 511 keV gamma emissions and determine whether or not both gamma rays from the annihilation event are detected in coincidence. The detection of coincident gamma rays by opposing detectors in a PET scanner defines a line of response (LOR) between them. While it cannot be inferred where along the LOR the annihilation occurred, the tracer concentration at any given point is proportional to the number of LOR’s that intersect at that point (figure 1).
Figure 1. Principles of Positron Emission Tomography.

A simple scheme for PET radiotracer synthesis involves the labeling of a precursor molecule with a chemical synthon (e.g. methyl iodide) containing the PET radionuclide (e.g. carbon-11) to produce the desired radiotracer (top). PET radionuclides such as carbon-11 decay by positron emission to a stable nuclide. A positron is emitted from the decaying nucleus with a characteristic energy spectrum and quickly loses energy by inelastic scattering with electrons in the surrounding medium. Once the positron is in thermal equilibrium with its surroundings (thermalization) it is most likely to interact with a free electron, briefly forming a bound state called positronium before both particles mutually annihilate. The annihilation of the electron and positron results in the conversion of the rest masses of both particles into energy, manifested as the emission of two gamma ray photons each having an energy of 511 kilo-electron volts (keV). The 511 keV annihilation photons travel in opposite directions along a line, forming the basis for PET imaging (middle). A PET scanner is comprised of rings of radiation detectors that are triggered by the coincident detection of two 511 keV gamma rays, forming a line-of-response (LORs) between the detectors in coincidence. LORs are collected and sorted by the scanner in order to reconstruct the radioactivity distribution within the source object (bottom).
The ability to quantitatively determine the spatial distribution of radioactivity is one of the major advantages of PET. By acquiring many serial PET images in the same subject over discrete time intervals, a dynamic series of PET images may be reconstructed as a representation of the time-varying radioactivity concentration in a tissue of interest by application of mathematical models of radiotracer kinetic behavior to the dynamic PET data set. Non-human primate studies may be particularly useful in the process of compartmental model formulation for a prospective PET radioligand, as they are phylogenetically proximal to humans and thus provide a reasonably good approximation of the expected kinetic behavior of the radiotracer in humans. For a more detailed discussion of methods applied to [11C]PK11195 data analyses the reader is directed to a comprehensive review of the subject (Venneti et al. 2006).
TSPO as a molecular target for PET imaging of M/μ
TSPO structure and function
The peripheral benzodiazepine receptor was discovered in 1977 when high affinity binding of diazepam binding was noted in the rat kidney. Since this differed from diazepam binding to the central benzodiazepine receptor expressed in GABAergic neurons, it was named peripheral benzodiazepine receptor (Braestrup et al. 1977). In 2006, a focus group renamed the peripheral benzodiazepine receptor to “translocator protein −18 kDa” (TSPO) to reflect: (i) its role in cholesterol translocation and (ii) the molecular weight of the protein. (Papadopoulos et al. 2006a). In contrast to the central benzodiazepine receptor located on the plasma membrane of GABA-ergic neurons (Stephenson 1995), TSPO is localized to the outer mitochondrial membrane of astrocytes and M/μ in the CNS (Casellas et al. 2002). TSPO has a putative five transmembrane helical structure forming a hetero-oligomeric complex with the voltage-dependent anion channel (VDAC) and the adenine nucleotide carrier (ANC) (figure 2). Together, these proteins are thought to constitute the mitochondrial permeability transition pore (Joseph-Liauzun et al. 1998; McEnery et al. 1992).
Figure 2. PET imaging of brain activated microglia/macrophages with ligands that bind TSPO.
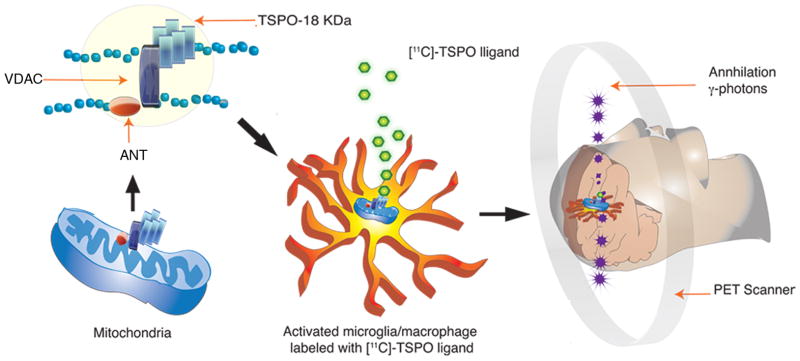
Translocator protein -18 kDa (TSPO) is located at the outer mitochondrial membrane and has a putative five transmembrane helical structure. It forms a hetero-oligomeric complex with the voltage dependent anion channel (VDAC) and the adenine nucleotide transporter (ANT) constituting the putative mitochondrial permeability transition pore. [11C]-labeled TSPO ligands (green) bind to TSPO located in activated microglia/macrophages. The [11C]-radionuclide decays by the emission of a positron, which combines with a free electron, resulting in the annihilation of both particles. Two gamma ray photons (purple) are simultaneously emitted at an angle of 180° from each other. The positron emission tomography (PET) scanner detects both gamma rays from the annihilation to help generate the 3-dimensional PET image.
The functions of TSPO in the CNS are not clearly known. Since TSPO is enriched in steroid synthesizing tissues such as the adrenal gland and the testis, the most well characterized function of TSPO is in steroid synthesis through regulation of cholesterol transport from the outer to the inner mitochondrial membranes (Papadopoulos et al. 1997). It is not known if M/μ synthesize steroids in the CNS. Intriguingly, emapunil (XBD173), a TSPO agonist, has anxiolytic effects in animal models and human subjects by enhancement of GABAergic neurotransmission hypothesized to be driven by TSPO regulated neurosteroid synthesis (Rupprecht et al. 2010; Rupprecht et al. 2009). While these findings need further elucidation, they raise the possibility that TSPO may regulate endogenous steroid synthesis in the CNS. Due to its association with the mitochondrial permeability transition pore, TSPO may regulate mitochondrial function (Hirsch et al. 1989, Sileikyte, 2011 #632). TSPO can also influence apoptosis by altering interactions of Bcl-2, Bcl-Xl and Bax with mitochondrial permeability transition pore proteins (Castedo et al. 2002; McEnery et al. 1992). TSPO overexpression in myxoma-infected macrophages and Jurkat cells blocks apoptosis (Everett et al. 2002; Everett and McFadden 2001; Stoebner et al. 2001). It is thus possible that TSPO regulates survival of M/μ in the chronically injured CNS, promoting neuroinflammation. Various functions have been attributed to TSPO outside the CNS and have been reviewed by others (Papadopolus). Despite an extensive literature describing TSPO ligands to image neuroinflammation, the role played by TSPO in both the physiologic and diseased CNS needs further elucidation and represents a significant gap in our knowledge.
Cellular localization of TSPO in neurological diseases
The cellular localization of increased TSPO expression in the diseased CNS is the subject of debate.. Animal models of disease and human postmortem tissues are used to address this question using immunohistochemistry and TSPO-ligand high-resolution emulsion autoradiography. These data suggest that the relative contributions of different cell types to the overall TSPO ligand binding vary depending on the specific animal model in question and the techniques used in localization of TSPO expression to specific cell types. [3H]PK11195 showed substantial localization to activated M/μ in brain tissues in animal models of cerebral ischemia (Myers et al. 1991; Stephenson et al. 1995), multiple sclerosis (Banati et al. 2000; Vowinckel et al. 1997), facial nerve axotomy (Banati et al. 1997), brain trauma (Raghavendra Rao et al. 2000), hippocampal axonal lesions (Pedersen et al. 2006), transgenic mouse models of Alzheimer’s disease (Venneti et al. 2008a) and SIV encephalitis in macaques (Mankowski et al. 2003; Venneti et al. 2008a; Venneti et al. 2004). TSPO expression co-localized mainly with activated M/μ, but only transiently in astrocytes in rats injected intracerebrally with ethanol (Maeda et al. 2007a). In contrast, TSPO immunostaining and ligand binding corresponded to astrocytosis with initial increases in M/μ in rodents treated with the neurotoxin trimethyltin (Kuhlmann and Guilarte 2000) and cuprizone (Chen et al. 2004). Similarly in rat models of focal ischemia, TSPO expression was initially high in M/μ followed by expression in reactive astrocytes (Martin et al. 2010). These data suggest a possible temporal relationship between TSPO expression in activated M/μ and reactive astrocytes. In line with these observations, Ji et. al. propose a model where in TSPO expression in astrocytes and activated M/μ is driven by the nature of neuronal injury with complex intercellular interactions between M/μ and reactive astrocytes (Ji et al. 2008).
Since TSPO expression in M/μ and reactive astrocytes in animal models vary depending on the specific neuronal insult, our laboratory and others have used human post-mortem tissues obtained from patients with various neurological disorders to determine cellular localization of TSPO ex vivo. In our hands, TSPO-radioligand binding correlated more with the abundance of activated M/μ as compared to the abundance of reactive gliosis in regions of pathology in postmortem brain tissues of cerebrovascular infarction, multiple sclerosis (MS), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD, figure 3) (Venneti et al. 2008b). Reactive astrocytes certainly bound [3H](R)-PK11195, but increases in the abundance of GFAP labeled astrocytes did not correlate with increases in [3H](R)-PK11195 binding in these diseases. TSPO-ligand binding showed similar results in brain regions with MS plaques (Banati et al. 2000). Further, immunostaining for TSPO co-localized with activated M/μ but not reactive astrocytes in brain tissues obtained from patients with AD, Pick’s disease and progressive supranuclear palsy (Maeda et al. 2011).
Figure 3. [3H](R)-PK11195 autoradiography overlaps mainly with CD68 labeled activated microglia/macrophages.
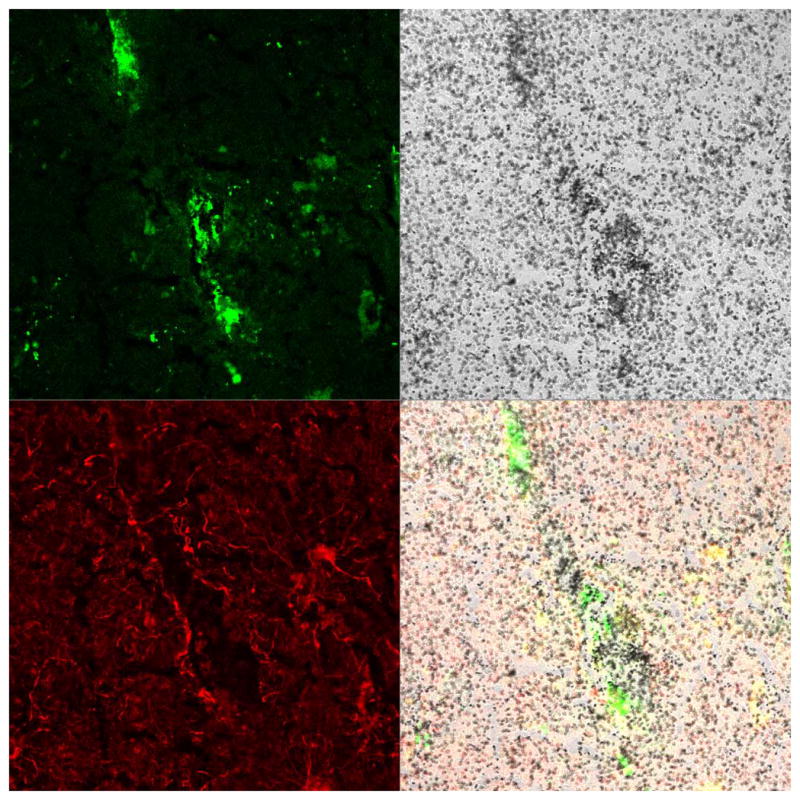
Combined autoradiography with [3H](R)-PK11195 and immunostaining was performed on a frozen brain section obtained from the frontal cortex of a patient with AD. Immunostaining was performed for astrocytes (GFAP, red) and activated microglia/macrophages (CD68, green). [3H](R)-PK11195 specific binding (black grains) overlapped mainly with CD68 labeled activated microglia/macrophages (merge).
Limitations of these studies are several. High-resolution emulsion autoradiography performed on frozen brain sections is compromised by relatively poorly preserved cellular morphology. Double-label immunohistochemistry in paraffin embedded sections overcomes this issue but should be interpreted with caution, as the binding sites of TSPO antibodies and TSPO ligands may be different (Cagnin et al. 2006). Further, the intimate association of astrocytic and M/μ processes, limitations of antibodies used to detect specific glial cell types in different model systems and sampling biases restricted to the brain sections used in the analyses, pose additional challenges for determining the relative contributions of specific cell types to TSPO radioligand binding. Finally, translating these data to a macroscopic scale to compare with in vivo PET imaging results brings additional complexities. While it is generally accepted that both astrocytes and M/μ express TSPO, there is no consensus on the relative increases in each cell type in response to CNS insults.
PET imaging with PK1195 and its limitations
PET imaging with racemic _R,S_-[N-methyl-11C]PK11195 was first reported in the hearts of dogs and humans in 1986 (Charbonneau et al. 1986). PK11195 is a pharmacological antagonist of TSPO, but in the tracer concentrations typical of a PET imaging study the ligand has no observable pharmacological effects. Both the racemic mixture and the more active R-enantiomer of PK11195 have been labeled with 3H for in vitro studies and 11C for in vivo PET imaging studies, producing a considerable body of literature in both human subjects and animal models of various neurological disorders. A complete discussion of this literature has been the subject of several recent reviews (Chauveau et al. 2008; Owen and Matthews 2011; Venneti et al. 2006). We summarize many of these studies, but more importantly discuss the major limitations of [11C]PK11195 imaging studies as well as limitations of the approach of targeting TSPO for in vivo imaging studies in general.
PK11195 labeled with 11C for PET imaging studies or 3H for in vitro autoradiographic studies and binding assays have shown varying degrees of success in both animal models and human subjects of neurological disorders that include Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, corticobasal degeneration, frontotemporal dementia, Huntington’s disease, stroke and multiple sclerosis (reviewed in (Banati 2002; Owen and Matthews 2011; Venneti et al. 2006)). Despite this considerable body of literature using [11C]PK11195 to investigate neuroinflammatory processes in vivo, several properties of the radioligand render it less than ideal for in vivo imaging studies. Although [11C]PK11195 readily enters and rapidly clears from normal mammalian brain (Cremer et al. 1992; Petit-Taboue et al. 1991; Price et al. 1990), difficulties in the interpretation of [11C]PK11195 PET scans were evident from a very early stage in the development of the radioligand. The R-enantiomer of PK11195 has been shown to have high affinity (KD ~ 2–20 nM) for TSPO from in vitro binding studies (Awad and Gavish 1991; Kurumaji et al. 1997; Rao and Butterworth 1997; Venneti et al. 2008b), however, the in vivo demonstration of elevated [11C]PK11195 specific binding to TSPO in pathologic CNS inflammation has been more elusive. Demonstration of in vivo specific binding is typically made by the blockade or displacement of the target receptors by a pharmacologically significant dose of a competing ligand, which causes the radioligand to bind less or clear more rapidly from tissue than without the competitor. In the case of [11C]PK11195, the administration of a pharmacological dose of unlabeled PK11195 was shown in baboons to result in a transient increase in brain radioactivity concentration likely due to an increase in the availability of free radioligand in plasma due to the blockade of far more abundant TSPO sites in peripheral organs by the unlabeled competitor (Petit-Taboue et al. 1991). Despite this transient increase, the authors noted afterward an apparent increase in the clearance rate of brain radioactivity that they ascribed to the displacement of [11C]PK11195 from a constitutive population of TSPO binding sites found throughout in normal brain. Indeed, this observation was consistent with the association between TSPO and glial cells (Casellas et al. 2002; Papadopoulos et al. 2006b; Rao and Butterworth 1997), although it is possible to attribute the accelerated clearance rate after pharmacological displacement to other phenomenon. In any case, the fact that a sizeable constitutive TSPO population exists in normal brain that binds PK11195 is not strictly a confound for TSPO imaging in disease, provided that a detectable increase in TSPO expression can be measured in the disease state that can be reasonably attributed to increased M/μ activation related to the underlying disease pathology. Indeed, in vitro saturation binding assays have demonstrated several-fold increases in the Bmax of binding sites for [3H]PK11195 in several neurodegenerative diseases as compared to healthy controls, and these increases have been shown to correlate with CD68 labeled activated M/μ but not GFAP staining of reactive gliosis (Venneti et al. 2008b).
Despite these encouraging in vitro data, in vivo imaging studies using [11C]PK11195 to indicate ongoing CNS inflammation in disease populations have been underwhelming, in part due to poor signal to noise characteristics. Many refinements to data analysis techniques have been explored and applied to human [11C]PK11195 studies in an effort to improve the detection of an in vivo specific binding signal. Fully quantitative analytic methods that relate radiotracer specific binding in brain to plasma concentrations of radioligand under equilibrium conditions have been validated for [11C]PK11195 (Kropholler et al. 2005). More commonly, simplified methods of analysis such as the Simplified Reference Tissue Model (SRTM) have been employed for the analysis of [11C]PK11195 PET data that do not require arterial sampling and input function determination (Gunn et al. 1997; Lammertsma and Hume 1996). The SRTM is an image-driven, model-based analytic method that assumes a particular underlying model configuration to describe the radiotracer kinetics, but implements simplifications of the model equations that allow radiotracer specific binding outcome measures such as the radiotracer binding potential (BP) to be directly estimated using the reference ligand kinetic in lieu of the true arterial input function (Innis et al. 2007). All reference region methods assume that a reference region characterized by negligible specific binding can be consistently identified. It may not be possible to meet this criterion in the case of TSPO, due to the ubiquitous constitutive TSPO receptor population in normal brain. Therefore, reference region methods are unable to detect concentrations of TSPO receptors at or below the concentration of the constitutive TSPO receptor population in the defined reference region. Based on the premise that the distribution of activated M/μ in brain is spatially variant and disease-specific, it has been argued that simplified methods of analysis that do not rely on any a priori assumptions regarding the selection of a reference tissue are most applicable across various diseases. Such methods rely on the kinetic segmentation of the dynamic PET image into clusters of brain voxels with indistinguishable kinetic behavior, and the identification of the kinetic cluster with the most rapid tissue clearance rate which is assumed to be most representative of the non-specific binding component (Cagnin et al. 2001) ((Anderson et al. 2007; Turkheimer et al. 2007). Interestingly, [11C]PK11195 BP outcomes determined using arterial- and reference tissue-based analyses were shown to be discordant, and may reflect assumptions made by the model, such as the rapid exchange between free and non-specifically bound tracer, that do not adequately describe the in vivo environment (Kropholler et al. 2006; Kropholler et al. 2005).
Another potential limitation regarding the use of [11C]PK11195 may be related to reports of its capacity to bind reversibly with low micromolar affinity to acute phase alpha-1 acid glycoprotein (AGP) (Lockhart et al. 2003), which is an immunomodulatory protein synthesized by the liver and present in human plasma at a concentration as high as 1 mg/ml (Gunnarsson et al. 2007). In general, plasma protein binding limits the free concentration of drug (or radiotracer) available for Blood-brain barrier (BBB) passage. While a significant component of plasma protein binding is common to most, if not all, compounds that are sufficiently lipophilic for diffusion across the BBB, it does not usually represent a problem for PET studies as the models employed for the analysis of PET neuroreceptor binding phenomenon account for variations in the delivery of radioligand to the brain in order to provide a measure of specifically bound ligand that is independent of delivery. It has been suggested that, in diseases such as MS that are characterized by increased BBB permeability, AGP has the potential to invade and accumulate in brain parenchyma. Despite the relatively low affinity of PK11195 (~ 1 uM) for AGP compared to TSPO (~ 10 nM), the potentially enormous Bmax of abundant AGP could represent a situation where a low affinity, high capacity interaction may compete effectively for PK11195 as has been demonstrated by Katzenellenbogen et al. for estradiol binding to human serum albumin (Katzenellenbogen et al. 1982). Extrahepatic expression of AGP has been reported in a range of tissues and cells, including brain, human monocytes, and activated polymorhonuclear leukocytes (Ceciliani and Pocacqua 2007). Further study of the interaction between PK11195 and AGP in CNS disease is warranted.
Challenges in designing newer ligands to image M/μ
In the process of radiopharmaceutical design, it is often advantageous to characterize the binding properties of a candidate radioligand using classical in vitro binding experiments to assess binding parameters such as the free receptor concentration (Bmax) and the ligand dissociation constant (KD). Under tracer conditions where the concentration of bound ligand can be negated, the ratio Bmax/KD provides a useful although idealized index for predicting the maximal bound/free (B/F) ratio in vivo. Often, the in vitro affinity of a candidate ligand is emphasized as a predictor of high in vivo specific binding. Consideration of the affinity alone can be misleading, as receptor binding phenomenon are generally bimolecular reactions that depend on the concentrations of both the ligand and receptor. Therefore, it is necessary to consider both the affinity of the ligand and the maximal concentration of the receptor to assess the potential binding capacity of a ligand-receptor system. For this reason, the ratio Bmax/KD is the preferred index of a putative radiotracer’s capacity to bind specifically in vivo under ideal conditions. In reality, factors such as non-specific binding, radioligand metabolism, delivery, etc. conspire to lower this ratio in vivo. As a consequence of these complicating factors, it is not possible to tightly define a range of in vitro ligand affinities that is predictive of high in vivo radioligand specific binding. However, some general rules are observed that suggest a ligand with an affinity higher than 10 nM is necessary to be useful for targeting proteins in vivo using PET, as the dissociation rate of less potent ligands may be too rapid to delineate specific from non-specific binding over the time periods needed to conduct a PET scan (typically 60–120 min post-injection). On the other hand, the off-rate of extremely high affinity ligands (< 10 pM) may be too slow to reach equilibrium in a convenient time frame. Eckleman and colleagues (2006) suggest a range of 10 pM to 10 nM as a target in vitro affinity range for screening candidate ligands as potential PET imaging agents, although it must be emphasized that factors affecting the in vivo specific to non-specific binding ratio can only be assessed in vivo. Other properties of the candidate ligand, such as the lipophilicity, can be assessed in vitro and may be useful as a screening tool for predicting brain uptake. For brain imaging, a relatively high lipophilicity is required for free diffusion of the radiotracer across the blood-brain barrier (BBB). Simple laboratory measures of the lipophilicity of a molecule, such as the octanol-water partition coefficient (log P), may be useful predictors of BBB passage. Based on brain uptake studies of radiolabeled compounds, brain uptake peaks around a log P of 2.5 and falls off with higher lipophilicity (Mathis et al. 2003; Waterhouse 2003). Very lipophilic compounds introduce another concern, in that they tend increasingly to bind to other proteins in plasma and tissue non-specifically and reduce the free ligand concentration available for BBB passage and specific binding to the target receptor. It is important to emphasize that the relationship between lipophilicity and brain uptake is not this simple, as other factors can influence the free radioligand concentration in vivo and thus affect overall brain uptake (e.g., the tissue and plasma free fraction). Like the in vitro affinity, lipophilicity is a parameter that is most useful for screening out radioligand candidates that are likely too polar to achieve significant BBB penetration or too lipophilic to result in an acceptable free ligand concentration. Beyond analysis of the theoretical strengths and weaknesses of particular ligands, some of the compounds have been assessed with in vivo PET in animal models and human diseases. Moving from in vitro studies to in vivo investigations introduces a number of uncontrolled variables. A note of caution needs to be interjected regarding the interpretation of these studies. Each biological model is a complex set of interactions between all cell elements in the nervous system along with complex alterations in systemic physiology (e.g. ligand metabolism) which need due consideration while interpreting the data.
PET imaging with novel TSPO ligands
Due to the low in vivo specific binding of [11C]PK11195, recent efforts have focused on identifying novel compounds that selectively bind to TSPO with high affinity in an effort to improve the delineation of in vivo specific binding. In this regard, the field continues to realize considerable success. Many new classes of compounds have been identified that have members that bind TSPO specifically with low nanomolar or sub-nanomolar affinity. A detailed discussion of the relative merits and drawbacks of these ligands is beyond the scope of this article and has been reviewed by others (Doorduin et al. 2008; Owen and Matthews 2011). It would be expected that higher affinity ligands such as these would yield improvements in the delineation of in vivo specific binding. Indeed, this seems to be the case as the in vivo binding of many of these radioligands has shown to be displaceable in vivo by a pharmacological dose of a competing ligand, typically 5 mg/kg of PK11195, in the non-human primate brain. Up to 80% of the radioactivity in brain could be displaced by competition with PK11195, providing a very convincing demonstration of in vivo specific binding to constitutive TSPO (Chauveau et al. 2008). Two agents among these classes, DAA1106 and PBR28, have been shown to have ~10-fold higher affinity (Ki=2.8±0.3 nM and 3.4±0.2 nM, respectively) compared to PK11195 (Ki=28.3±4 nM) (Owen et al., 2010a, b). These agents have been labeled with carbon-11 and characterized in preclinical and human studies. Despite the improved pharmacological properties of these agents, the demonstration of increased in vivo sensitivity to TSPO in disease remains questionable. The lone report of [11C]PBR28 in a clinical population shows a modest but statistically significant (p=0.019) increase in the relative retention of the radioligand in white matter vs grey matter between MS patients and controls (~0.7 vs ~0.6) (Oh et al. 2010). Increases of similar magnitude and significance have been reported in previous [11C]PK11195 studies in MS (Banati et al., 2000; Debruyne et al. 2003; Vas et al., 2008).
Our laboratory has characterized DAA1106, a TSPO ligand with more favorable pharmacological characteristics compared to PK11195. [3H]DAA1106 labeled activated M/μ exhibit higher binding affinity compared to [3H]PK11195 in animal models and human tissues from various neurological disorders (Ji et al. 2008; Maeda et al. 2011; Venneti et al. 2007a; Venneti et al. 2007b; Venneti et al. 2008c). [18F]-FEDAA1106 showed higher brain retention compared to controls in animal models of Alzheimer’s disease and traumatic brain injury (Maeda et al. 2007b; Yu et al. 2010). Ex-vivo, but not in vivo PET [11C]DAA1106 retention was higher in animal models of herpes encephalitis, which the authors attribute to limited spatial resolution of the PET scanner (Doorduin et al. 2010). In vivo imaging with [11C]DAA1106 in human subjects to date has been performed in Alzheimer’s disease (Yasuno et al. 2008), schizophrenia (Takano et al. 2010) and patients with frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) mutations (Miyoshi et al. 2010). These data are interesting but require further evaluation, for example in vivo data from 10 human subjects with mild to moderate Alzheimer’s show [11C]DAA1106 retention in all measured regions such as the dorsal and medial prefrontal cortex, lateral temporal cortex, parietal cortex, occipital cortex, anterior cingulate cortex, including some regions not typically associated with Alzheimer’s disease pathology such the striatum, and cerebellum (Yasuno et al. 2008).
While the authors consider various possibilities, it is conceivable that the higher binding affinity of DAA1106 compared with PK11195 may compromise key PET characteristics. On similar lines, no significant differences in [11C]DAA1106 retention were noted in the brains of 14 patients each with schizophrenia and those of normal control subjects, but [11C]DAA1106 binding in patients with schizophrenia correlated with positive symptoms, disease duration and age (Takano et al. 2010).
Studies reporting significant differences in [11C]PK11195 binding between AD subjects and controls have also yielded curious findings, showing that elevated [11C]PK11195 retention were elevated in regions not generally associated with amyloid pathology (e.g. cerebellum, thalamus) (Tomasi et al., 2008). Okello et al. (2009) demonstrated significantly increased [11C]PK11195 binding in frontal cortex and anterior and posterior cingulate gyri in AD and MCI subjects showing evidence of cerebral amyloid load as assessed by [11C]PiB. Curiously, the level of [11C]PK11195 binding did not correlate with amyloid load, and in some subjects was elevated in the absence of demonstrable Aβ burden.
Challenges in translating tissue based studies to in vivo PET imaging
Despite a large body of literature demonstrating increased expression of TSPO on activated M/u in response to pathological insult to the brain, more than two decades of aggressive development of TSPO-specific radioligands for PET imaging have yielded mixed results. Based on saturation and/or competition binding experiments in tissues, several members of these new classes bind to TSPO in vitro with low nanomolar or subnanomolar affinity. From these same experiments, it also appears that the concentration of constitutive TSPO in the brain is quite high and increased several-fold in disease states (Banati et al. 2000; Venneti et al. 2008b). Saturation binding experiments using [3H]PK11195 in normal human brain cortex tissue reveal that TSPO is present in concentrations of 500–1000 fmol/mg protein (50–100 nM assuming 10% protein per gram of brain tissue), with a KD of ~ 10 nM (Kurumaji et al. 1997; Rao and Butterworth 1997; Venneti et al. 2008b). As in vitro Bmax/KD values are useful predictors of the in vivo ligand bound/free ratio, one can easily calculate this ratio to be on the order of 5–10 with the caveat that mitigating factors such as non-specific binding, ligand metabolism, free fraction, etc., reduce the in vivo ratio of specific:non-specific binding considerably. Eckelman and colleagues (Eckelman et al. 2006) point out the disparities between in vitro Bmax/KD ratio and the observed in vivo specific to non-specific distribution volume ratio (DVR) for a high affinity (0.03 nM) dopamine D2 receptor antagonist (fallypride) under equilibrium conditions. These studies demonstrate that, for fallypride, maximal in vitro Bmax/KD ratios of 900 are found in the putamen where D2 receptor concentrations are highest (~ 27 nM). In vivo studies using [18F]fallypride reveal that, in these same regions, the DVR is reduced by a factor of ~40 to 22. In other brain regions where D2 receptors exist, albeit in lower concentration, a similar phenomenon is observed. For example, in the amygdala, where D2 receptors are present in relatively low concentrations (~ 1 nM), an in vitro Bmax/KD ratio of 30 translates to an in vivo DVR of only 2. Thus, the observed in vitro Bmax/KD ratio for PK11195 of 5–10 would seem to predict a very weak in vivo specific to non-specific binding ratio. This prediction seems to be in agreement with the vast majority of published human studies using [11C]PK11195 to assess CNS inflammation in disease, which in general show modest increases in in vivo signal that could not be positively attributed to increased M/μ activation.
There are several possible explanations for the lack of improved in vivo specific signal. One is that the in vitro binding experiments grossly overestimate the concentration of TSPO receptors available to the radioligand in the intact cell. As these receptors are located intracellularly on mitochondrial membranes, it may be the case that only those proximal to the cell surface can be labeled in vivo with TSPO radioligands. Indeed, the binding of other PET radioligands to neuroreceptor populations (notably the dopamine D2 system) are inhibited by receptor internalization following the administration of a pharmacological stimulus such as amphetamine (Ginovart et al. 2004; Scott et al. 2007) (Skinbjerg et al. 2010). However, in animal studies the majority of the radioactivity in brain after the injection of these high affinity TSPO radioligands can be displaced in vivo by a pharmacological dose of a competing ligand such as PK11195, thus not supporting TSPO receptor internalization. Other explanations for this discrepancy can be postulated, such as mixed cellular sources of TSPO expression in the CNS, differences between the in vitro and in vivo affinity of these ligands for TSPO, although this would be challenging to demonstrate. As high-affinity TSPO radioligands to date show at best modest increases in sensitivity to detect TSPO expression in vivo using PET, it seems unlikely that further pharmacological manipulation of these novel classes will yield dramatic improvements in the delineation of specific radiortracer binding in vivo. We suggest the possibility that the failure of ongoing PET radioligand development efforts to realize improved in vivo specific binding to TSPO is perhaps more attributable to the behavior of the target and not the tools developed to probe it.
TSPO polymorphisms and effects on PET imaging
Initial human studies using the putative TSPO radioligand [11C]PBR-28 revealed that approximately 10% of subjects studied showed no apparent specific binding of the radioligand to TSPO binding sites in brain or peripheral organs (Fujita et al. 2008; Kreisl et al. 2010). Binding assays from leukocyte membranes collected from groups of binders and non-binders identified by [11C]PBR-28 whole body PET scans revealed that [11C]PBR-28 exhibited greater than 10-fold lower affinity for TSPO in non-binders compared to binders, suggesting a possible polymorphism in the TSPO binding site (Kreisl et al. 2010). Autoradiographic studies of human brain tissues conducted using [3H]PBR-28 extended these observations by identifying classes of high affinity (Ki ~ 3.4±0.5 nM) low-affinity binders (Ki ~ 188±15.6 nM), as well as a class of mixed-affinity binders (approximately 40% of subjects) for whom a two-site binding model was required. The affinities (Ki) of the two binding sites were determined to be 4.0±2.4 nM (high affinity site) and 313±77 nM (low affinity site), closely corresponding to the high and low affinity binding sites characterized by a single site model in the binder and non-binder groups (Owen et al. 2010a; Owen et al. 2010b; Owen and Matthews 2011). A genetic association study of known TSPO polymorphisms reported by Owen and colleagues revealed that the rs6971 polymorphism showed complete concordance with the PBR28 binding phenotype (low-affinity, high-affinity, mixed affinity) (Owen et al. 2010a; Owen et al. 2010b; Owen and Matthews 2011). This polymorphism occurs at position 147 within exon 4 of the TSPO gene on chromosome 22. The identified polymorphism is an amino acid substitution of alanine for threonine (Ala147Thr) in the fifth transmembrane domain of TSPO, having a co-dominant pattern of inheritance. The major allele is alanine at position 147 (Ala147), while the minor allele is the threonine substitution (Thr147). Therefore, high affinity binders are homozygous for Ala147, low affinity binders are homozygous for Thr147, while mixed-affinity binders are heterozygous (Ala147/Thr147). Owen and colleagues also reports that there exists a substantial disparity in the prevalence of the minor allele between ethnic groups. The greatest prevalence of the minor allele (~30%) is in Caucasians, while Asian populations such as the Han Chinese and Japanese have a comparatively low prevalence of the Thr 147 substitution (2–4%). In another recent work, Owen and colleagues report that the sensitivity of binding to the rs6971 polymorphism is not limited to [11C]PBR-28, but also to other putative TSPO ligands under development as PET TSPO radioligands, which include DAA1106, PBR06, PBR111, XBD173, and DPA713 (Owen et al. 2010a; Owen and Matthews 2011). The differences in the affinity of these ligands between high-affinity binders and low affinity binders ranges from a ratio of 55:1 for PBR28 to approximately 4:1 for PBR111, DPA713, and DAA1106. Interestingly, PK11195 binding in brain did not exhibit a sensitivity to the rs6971 polymorphism (Kreisl et al. 2010; Owen et al. 2010a), although differences in [11C]PK11195 binding were observed in peripheral organs such as the heart and lung, where TSPO expression is relatively high compared to brain (Kreisl et al. 2010). A possible explanation for the failure to observe this phenomenon with [11C]PK11195 despite nearly three decades of investigational use of the radioligand in human subjects is the relatively low ratio of specific to non-specific binding of [11C]PK11195 in brain. Future studies using TSPO ligands sensitive to the rs6791 polymorphism will have to control for this confound by identifying potential subjects who are low- and mixed-affinity binders through a priori genetic testing or leukocyte binding assays.
New M/μ molecular targets PET imaging
The extensive use of TSPO as a molecular target in PET imaging has yielded significant interest in PET imaging of neuroinflammation. Further, due to the several limitations of TSPO binding ligands discussed above, much promise lies in identifying newer targets for PET imaging of M/μ. Recent studies take advantage of two aspects of M/μ biology to identify new molecular targets for PET imaging. The type 2 cannabinoid receptor (CB2R) is part of the endocannabinoid system expressed in peripheral immune cells and in M/μ in neurological disorders like AD (Ramirez et al. 2005). 1-bromo-2-[18F]fluoroethane labelled -GW405833 is a ligand that binds CB2R and showed increased uptake in mice expressing CB2R in the striatum (Evens and Bormans 2010; Evens et al. 2011). Another molecular target is cyclooxygenase (COX), which converts arachidonic acid to prostaglandin in the prostaglandin pathway. [11C]ketoprofen methyl ester targets COX-1 and showed increased uptake in rats injected with LPS but not COX-1 deficient rats. Further, temporal changes in [11C]ketoprofen methyl ester uptake paralleled immunohistochemical assessment of M/μ in rat brain tissues (Shukuri et al. 2011). Other non-TSPO binding agents used in imaging neuroinflammation include 2-[18F]-fluoroacetate targeted at the citric acid cycle in glial metabolism and [11C]-L-deprenyl labeling monoamine oxidase B are hypothesized to be enriched in astrocytes (Gulyas et al. 2010; Marik et al. 2009). While these approaches have shown initial promise, more extensive characterization of the cellular signal source is required before human studies can be conducted.
MR imaging of M/μ
Beyond photon emitters, cells can be labeled with a wider variety of molecules that can be detected with other modalities. Of particular relevance to cell tracking in vivo, magnetic resonance imaging (MRI) has been extensively employed in biological studies (Muja and Bulte 2009). Avoiding potentially harmful radiation, MRI is based on the principle of detecting proton relaxation times in a strong magnetic field disturbed by a radio-frequency pulse. As biological structures are mostly composed of water, the MRI signal principally uses the water proton environment to define anatomical structure. However paramagnetic and super paramagnetic compounds can be used to label cells and generate MR contrast. Coupled with novel cell-labeling techniques MRI has proven very useful in safely and continuously assessing cell trafficking at high resolution in living organisms.
For MRI most macrophage labeling techniques take advantage of their strong propensity to phagocytose particles. At the outset it is important to recognize that phagocytosis is not exclusively a property of macrophages. It occurs to a more limited extent in a wide variety of cells including neurons. Because of this shared property many MRI macrophage tracing experiments utilize ex vivo labeling followed by re-introduction of the pre-labeled cells to the vasculature. This imposes several critical limitations: First; label introduced into a cell ex vivo does not remain limited to that cell after re-introduction into the animal. Labels can leach from living cells or the cell can die and release label into the circulation or host phagocytic cells. In the case of studying M/μ this is a double-edged sword. The same label introduced into a peripheral monocytic element has the potential to be phagocytosed and appear in CNS resident microglia. Second, while it is fortunate to be able to take advantage of macrophages’ propensity to phagocytose particles, it is important to recognize that in studying cell trafficking, it is the macrophage precursor, the monocyte, that traffics through the circulatory system. The monocyte’s capacity to phagocytose or pinocytose particles is much more limited than the macrophage. Indeed it is a very fine balance to label a monocyte without causing it to differentiate into a macrophage that would not be expect to traffic through the vascular system. Finally other trafficking immune cells such as T-cells and polymorphonuclear cells can also take up label injected into the host.
MR contrast can be generated in two directions, positive and negative. Iron oxide in its Fe2O3 and Fe3O4 is the most commonly used negative contrast agent (i.e. creating decreased signal). Iron oxides can be coated and stabilized with a variety of larger molecules designed to augment cellular uptake. Several of these contrast agents are available for both animal and human use. Variation in structure and size permits tailoring their use to labeling different cellular elements. Labeling with large polystyrene microsphere containing iron oxides has permitted single cell detection in high field ex vivo MRI. This has raised the possibility of using such labeled cells to track individual cells in vivo (Wu et al. 2006). When individual cells are loaded with ~10pg of iron oxide, current commonly used experimental MR devices permit detection of 100–500 cells (Muja and Bulte 2009). Loading with even large particles can load the cell with ~100pg of iron oxide and potentially permit imaging individual cells. Unfortunately such high concentrations can severely compromise not only cell physiology but cell viability. Whether advances in magnetic field strength or instrumentation eventually break through this barrier and allow sensitive MR imaging of individual cells remains to be seen.
Paramagnetic metals with unpaired electrons provide the most useful positive MR contrast agents. The most widely used of these, gadolinium, effects T1 relaxation but at a weaker level than super paramagnetic iron oxide nanoparticles. MR is not limited to detection of hydrogen nuclei of water. Protons of other naturally occurring isotopes (e.g. 19F and 13C) can be assessed in vivo using spectroscopy. Nanoparticles containing tracers with multiple fluorine nuclei have achieved label efficiencies such that as few as 200 cells can be detected in a MR voxel (Flogel et al. 2008). In vivo studies of neuroinflammation utilizing 19F are very new and the data limited.
Using MRI to assess M/μ in vivo
The different labeling techniques have been used to monitor neuroinflammation in general and macrophage presence in particular in a variety of diseases. Both ex vivo and in vivo labeling have been employed in MR studies of the human disease and animals models of stroke and demyelination (Jander et al. 2007; Stoll and Bendszus 2010). Ex vivo labeling most frequently consists of removing peripheral blood, exposing mononuclear cells to iron oxide nanoparticles, rinsing away excess label and re-introducing the now labeled monocytes into the vasculature. Alternatively iron oxide nanoparticles can be directly injected into the blood stream for in vivo labeling. Moderate size particles (50–200 nm in diameter) are avidly taken up by the host reticuloendothelial system (mostly liver, spleen and lymph node histiocytes) with a small portion taken up by circulating mononuclear elements. To circumvent this dilution of label, smaller iron oxide nanoparticles (ultra small super paramagnetic USPIO, 35 nm in diameter) can be used. A larger fraction of the UPSIO nanoparticles are taken up by circulating cells and thus available for trafficking studies. From the CNS perspective an important caveat to the in vivo labeling is that USPIO particles are excluded from the CNS by the blood brain barrier (BBB). However in many experimental models the BBB is variably or not at all intact leading to capricious availability of the label to phagocytic cells in the CNS. Interpretation of experiments based upon in vivo labeling is thus substantially compromised.
Recently creative experimentation has been designed to address this deficiency. Sequentially measuring deficiencies in the BBB using blood contrast agents like gadolinium coupled with in vivo labeling of phagocytic cells has been attempted in several experimental systems including stroke and EAE (reviewed in (Stoll and Bendszus 2010)). In these systems it has been possible to distinguish areas of BBB break down showing gadolinium contrast enhancement, from areas without BBB that contain USPIO enhancement secondary to trafficking of phagocytic mononuclear elements.
Summary
Non-invasive imaging of M/μ serves as an important tool to detect neuroinflammation in vivo. Designing therapies for neurological diseases must include a means to monitor their efficacy. As newer therapies emerge targeting neuroinflammation in various neurological diseases, the need for a means to monitor the inflammatory pathologic process and assessment of therapeutic efficacies of anti-inflammatory and other potentially neuroprotective drugs is paramount. We have reviewed techniques to monitor M/μ in vivo. The techniques of MRI and PET have complimentary sensitivity and spatial resolution limits. Compared to PET, MRI has excellent spatial resolution (50 microns compared to 1–3 mm) but with respect to sensitivity, PET exceeds MRI by a thousand fold (less than 10−8 mol/L for PET). PET imaging of M/μ has come a long way since the first description of the PK11195 as a TSPO ligand. While PK11195 has been extensively used, many limitations of the ligand have emerged. Recent years have yielded new TSPO ligands that show promise but require further evaluation. There are many challenges before TSPO ligands could be routinely used in the clinic. These include finding the optimal ligand that shows required sensitivity and specificity and understanding how TSPO polymorphisms can alter uptake of these ligands. From a biological perspective, the function of TSPO in M/μ in neurological disease remains to be elucidated. Further, newer non-TSPO ligands that target other aspects of M/μ biology (e.g. the innate immune response) are emerging. Because therapies for many neurological diseases require modulating neuroinflammation, there is great need to continue to develop techniques to non-invasively image M/μ in vivo to monitor progression and severity of disease, and help determine therapeutic efficacies.
References
- Anderson AN, Pavese N, Edison P, Tai YF, Hammers A, Gerhard A, Brooks DJ, Turkheimer FE. A systematic comparison of kinetic modelling methods generating parametric maps for [(11)C]-(R)-PK11195. Neuroimage. 2007;36(1):28–37. doi: 10.1016/j.neuroimage.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Awad M, Gavish M. Peripheral-type benzodiazepine receptors in human cerebral cortex, kidney, and colon. Life Sci. 1991;49(16):1155–61. doi: 10.1016/0024-3205(91)90562-p. [DOI] [PubMed] [Google Scholar]
- Banati RB. Visualising microglial activation in vivo. Glia. 2002;40(2):206–17. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine’)--binding sites in the CNS indicate early and discrete brain lesions: microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol. 1997;26(2):77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, Price G, Wegner F, Giovannoni G, Miller DH, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123 (Pt 11):2321–37. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Braestrup C, Albrechtsen R, Squires RF. High densities of benzodiazepine receptors in human cortical areas. Nature. 1977;269(5630):702–4. doi: 10.1038/269702a0. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–7. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Kassiou M, Meikle SR, Banati RB. In vivo evidence for microglial activation in neurodegenerative dementia. Acta Neurol Scand Suppl. 2006;185:107–14. doi: 10.1111/j.1600-0404.2006.00694.x. [DOI] [PubMed] [Google Scholar]
- Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40(6):475–86. doi: 10.1016/s0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Kroemer G. Mitochondrial apoptosis and the peripheral benzodiazepine receptor: a novel target for viral and pharmacological manipulation. J Exp Med. 2002;196(9):1121–5. doi: 10.1084/jem.20021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceciliani F, Pocacqua V. The acute phase protein alpha1-acid glycoprotein: a model for altered glycosylation during diseases. Curr Protein Pept Sci. 2007;8(1):91–108. doi: 10.2174/138920307779941497. [DOI] [PubMed] [Google Scholar]
- Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia: new concepts. Brain Res Rev. 2007;53(2):344–54. doi: 10.1016/j.brainresrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149(8):2736–41. [PubMed] [Google Scholar]
- Chao CC, Hu S, Peterson PK. Modulation of human microglial cell superoxide production by cytokines. J Leukoc Biol. 1995a;58(1):65–70. doi: 10.1002/jlb.58.1.65. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Sheng WS, Peterson PK. Tumor necrosis factor-alpha production by human fetal microglial cells: regulation by other cytokines. Dev Neurosci. 1995b;17(2):97–105. doi: 10.1159/000111278. [DOI] [PubMed] [Google Scholar]
- Charbonneau P, Syrota A, Crouzel C, Valois JM, Prenant C, Crouzel M. Peripheral-type benzodiazepine receptors in the living heart characterized by positron emission tomography. Circulation. 1986;73(3):476–83. doi: 10.1161/01.cir.73.3.476. [DOI] [PubMed] [Google Scholar]
- Chauveau F, Boutin H, Van Camp N, Dolle F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35(12):2304–19. doi: 10.1007/s00259-008-0908-9. [DOI] [PubMed] [Google Scholar]
- Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain. 2004;127(Pt 6):1379–92. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- Colton CA, Keri JE, Chen WT, Monsky WL. Protease production by cultured microglia: substrate gel analysis and immobilized matrix degradation. J Neurosci Res. 1993;35(3):297–304. doi: 10.1002/jnr.490350309. [DOI] [PubMed] [Google Scholar]
- Cremer JE, Hume SP, Cullen BM, Myers R, Manjil LG, Turton DR, Luthra SK, Bateman DM, Pike VW. The distribution of radioactivity in brains of rats given [N-methyl-11C]PK 11195 in vivo after induction of a cortical ischaemic lesion. Int J Rad Appl Instrum B. 1992;19(2):159–66. doi: 10.1016/0883-2897(92)90003-h. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Del Rio Hortega P. Cytology and Cellular Pathology of the Nervous system. Hoeber; New York: 1932. pp. 481–534. [Google Scholar]
- Doorduin J, de Vries EF, Dierckx RA, Klein HC. PET imaging of the peripheral benzodiazepine receptor: monitoring disease progression and therapy response in neurodegenerative disorders. Curr Pharm Des. 2008;14(31):3297–315. doi: 10.2174/138161208786549443. [DOI] [PubMed] [Google Scholar]
- Doorduin J, Klein HC, de Jong JR, Dierckx RA, de Vries EF. Evaluation of [11C]-DAA1106 for imaging and quantification of neuroinflammation in a rat model of herpes encephalitis. Nucl Med Biol. 2010;37(1):9–15. doi: 10.1016/j.nucmedbio.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Eckelman WC, Kilbourn MR, Mathis CA. Discussion of targeting proteins in vivo: in vitro guidelines. Nucl Med Biol. 2006;33(4):449–51. doi: 10.1016/j.nucmedbio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Evens N, Bormans GM. Non-invasive imaging of the type 2 cannabinoid receptor, focus on positron emission tomography. Curr Top Med Chem. 2010;10(15):1527–43. doi: 10.2174/156802610793176819. [DOI] [PubMed] [Google Scholar]
- Evens N, Vandeputte C, Muccioli GG, Lambert DM, Baekelandt V, Verbruggen AM, Debyser Z, Van Laere K, Bormans GM. Synthesis, in vitro and in vivo evaluation of fluorine-18 labelled FE-GW405833 as a PET tracer for type 2 cannabinoid receptor imaging. Bioorg Med Chem. 2011;19(15):4499–505. doi: 10.1016/j.bmc.2011.06.033. [DOI] [PubMed] [Google Scholar]
- Everett H, Barry M, Sun X, Lee SF, Frantz C, Berthiaume LG, McFadden G, Bleackley RC. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J Exp Med. 2002;196(9):1127–39. doi: 10.1084/jem.20011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett H, McFadden G. Viruses and apoptosis: meddling with mitochondria. Virology. 2001;288(1):1–7. doi: 10.1006/viro.2001.1081. [DOI] [PubMed] [Google Scholar]
- Flogel U, Ding Z, Hardung H, Jander S, Reichmann G, Jacoby C, Schubert R, Schrader J. In vivo monitoring of inflammation after cardiac and cerebral ischemia by fluorine magnetic resonance imaging. Circulation. 2008;118(2):140–8. doi: 10.1161/CIRCULATIONAHA.107.737890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Imaizumi M, Zoghbi SS, Fujimura Y, Farris AG, Suhara T, Hong J, Pike VW, Innis RB. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. Neuroimage. 2008;40(1):43–52. doi: 10.1016/j.neuroimage.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginovart N, Wilson AA, Houle S, Kapur S. Amphetamine pretreatment induces a change in both D2-Receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol Psychiatry. 2004;55(12):1188–94. doi: 10.1016/j.biopsych.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ, Shih LC, Lachman LB. Interleukin 1 of the central nervous system is produced by ameboid microglia. J Exp Med. 1986;164(2):594–604. doi: 10.1084/jem.164.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas B, Pavlova E, Kasa P, Gulya K, Bakota L, Varszegi S, Keller E, Horvath MC, Nag S, Hermecz I, et al. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem Int. 2010;58(1):60–8. doi: 10.1016/j.neuint.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ. Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. Neuroimage. 1997;6(4):279–87. doi: 10.1006/nimg.1997.0303. [DOI] [PubMed] [Google Scholar]
- Gunnarsson P, Levander L, Pahlsson P, Grenegard M. The acute-phase protein alpha 1-acid glycoprotein (AGP) induces rises in cytosolic Ca2+ in neutrophil granulocytes via sialic acid binding immunoglobulin-like lectins (siglecs) FASEB J. 2007;21(14):4059–69. doi: 10.1096/fj.07-8534com. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320 (Pt 2):595–7. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JD, Beyer CF, Malkowitz L, Beer B, Blume AJ. Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Mol Pharmacol. 1989;35(1):157–63. [PubMed] [Google Scholar]
- Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27(9):1533–9. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- Jander S, Schroeter M, Saleh A. Imaging inflammation in acute brain ischemia. Stroke. 2007;38(2 Suppl):642–5. doi: 10.1161/01.STR.0000250048.42916.ad. [DOI] [PubMed] [Google Scholar]
- Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, Zhang MR, Suzuki K, Ando K, Staufenbiel M, et al. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer’s and other CNS pathologies. J Neurosci. 2008;28(47):12255–67. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph-Liauzun E, Delmas P, Shire D, Ferrara P. Topological analysis of the peripheral benzodiazepine receptor in yeast mitochondrial membranes supports a five-transmembrane structure. J Biol Chem. 1998;273(4):2146–52. doi: 10.1074/jbc.273.4.2146. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen JA, McElvany KD, Senderoff SG, Carlson KE, Landvatter SW, Welch MJ. 16 alpha-[77Br]bromo-11 beta-methoxyestradiol-17 beta: a gamma-emitting estrogen imaging agent with high uptake and retention by target organs. J Nucl Med. 1982;23(5):411–9. [PubMed] [Google Scholar]
- Kofler J, Wiley CA. Microglia: key innate immune cells of the brain. Toxicol Pathol. 2011;39(1):103–14. doi: 10.1177/0192623310387619. [DOI] [PubMed] [Google Scholar]
- Kreisl WC, Fujita M, Fujimura Y, Kimura N, Jenko KJ, Kannan P, Hong J, Morse CL, Zoghbi SS, Gladding RL, et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: Implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage. 2010;49(4):2924–32. doi: 10.1016/j.neuroimage.2009.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropholler MA, Boellaard R, Schuitemaker A, Folkersma H, van Berckel BN, Lammertsma AA. Evaluation of reference tissue models for the analysis of [(11)C](R)-PK11195 studies. J Cereb Blood Flow Metab. 2006 doi: 10.1038/sj.jcbfm.9600289. [DOI] [PubMed] [Google Scholar]
- Kropholler MA, Boellaard R, Schuitemaker A, van Berckel BN, Luurtsema G, Windhorst AD, Lammertsma AA. Development of a tracer kinetic plasma input model for (R)-[11C]PK11195 brain studies. J Cereb Blood Flow Metab. 2005;25(7):842–51. doi: 10.1038/sj.jcbfm.9600092. [DOI] [PubMed] [Google Scholar]
- Kuhlmann AC, Guilarte TR. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J Neurochem. 2000;74(4):1694–704. doi: 10.1046/j.1471-4159.2000.0741694.x. [DOI] [PubMed] [Google Scholar]
- Kurumaji A, Wakai T, Toru M. Decreases in peripheral-type benzodiazepine receptors in postmortem brains of chronic schizophrenics. J Neural Transm. 1997;104(11–12):1361–70. doi: 10.1007/BF01294737. [DOI] [PubMed] [Google Scholar]
- Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4(3 Pt 1):153–8. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- Levin CS, Hoffman EJ. Calculation of positron range and its effect on the fundamental limit of positron emission tomography system spatial resolution. Phys Med Biol. 1999;44(3):781–99. doi: 10.1088/0031-9155/44/3/019. [DOI] [PubMed] [Google Scholar]
- Lockhart A, Davis B, Matthews JC, Rahmoune H, Hong G, Gee A, Earnshaw D, Brown J. The peripheral benzodiazepine receptor ligand PK11195 binds with high affinity to the acute phase reactant alpha1-acid glycoprotein: implications for the use of the ligand as a CNS inflammatory marker(1) Nucl Med Biol. 2003;30(2):199–206. doi: 10.1016/s0969-8051(02)00410-9. [DOI] [PubMed] [Google Scholar]
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007a;1157:100–11. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, Okauchi T, Staufenbiel M, Iwata N, Ono M, Saido TC, et al. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci. 2007b;27(41):10957–68. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Zhang MR, Okauchi T, Ji B, Ono M, Hattori S, Kumata K, Iwata N, Saido TC, Trojanowski JQ, et al. In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. J Neurosci. 2011;31(12):4720–30. doi: 10.1523/JNEUROSCI.3076-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marik J, Ogasawara A, Martin-McNulty B, Ross J, Flores JE, Gill HS, Tinianow JN, Vanderbilt AN, Nishimura M, Peale F, et al. PET of glial metabolism using 2-18F-fluoroacetate. J Nucl Med. 2009;50(6):982–90. doi: 10.2967/jnumed.108.057356. [DOI] [PubMed] [Google Scholar]
- Martin A, Boisgard R, Theze B, Van Camp N, Kuhnast B, Damont A, Kassiou M, Dolle F, Tavitian B. Evaluation of the PBR/TSPO radioligand [(18)F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30(1):230–41. doi: 10.1038/jcbfm.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46(13):2740–54. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A. 1992;89(8):3170–4. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi M, Shinotoh H, Wszolek ZK, Strongosky AJ, Shimada H, Arakawa R, Higuchi M, Ikoma Y, Yasuno F, Fukushi K, et al. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: a PET and MRI study. Parkinsonism Relat Disord. 2010;16(6):404–8. doi: 10.1016/j.parkreldis.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Muja N, Bulte JW. Magnetic resonance imaging of cells in experimental disease models. Prog Nucl Magn Reson Spectrosc. 2009;55(1):61–77. doi: 10.1016/j.pnmrs.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers R, Manjil LG, Cullen BM, Price GW, Frackowiak RS, Cremer JE. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J Cereb Blood Flow Metab. 1991;11(2):314–22. doi: 10.1038/jcbfm.1991.64. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Oh U, Fujita M, Ikonomidou VN, Evangelou IE, Matsuura E, Harberts E, Fujimura Y, Richert ND, Ohayon J, Pike VW, et al. Translocator protein PET imaging for glial activation in multiple sclerosis. J Neuroimmune Pharmacol. 2010;6(3):354–61. doi: 10.1007/s11481-010-9243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, Innis RB, Pike VW, Reynolds R, Matthews PM, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2010a;52(1):24–32. doi: 10.2967/jnumed.110.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Howell OW, Tang SP, Wells LA, Bennacef I, Bergstrom M, Gunn RN, Rabiner EA, Wilkins MR, Reynolds R, et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab. 2010b;30(9):1608–18. doi: 10.1038/jcbfm.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Matthews PM. Imaging brain microglial activation using positron emission tomography and translocator protein-specific radioligands. Int Rev Neurobiol. 2011;101:19–39. doi: 10.1016/B978-0-12-387718-5.00002-X. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Amri H, Li H, Boujrad N, Vidic B, Garnier M. Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem. 1997;272(51):32129–35. doi: 10.1074/jbc.272.51.32129. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006a;27(8):402–9. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Lecanu L, Brown RC, Han Z, Yao ZX. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience. 2006b;138(3):749–56. doi: 10.1016/j.neuroscience.2005.05.063. [DOI] [PubMed] [Google Scholar]
- Pedersen MD, Minuzzi L, Wirenfeldt M, Meldgaard M, Slidsborg C, Cumming P, Finsen B. Up-regulation of PK11195 binding in areas of axonal degeneration coincides with early microglial activation in mouse brain. Eur J Neurosci. 2006;24(4):991–1000. doi: 10.1111/j.1460-9568.2006.04975.x. [DOI] [PubMed] [Google Scholar]
- Petit-Taboue MC, Baron JC, Barre L, Travere JM, Speckel D, Camsonne R, MacKenzie ET. Brain kinetics and specific binding of [11C]PK 11195 to omega 3 sites in baboons: positron emission tomography study. Eur J Pharmacol. 1991;200(2–3):347–51. doi: 10.1016/0014-2999(91)90594-g. [DOI] [PubMed] [Google Scholar]
- Price GW, Ahier RG, Hume SP, Myers R, Manjil L, Cremer JE, Luthra SK, Pascali C, Pike V, Frackowiak RS. In vivo binding to peripheral benzodiazepine binding sites in lesioned rat brain: comparison between [3H]PK11195 and [18F]PK14105 as markers for neuronal damage. J Neurochem. 1990;55(1):175–85. doi: 10.1111/j.1471-4159.1990.tb08836.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000;161(1):102–14. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25(8):1904–13. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rao VL, Butterworth RF. Characterization of binding sites for the omega3 receptor ligands [3H]PK11195 and [3H]RO5-4864 in human brain. Eur J Pharmacol. 1997;340(1):89–99. doi: 10.1016/s0014-2999(97)01395-2. [DOI] [PubMed] [Google Scholar]
- Righi M, Mori L, De Libero G, Sironi M, Biondi A, Mantovani A, Donini SD, Ricciardi-Castagnoli P. Monokine production by microglial cell clones. Eur J Immunol. 1989;19(8):1443–8. doi: 10.1002/eji.1830190815. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9(12):971–88. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Rammes G, Eser D, Baghai TC, Schule C, Nothdurfter C, Troxler T, Gentsch C, Kalkman HO, Chaperon F, et al. Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science. 2009;325(5939):490–3. doi: 10.1126/science.1175055. [DOI] [PubMed] [Google Scholar]
- Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH, Grant I. Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev. 2007;17(3):275–97. doi: 10.1007/s11065-007-9031-0. [DOI] [PubMed] [Google Scholar]
- Shukuri M, Takashima-Hirano M, Tokuda K, Takashima T, Matsumura K, Inoue O, Doi H, Suzuki M, Watanabe Y, Onoe H. In vivo expression of cyclooxygenase-1 in activated microglia and macrophages during neuroinflammation visualized by PET with 11C-ketoprofen methyl ester. J Nucl Med. 2011;52(7):1094–101. doi: 10.2967/jnumed.110.084046. [DOI] [PubMed] [Google Scholar]
- Skinbjerg M, Liow JS, Seneca N, Hong J, Lu S, Thorsell A, Heilig M, Pike VW, Halldin C, Sibley DR, et al. D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: a PET study in a receptor internalization-deficient mouse model. Neuroimage. 2010;50(4):1402–7. doi: 10.1016/j.neuroimage.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulet D, Rivest S. Bone-marrow-derived microglia: myth or reality? Curr Opin Pharmacol. 2008;8(4):508–18. doi: 10.1016/j.coph.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15(7 Pt 2):5263–74. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson FA. The GABAA receptors. Biochem J. 1995;310 (Pt 1):1–9. doi: 10.1042/bj3100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoebner PE, Carayon P, Casellas P, Portier M, Lavabre-Bertrand T, Cuq P, Cano JP, Meynadier J, Meunier L. Transient protection by peripheral benzodiazepine receptors during the early events of ultraviolet light-induced apoptosis. Cell Death Differ. 2001;8(7):747–53. doi: 10.1038/sj.cdd.4400861. [DOI] [PubMed] [Google Scholar]
- Stoll G, Bendszus M. New approaches to neuroimaging of central nervous system inflammation. Curr Opin Neurol. 2010;23(3):282–6. doi: 10.1097/WCO.0b013e328337f4b5. [DOI] [PubMed] [Google Scholar]
- Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, Okubo Y, Suhara T. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol. 2010;13(7):943–50. doi: 10.1017/S1461145710000313. [DOI] [PubMed] [Google Scholar]
- Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A, Gerhard A, Hinz R, Tai YF, Brooks DJ. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med. 2007;48(1):158–67. [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, Apte UM, Wiley CA. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol Aging. 2008a doi: 10.1016/j.neurobiolaging.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wang G, Slagel SL, Mason NS, Mathis CA, Fischer ML, Larsen NJ, Mortimer AD, Hastings TG, et al. A comparison of the high-affinity peripheral benzodiazepine receptor ligands DAA1106 and (R)-PK11195 in rat models of neuroinflammation: implications for PET imaging of microglial activation. J Neurochem. 2007a;102(6):2118–31. doi: 10.1111/j.1471-4159.2007.04690.x. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: From pathology to imaging. Prog Neurobiol. 2006 doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Wagner AK, Wang G, Slagel SL, Chen X, Lopresti BJ, Mathis CA, Wiley CA. The high affinity peripheral benzodiazepine receptor ligand DAA1106 binds specifically to microglia in a rat model of traumatic brain injury: implications for PET imaging. Exp Neurol. 2007b;207(1):118–27. doi: 10.1016/j.expneurol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Wang G, Nguyen J, Wiley CA. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol. 2008b;67(10):1001–10. doi: 10.1097/NEN.0b013e318188b204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Wang G, Wiley CA. The high affinity peripheral benzodiazepine receptor ligand DAA1106 binds to activated and infected brain macrophages in areas of synaptic degeneration: implications for PET imaging of neuroinflammation in lentiviral encephalitis. Neurobiol Dis. 2008c;29(2):232–41. doi: 10.1016/j.nbd.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T, Antel JP. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50(2):345–53. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5(6):376–89. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Wu YL, Ye Q, Foley LM, Hitchens TK, Sato K, Williams JB, Ho C. In situ labeling of immune cells with iron oxide particles: an approach to detect organ rejection by cellular MRI. Proc Natl Acad Sci U S A. 2006;103(6):1852–7. doi: 10.1073/pnas.0507198103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuno F, Ota M, Kosaka J, Ito H, Higuchi M, Doronbekov TK, Nozaki S, Fujimura Y, Koeda M, Asada T, et al. Increased Binding of Peripheral Benzodiazepine Receptor in Alzheimer’s Disease Measured by Positron Emission Tomography with [(11)C]DAA1106. Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Yu I, Inaji M, Maeda J, Okauchi T, Nariai T, Ohno K, Higuchi M, Suhara T. Glial cell-mediated deterioration and repair of the nervous system after traumatic brain injury in a rat model as assessed by positron emission tomography. J Neurotrauma. 2010;27(8):1463–75. doi: 10.1089/neu.2009.1196. [DOI] [PubMed] [Google Scholar]