Transcription Factor Foxo3a Prevents Apoptosis by Regulating Calcium through the Apoptosis Repressor with Caspase Recruitment Domain (original) (raw)
Background: Foxo3a plays a pivotal role in regulating apoptosis; however, its role in controlling cardiac apoptosis remains to be fully elucidated.
Results: Foxo3a prevents cardiac apoptosis by regulating calcium through ARC.
Conclusion: Foxo3a and ARC constitute an anti-apoptotic pathway that regulates calcium homeostasis in the heart.
Significance: Our results provide important information for exploring the beneficial effects of this pathway on apoptosis-related cardiac diseases.
Keywords: Apoptosis, Calcium Signaling, Cardiovascular, Cell Biology, Transcription Regulation
Abstract
Apoptosis can occur in the myocardium under a variety of pathological conditions, including myocardial infarction and heart failure. The forkhead family of transcription factor Foxo3a plays a pivotal role in apoptosis; however, its role in regulating cardiac apoptosis remains to be fully elucidated. We showed that enforced expression of Foxo3a inhibits cardiomyocyte apoptosis, whereas knockdown of endogenous Foxo3a sensitizes cardiomyocytes to undergo apoptosis. The apoptosis repressor with caspase recruitment domain (ARC) is a potent anti-apoptotic protein. Here, we demonstrate that it attenuates the release of calcium from the sarcoplasmic reticulum and inhibits calcium elevations in the cytoplasm and mitochondria provoked by oxidative stress in cardiomyocytes. Furthermore, Foxo3a is shown to maintain cytoplasmic and mitochondrial calcium homeostasis through ARC. We observed that Foxo3a knock-out mice exhibited enlarged myocardial infarction sizes upon ischemia/reperfusion, and ARC transgenic mice demonstrated reduced myocardial infarction and balanced calcium levels in mitochondria and sarcoplasmic reticulum. Moreover, we showed that Foxo3a activates ARC expression by directly binding to its promoter. This study reveals that Foxo3a maintains calcium homeostasis and inhibits cardiac apoptosis through trans-activation of the ARC promoter. These findings provided novel evidence that Foxo3a and ARC constitute an anti-apoptotic pathway that regulates calcium homeostasis in the heart.
Introduction
Apoptosis can occur in the myocardium under a variety of pathological conditions (1, 2). For example, myocyte apoptosis is increased in myocardium from patients with myocardial infarction and heart failure and from experimental models of hypertrophy and heart failure. Thus, it necessitates the identification of the molecules that are able to regulate cardiac apoptosis.
The forkhead family of transcription factors participates in regulating diverse cellular functions such as apoptosis, differentiation, metabolism, proliferation, and survival (3). Foxo3a can either induce or prevent apoptosis. For example, its activation in hematopoietic (4, 5) and neuronal cells (6) results in the induction of apoptosis. In contrast, Foxo3a is necessary for the maintenance of neutrophil survival. Such a differential cellular response of Foxo3a activation can be related to the cell type-specific regulation of pro- and anti-apoptotic genes. Foxo3a is expressed in the heart and skeletal muscle (7–9). We and others have shown that Foxo3a can negatively regulate cardiac hypertrophy (8, 10, 11). It is not yet clear whether Foxo3a participates in the regulation of cardiac apoptosis.
Mitochondrial Ca2+ homeostasis plays a critical role in maintaining cell survival. Its disruption by Ca2+ overload can lead to apoptosis (12). For example, Ca2+ overload promotes the opening of the mitochondrial permeability transition pore, and agents that block the mitochondrial permeability transition pore opening inhibit apoptosis (12). Oxidative stress induces a significant change in mitochondrial Ca2+ flux leading to mitochondrial destabilization and apoptosis (13). Post-ischemic dopamine treatment of contractile dysfunction activates pro-apoptotic signal cascades via Ca2+-dependent mitochondrial damage (14). Sarcoplasmic reticulum (SR)3 and mitochondria locate close to each other in cardiomyocytes (15). There is a tight coupling of Ca2+ signaling between SR release sites and nearby mitochondria, because the focal SR Ca2+ release results in Ca2+ microdomains sufficient to promote local mitochondrial Ca2+ uptake (16, 17). Thus far, it remained unknown as to whether Foxo3a can regulate mitochondrial Ca2+ homeostasis.
The heart has evolutionarily developed a highly expressed anti-apoptotic protein, ARC (18, 19). It was originally identified to be a caspase-inhibiting protein and can specifically inhibit the activation of caspase-2 and -8, thereby blocking apoptosis induced by a variety of stimuli requiring the engagement of these caspases (18). Further studies revealed that ARC also may elicit its anti-apoptotic function by other means. It can interact with Fas, FADD, and Bax (20, 21), inhibit cytochrome c release (22), and maintain mitochondrial membrane potential (23, 24). Despite ARC's abundant expression and blocking apoptosis induced by either intrinsic or extrinsic stimulus, cardiomyocytes still undergo apoptosis under pathological conditions such as oxidative stress (23, 25) and hypoxia (26–28). Apoptosis is controlled by a complex interplay between pro- and anti-apoptotic factors. The occurrence of apoptosis under pathological conditions indicates that this interplay is imbalanced. It remains to be further elucidated as to how ARC is dysregulated under the pathological conditions. Also, it is not yet clear whether ARC is involved in the maintenance of mitochondrial Ca2+ homeostasis.
This work aimed to elucidate the role of Foxo3a in cardiac apoptosis. Our results show that Foxo3a can inhibit apoptosis induced by oxidative stress. Foxo3a knock-out mice exhibit accelerated myocardial infarction upon ischemia/reperfusion. Furthermore, ARC can maintain calcium homeostasis, and Foxo3a regulates calcium through ARC. Finally, our results revealed that ARC is a transcription target of Foxo3a. Taken together, our study revealed a novel anti-apoptotic pathway in which Foxo3a regulates calcium through transactivating ARC.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
Cardiomyocytes were isolated from 1- to 2-day-old Wistar rats as we described previously (10, 29). In brief, after the dissected hearts were washed, they were minced in HEPES-buffered saline solution containing 130 mmol/liter NaCl, 3 mmol/liter KCl, 1 mmol/liter NaH2PO4, 4 mmol/liter glucose, and 20 mmol/liter HEPES (pH adjusted to 7.35 with NaOH). Tissues were then dispersed in a series of incubations at 37 °C in HEPES-buffered saline solution containing 1.2 mg/ml pancreatin and 0.14 mg/ml collagenase (Worthington). After centrifugation, cells were resuspended in Dulbecco's modified Eagle's medium/F-12 (Invitrogen) containing 5% heat-inactivated horse serum, 0.1 mmol/liter ascorbate, insulin-transferring sodium selenite medium supplement, 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.1 mmol/liter bromodeoxyuridine. The dissociated cells were pre-plated at 37 °C for 1 h. The cells were then diluted to 1 × 106 cells/ml and plated in 10 μg/ml laminin-coated separate culture dishes according to the specific experimental requirements. Neonatal mouse cardiomyocytes were cultured as described elsewhere (30, 31). In brief, the hearts from wild type and Foxo3a knock-out mice were harvested, minced, and dispersed by 1.2 mg/ml pancreatin and 0.625 mg/ml collagenase (Worthington). Myocytes and nonmyocytes were separated by pre-plating for 1 h. Treatment of cells with hydrogen peroxide was carried out as we described previously (32). 10 μm Ca2+ chelator BAPTA-AM (Invitrogen) was administered 1 h before hydrogen peroxide or anoxia/reoxygenation treatment. Anoxia/reoxygenation was performed as described elsewhere (33).
ARC Transgenic Mice
For creating the ARC transgenic mice, rat ARC coding sequence (GenBankTM accession number NM_053516) was cloned to the vector, pαMHC-clone26 (kindly provided by Dr. Zhongzhou Yang), under the control of the α-myosin heavy chain promoter. Microinjection was performed following standard protocols. The primers for genotyping ARC transgenic mice include the following: forward primer in the α-MHC promoter, 5′-CACATAGAAGCCTAGCCCACA-3′, and the reverse primer in the ARC coding sequence, 5′-TTAGGTGTTCTCACAACCTTC-3′.
Genotyping of Foxo3a Knock-out Mice
Foxo3a knock-out (KO) mice were purchased from the Mutant Mouse Regional Resource Center. Foxo3a+/− mice were interbred to give knock-out mice (Foxo3a−/−), which were used for further studies. Mice were genotyped by multiplex PCR (primers and conditions are available from Mutant Mouse Regional Resource Center). All experiments were performed on Foxo3a−/− mice and their wild type littermates (Foxo3a+/+) and were approved by government authorities.
Adenoviral Vector Construction and Infection
The adenoviruses harboring ARC, wild type Foxo3a (WTFoxo3a), and the constitutively active form of human Foxo3a (caFoxo3a) were as we described previously (10, 34). The adenoviruses harboring rat Foxo3a and ARC RNAi were constructed using pSilencerTM adeno 1.0-CMV system. The rat Foxo3a RNAi target sequence is 5′-CAAGTACACCAAGAGCCGA-3′. A nonrelated and scrambled RNAi without any other match in the rat genomic sequence was used as a control (5′-TCAGACAGACAGACAGACC-3′). The rat ARC RNAi target sequence is 5′-ACTGTGAGCATGCCAGACC-3′, and the scrambled RNAi sequence is 5′-GTGCATCAGACTACCAGGC-3′. All viruses were amplified in HEK-293 cells. Cells were infected at the indicated multiplicity of infection (m.o.i.) for 60 min. After washing with phosphate-buffered saline (PBS), culture medium was added, and cells were cultured until the indicated time.
Preparations of Subcellular Fractionation
Mitochondrion-enriched heavy membranes and cytosolic fractions were prepared as described previously (35). Briefly, cells were washed twice with PBS, and the pellet was suspended in 0.5 ml of buffer (20 mmol/liter HEPES, pH 7.5, 10 mmol/liter KCl, 1.5 mmol/liter MgCl2, 1 mmol/liter EGTA, 1 mmol/liter EDTA, 1 mmol/liter DTT, 0.1 mmol/liter PMSF, 10 mg/ml each of leupeptin, aprotinin, and pepstatin A) containing 250 mmol/liter sucrose. The cells were homogenized by 10 strokes in a Dounce homogenizer. The homogenates were centrifuged twice at 750 × g for 5 min at 4 °C. The supernatants were centrifuged at 10,000 × g for 15 min at 4 °C to collect mitochondrion-enriched heavy membranes. The final supernatants are referred to as cytosolic fractions. The isolation of SR-enriched membrane fractions were carried out as described previously (36, 37). In brief, the cells were homogenized and centrifuged at 12,000 × g for 10 min. The supernatants were centrifuged at 100,000 × g for 45 min. The pellets are referred to as SR fractions.
Immunoblotting Analysis
Cells were lysed for 1 h at 4 °C in a lysis buffer (20 mmol/liter Tris, pH 7.5, 2 mmol/liter EDTA, 3 mmol/liter EGTA, 2 mmol/liter dithiothreitol (DTT), 250 mmol/liter sucrose, 0.1 mmol/liter phenylmethylsulfonyl fluoride, 1% Triton X-100) containing a protease inhibitor mixture (Sigma). Samples were subjected to 12% SDS-PAGE and transferred to nitrocellulose membranes. Equal protein loading was controlled by Ponceau Red staining of membranes. Blots were probed using the following primary antibodies: the anti-ARC antibody (Millipore, 1:1000), the anti-Foxo3a antibody (Cell Signaling, 1:1000), the anti-COXIV antibody (Abcam, 1:1000), the anti-calnexin antibody (Santa Cruz Biotechnology, 1:500), and the anti-cytochrome c antibody (Santa Cruz Biotechnology, 1:1000). After four washes with PBS/Tween 20, horseradish peroxidase-conjugated secondary antibodies were added. Antigen-antibody complexes were visualized by enhanced chemiluminescence.
Construction of Rat ARC Promoter and Luciferase Assays
ARC promoters were amplified from rat genome using PCR. The full-length fragment containing three Foxo3a potential binding sites (ARC promoter-1, 3138 bp) was amplified using the forward primer 5′-CAACCAAAGATCACTAGAGTCGCG-3′. The other two shorter fragments were amplified using the following forward primers: 5′-AACAGATTGGGCAGAATCCTGGGC-3′ (ARC promoter-2, 2448 bp) and 5′-GGGTGGAGTGTGTGAGAAGTACTC-3′ (ARC promoter-3, 1440 bp), respectively. All fragments were amplified using the reverse primer 5′-CATTTGGGCTATATCAAGAAGGAG-3′. These promoters were cloned into the reporter plasmid pGL4.17 (Promega).
Luciferase activity assay was performed using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions. Cells were lysed and assayed for luciferase activity 24 h after transfection. 20 μl of protein extracts were analyzed in a luminometer. Firefly luciferase activities were normalized to Renilla luciferase activity.
Quantitative Real Time-PCR
Quantitative RT-PCR was performed as we described previously (32). In brief, total RNA was isolated using TRIzol (Invitrogen). RNA was reverse-transcribed using Oligo(dT) and amplified using a TaqMan assay kit (TOYOBO). The samples were run in triplicate using the Applied Biosystems 7000 sequence detector according to the manufacturer's instructions. The results were standardized to control values of GAPDH. The sequences of ARC primers were as follows: forward 5′-ATGGGTAACATGCAGGAGCGC-3′ and reverse 5′-GTCCAGCAGCAACCCAGAGTC-3′; GAPDH forward primer 5′-GCTAACATCAAATGGGGTGATGCTG-3′ and reverse primer 5′-GAGATGATGACCCTTTTGGCCCCAC-3′. PCR was run under the following conditions: 95 °C for 60 s for 1 cycle; 95 °C for 15 s, 55 °C for 15 s, and 72 °C for 45 s for 40 cycles. The specificity of the PCR amplification was confirmed by agarose gel electrophoresis.
Chromatin Immunoprecipitation (ChIP) Analysis
ChIP was performed as we described elsewhere (10). PCRs were performed with the primers that encompass the three Foxo3a potential binding sites in the rat ARC promoter. The oligonucleotides were as follows: BS1 (corresponding to a 317-bp fragment), forward 5′-CAAGTGTTCAGATGTTCGGAC-3′ and reverse 5′-TCACTTGTCCAGCCCTCTCTC-3′; BS2 (corresponding to a 338-bp fragment), forward 5′-GGCTTGTCCACTGATGCACTG-3′ and reverse 5′-CCATGTCTATCCCAATTCCTG-3′; and BS3 (corresponding to a 329-bp fragment), forward 5′-GGGGTGCATGAAGTATGTGTG-3′ and reverse 5′-TTTCAGAGCTGGGGACCGAAC-3′.
Immunofluorescence
Immunofluorescence staining was performed as we and others described previously (35, 38, 39). Cells were fixed in methanol (−20 °C), washed, permeabilized (PBS, 0.3% Triton X-100), and blocked. Specimens were incubated overnight (4 °C) with primary antibodies as follows: ARC (Chemicon, 1:100) or cytochrome c (Santa Cruz Biotechnology, 1:200). Immunoreactive antigens were visualized after staining with Alexa Fluor® 594-conjugated goat anti-mouse IgG (1:500) or Alexa Fluor ® 488-conjugated goat anti-rabbit IgG (1:500). Nuclear staining was achieved using DAPI (1:1000 in PBS; 5 min). Images from fluorescence-labeled cells were obtained with a Zeiss LSM510 confocal laser scanning microscope.
Measurement of [Ca2+]c
Cytosolic calcium was measured as described previously (40–42). In brief, the cardiomyocytes cultured on a glass coverslip were loaded with 10 μm Fluo-3-AM (Molecular Probes), in the HEPES solution without Ca2+ at 37 °C for 30 min, then washed twice with dye-free HEPES solution, and placed in a chamber on the stage of a laser scanning confocal microscope (Zeiss LSM510). Fluo-3 in cells was excited with light at 488 nm, and emitted fluorescence was detected at >500 nm. For fluorescence analysis, the regions of interest identified in the average fluorescence intensities over 20 regions of interest minus background were calculated for each frame and normalized for comparative purposes. The [Ca2+]c values were calculated by Mn2+ quenching.
Detections of [Ca2+]m
The mitochondrial calcium indicator Rhod-2 AM (Molecular Probes) was used to measure changes of [Ca2+]m as described previously (43, 44). Briefly, cardiomyocytes were loaded with 5 μm Rhod-2 AM for 2 h at 4 °C and further incubated for 2 h at 37 °C in the culture medium. This two-step cold loading/warm incubation protocol achieves exclusive loading of Rhod-2 into the mitochondria. The Rhod-2 images were captured with excitation at 543 nm and detected at >560 nm. Rhod-2 fluorescence intensities (F) in each experiment were normalized to the average base-line fluorescence for the same region (_F_0).
Detections of [Ca2+]SR
The low affinity calcium indicator, Fluo-5N-AM (Molecular Probes), was used to assess changes in the intraluminal SR Ca2+ concentration as described previously (45, 46). In brief, the cardiomyocytes were loaded with 5 μm Fluo-5N-AM at 37 °C for 2 h, rinsed, and incubated in culture medium for a further 1.5 h at 37 °C to allow de-esterification and outward leak of the cytosolic indicator. Fluo-5N was excited with light at 488 nm, and the emitted fluorescence was detected at >500 nm. Fluo-5N fluorescence intensities (F) in each experiment were normalized to the average base-line fluorescence for the same region (_F_0).
Detection of Mitochondrial Membrane Potential (ΔΨm)
ΔΨ_m_ was measured using tetramethylrhodamine methyl ester (TMRM) as described previously (39). In brief, the cells were stained with 10 nm TMRM (Molecular Probes) at 37 °C for 30 min. TMRM images were captured using a laser scanning confocal microscope (Zeiss LSM510) with excitation at 543 nm and emission at >560 nm.
Ischemia/Reperfusion (I/R), Hemodynamic Assessment, LDH Release Assay, and Analysis of Infarction Sizes
Mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg) and fixed in the supine position, and tracheotomy was performed to provide artificial ventilation (0.2-ml tidal volume, 110 breaths/min) with a rodent ventilator supplemented with 100% oxygen. The left anterior descending coronary artery was identified and ligated with a slipknot using 8-0 silk suture at the inferior border of the left auricle. Myocardial ischemia was confirmed by the obvious cyanotic appearance of the left ventricle and S-T segment elevation on the electrocardiogram. After 30 min of ischemia, the knot was released to permit reperfusion of the heart, which was also confirmed by obvious S-T segment change. After removal of air and blood, the chest was closed, and the animal was removed from the respirator and transferred back to its cage. Sham-operated mice were prepared identically without undergoing the occlusion of the silk suture. 24 h after reperfusion, the mice were anesthetized as described above, and the LV hemodynamic measurements were conducted by an experienced investigator, who was blind to the treatment, through a closed-chest catheterization. A 1.4-F microtipped catheter (SPR-839, Millar Instruments) was inserted into the right carotid artery and advanced into the left ventricle. Hemodynamic parameters such as left ventricular end-diastolic pressure, left ventricular maximum first derivative of pressure (LV d_P_/d_t_max), minimum first derivative of pressure (LV d_P_/d_t_min), and LV ejection fraction were computed. The heparin-blood at the end of the hemodynamic assessment was collected from all groups, and the concentration of LDH in the plasma was assayed using an LDH ELISA kit (Adlitteram Diagnostic Laboratories). To determine myocardial infarct sizes, the thoracotomy was reopened; the suture was reoccluded, and 2% of Evans blue (Sigma) was injected into the left ventricular cavity to delineate the ischemic zone from the nonischemia zone. The heart was immediately removed and flushed with ice-cold saline and frozen in a −80 °C freezer. Each heart was then horizontally cut into five slices that were incubated in 1.0% 2,3,5-triphenyltetrazolium chloride (Sigma) for 15 min at 37 °C for demarcation of the viable and nonviable myocardium within AAR. Infarct myocardium appears yellowish white, and viable myocardium stains brick red. The staining was stopped by ice-cold PBS, and the slices were fixed in 10% neutral buffered formaldehyde and individually weighed. Both sides of each slice were photographed. The areas of infarction (INF), AAR, and LV were assessed by computerized planimetry, and the ratios of AAR/LV, INF/AAR, and INF/LV were analyzed by an observer blinded to the sample identity.
Evaluation of Apoptosis
To determine apoptosis in the heart sections, we used In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics). The detections were performed as we described elsewhere (32, 47).
Detection of Caspase-3 Activity
Caspase-3 activity was detected using an assay kit (R&D Systems). The assay procedures were according to the kit instructions.
Statistical Analysis
Paired data were evaluated by Student's t test. A one-way analysis of variance was used for multiple comparisons. A p value of <0.05 was considered significant.
RESULTS
Foxo3a Can Suppress Cytosolic Ca2+ Elevations and Apoptosis
Reactive oxygen species play an important role in mediating apoptosis in cardiomyocytes. In this study, we observed that hydrogen peroxide or anoxia/reoxygenation treatment leads to the increase of cytosolic Ca2+ levels [Ca2+]c in cardiomyocytes (Fig. 1A). Administration of the Ca2+ chelator, BAPTA-AM, inhibits apoptosis (Fig. 1B), suggesting that Ca2+ is necessary for the initiation of apoptosis.
FIGURE 1.

Foxo3a suppresses Ca2+ elevations and apoptosis. A, hydrogen peroxide or anoxia/reoxygenation (A/R) induces an elevation of [Ca2+]c. Neonatal rat cardiomyocytes were treated with hydrogen peroxide or anoxia/reoxygenation. The confocal images show representative images. The histograms show the summary of [Ca2+]c levels. *, p < 0.05. B, Ca2+ chelator BAPTA-AM inhibits apoptosis. Neonatal rat cardiomyocytes were exposed to the Ca2+ chelator BAPTA-AM (10 μm, 1 h) followed by H2O2 (24 h) or anoxia/reoxygenation treatment. *, p < 0.05. C, Foxo3a prevents apoptosis triggered by hydrogen peroxide. Neonatal rat cardiomyocytes were infected with the adenoviruses harboring constitutively active Foxo3a (caFoxo3a) or β-galactosidase cDNA _(β_-gal) at an m.o.i. of 80. 24 h after infection, cells were treated with hydrogen peroxide. The upper panel shows Foxo3a levels analyzed by immunoblotting. Apoptosis was detected 24 h after hydrogen peroxide treatment (lower panel). *, p < 0.05. D, caFoxo3a attenuates [Ca2+]c elevations triggered by hydrogen peroxide. Neonatal rat cardiomyocytes were treated as described for C. The histograms show the summary of [Ca2+]c levels. *, p < 0.05. E and F, inhibition of endogenous Foxo3a sensitizes [Ca2+]c elevations (E) and apoptosis (F) in response to hydrogen peroxide treatment. Neonatal rat cardiomyocytes were infected with the adenoviral Foxo3a RNAi or its scrambled form (Foxo3a-S-RNAi) at an m.o.i. of 50. 24 h after infection, cells were treated with 10 μm H2O2. The upper panel in E shows Foxo3a levels analyzed by immunoblotting. [Ca2+]c levels are shown in the lower panel of E. Apoptosis is shown in F. *, p < 0.05. G and H, cardiomyocytes from Foxo3a knock-out mice show a higher level of [Ca2+]c (G) and apoptosis (H) in response to hydrogen peroxide stimulation. Cardiomyocytes isolated from Foxo3a knock-out mice were infected with adenoviral caFoxo3a or β-gal for 24 h and then treated with hydrogen peroxide. *, p < 0.05. Data are expressed as mean ± S.E. from three independent experiments.
Foxo3a can trigger or inhibit apoptosis depending on the cell types or cellular context (6, 48). We tested whether Foxo3a can influence apoptosis in cardiomyocytes. Enforced expression of the constitutively active form of Foxo3a (caFoxo3a) itself did not induce apoptosis; however, it could prevent apoptosis triggered by hydrogen peroxide (Fig. 1C). Because Ca2+ is necessary for the initiation of apoptosis as shown in Fig. 1B, we thus tested whether Foxo3a can influence [Ca2+]c levels in cells treated with hydrogen peroxide. Enforced expression of caFoxo3a could attenuate [Ca2+]c elevations induced by hydrogen peroxide (Fig. 1D). We then tested whether endogenous Foxo3a participates in the regulation of [Ca2+]c and apoptosis in cardiomyocytes. Hydrogen peroxide at a low dose of 10 μm induced a slight alteration of [Ca2+]c levels. In contrast, knockdown of endogenous Foxo3a led to a significant elevation of [Ca2+]c in response to the treatment with the same low dose of hydrogen peroxide (Fig. 1E). Concomitantly, a significant amount of cells underwent apoptosis (Fig. 1F). Furthermore, we isolated cardiomyocytes from Foxo3a knock-out mice and observed a higher level of [Ca2+]c in these cells upon treatment with hydrogen peroxide or anoxia/reoxygenation, and noticeably, re-expression of caFoxo3a attenuated [Ca2+]c levels (Fig. 1G). Hydrogen peroxide or anoxia/reoxygenation induced more cardiomyocytes from Foxo3a knock-out mice to undergo apoptosis, and this can be inhibited by re-expression of caFoxo3a (Fig. 1H). Finally, we further characterized whether apoptosis indeed occurred under our experimental condition. Enforced expression of caFoxo3a suppressed the activation of caspase-3 in neonatal rat cardiomyocytes (Fig. 2, A and B) or cardiomyocytes from Foxo3a-deficient mice (Fig. 2C). Pre-treatment of the cells with caspase inhibitor Z-VAD-fmk suppressed both apoptosis (Fig. 2, D and E) and caspase-3 activation (Fig. 2, F and G) triggered by hydrogen peroxide or anoxia/reoxygenation. Taken together, these data indicate that Foxo3a is able to suppress [Ca2+]c elevations and apoptosis.
FIGURE 2.

Caspase-3 activation is inhibited by Foxo3a or Z-VAD-fmk. A and B, enforced expression of caFoxo3a attenuates caspase-3 activation triggered by hydrogen peroxide (A) or anoxia/reoxygenation (A/R) (B). Neonatal rat cardiomyocytes were infected with the adenoviruses harboring constitutively active Foxo3a (caFoxo3a) or β-galactosidase cDNA (β_-gal_) at an m.o.i. of 80. 24 h after infection, cells were subjected to hydrogen peroxide or anoxia/reoxygenation. Caspase-3 catalytic activity was measured after stimulation. *, p < 0.05. C, cardiomyocytes from Foxo3a knock-out mice show a higher level of caspase-3 activity in response to anoxia/reoxygenation. Cardiomyocytes isolated from Foxo3a knock-out mice were infected with adenoviral caFoxo3a or β-gal for 24 h and then subjected to anoxia/reoxygenation. *, p < 0.05. Data are expressed as mean ± S.E. from three independent experiments. D and E, analysis of cell death triggered by hydrogen peroxide (D) or anoxia/reoxygenation (E). The neonatal rat cardiomyocytes were pre-treated with or without caspase inhibitor Z-VAD-fmk (100 μm, 1 h) and subjected to hydrogen peroxide or anoxia/reoxygenation treatment. Apoptosis was detected 24 h after stimulation. *, p < 0.05. F and G, analysis of caspase-3 activation in cells stimulated with hydrogen peroxide (F) or anoxia/reoxygenation (G). The neonatal rat cardiomyocytes were treated as described for D and E. The caspase-3 activity was analyzed. *, p < 0.05. Data are expressed as mean ± S.E. from three independent experiments.
Foxo3a Knock-out Mice Exhibit an Enlarged Myocardial Infarction Size
Apoptosis is a kind of death form in myocardial infarction. We tested whether Foxo3a plays a role in regulating myocardial infarction. Foxo3a deficient mice exhibited a larger infarction size upon I/R (Fig. 3A), and a more severe injury in cardiac function (Fig. 3B). We analyzed apoptosis and observed more apoptotic cells in Foxo3a deficient mice upon I/R (Fig. 3C).
FIGURE 3.

Foxo3a knock-out mice demonstrate an enlarged myocardial infarction size. A, mice deficient in Foxo3a show an enlarged myocardial infarction size upon I/R injury. Foxo3a knock-out mice and wild type (WT) littermates were subjected to 30 min of ischemia and 24 h of reperfusion. The ratio of area-at-risk (AAR)/left ventricle (LV), INF myocardium/AAR, and INF/LV were calculated. (n = 6). *, p < 0.05. B, mice deficient in Foxo3a present worsened hemodynamics in response to I/R injury. Mice were treated as described for A. 24 h after reperfusion, the LV hemodynamic assessments were performed (n = 6). *, p < 0.05. C, TUNEL assay illustrates more apoptosis in Foxo3a knock-out mice than the wild type littermates. Representative myocardial sections stained by the TUNEL method are shown in the left panel. Apoptotic nuclei are shown by the bright green nuclear fluorescence. Sections were counterstained with anti-α-actinin antibody and DAPI to identify cardiac myocytes and nuclei. Bar, 20 μm. Foxo3a and ARC levels in the hearts were analyzed by immunoblotting (right lower panel). Quantification of apoptosis by the TUNEL method (right upper panel, n = 5). *, p < 0.05. D, Foxo3a null mice show increased LDH release induced by I/R injury. Plasma LDH levels at the end of 24 h of reperfusion in vivo were measured. (n = 5). *, p < 0.05. Data are expressed as mean ± S.E. from three independent experiments.
These results suggest that Foxo3a participates in inhibiting apoptosis and myocardial infarction in the animal model. Furthermore, our data shows that LDH release was significantly increased in hearts from Foxo3a deficient mice (Fig. 3D), which suggests that loss of Foxo3a exacerbated myocardial injury induced by I/R.
ARC Attenuates Cytosolic Ca2+ Elevations
To understand the relationship between ARC and Ca2+ in the apoptotic program of cardiomyocytes, we tested whether ARC could influence [Ca2+]c. ARC levels were decreased upon hydrogen peroxide treatment (Fig. 4A). Knockdown of endogenous ARC could sensitize hydrogen peroxide to induce [Ca2+]c elevations (Fig. 4B), apoptosis (Fig. 4C), and the activation of caspase-3 (Fig. 4D). Enforced expression of ARC attenuated [Ca2+]c elevations induced by hydrogen peroxide (Fig. 4E). Concomitantly, apoptosis (Fig. 4F) and caspase-3 activation could be inhibited (Fig. 4G). Furthermore, we isolated the cardiomyocytes from ARC transgenic mice and observed that the [Ca2+]c increases evoked by stimulation with hydrogen peroxide were significantly less than that from wild type mice (Fig. 4H). Thus, it appears that ARC is able to attenuate cytoplasmic calcium elevation induced by hydrogen peroxide in cardiomyocytes.
FIGURE 4.

ARC attenuates [Ca2+]c elevations. A, hydrogen peroxide reduces Foxo3a and ARC levels. Neonatal rat cardiomyocytes were treated with hydrogen peroxide. Foxo3a and ARC were analyzed by immunoblotting. The protein loading was illustrated by actin. A representative result of three independent experiments is shown. B–D, knockdown of endogenous ARC sensitizes hydrogen peroxide to induce [Ca2+]c elevations (B), apoptosis (C), and caspase-3 activation (D). Neonatal rat cardiomyocytes were infected with the adenoviral ARC RNAi or its scrambled form (ARC-S-RNAi) at an m.o.i. of 50. 24 h after infection cells, were treated with hydrogen peroxide. ARC levels analyzed by immunoblotting are shown in the upper panel in Fig. 3B. *, p < 0.05. E–G, enforced expression of ARC suppresses [Ca2+]c rises (E), apoptosis (F), and caspase-3 activation (G) induced by hydrogen peroxide. Neonatal rat cardiomyocytes were infected with the adenoviral ARC or β-gal at an m.o.i. of 80. 24 h after infection, cells were treated with hydrogen peroxide. *, p < 0.05. H, cardiomyocytes from ARC transgenic mice show a lower level of [Ca2+]c in response to hydrogen peroxide stimulation. The cardiomyocytes from ARC transgenic mice were treated with hydrogen peroxide. *, p < 0.05. Data are expressed as mean ± S.E. from three independent experiments.
ARC Participates in the Control of Ca2+ Release from Sarcoplasmic Reticulum
How can ARC lead to a reduction of [Ca2+]c? Sarcoplasmic reticulum release of Ca2+ has been proved to contribute to [Ca2+]c elevations during apoptosis. We tested whether ARC can influence sarcoplasmic reticulum release of Ca2+. ARC has been shown previously to be localized in the cytoplasm and mitochondria (20, 35), but it is not yet clear whether ARC is distributed in the sarcoplasmic reticulum. We analyzed ARC distributions in the subcellular organelles and observed that a portion of ARC was distributed in the sarcoplasmic reticulum as analyzed by immunoblotting and immunofluorescence (Fig. 5A). Subsequently, we investigated whether ARC can influence Ca2+ within the sarcoplasmic reticulum ([Ca2+]SR). Hydrogen peroxide induced a reduction of [Ca2+]SR. Enforced expression of ARC elevated [Ca2+]SR upon treatment with hydrogen peroxide (Fig. 5B). We attempted to understand whether endogenous ARC plays a role in regulating [Ca2+]SR, and we observed that knockdown of ARC could sensitize the reduction of [Ca2+]SR upon treatment with 10 μm hydrogen peroxide (Fig. 5C). These data indicate that ARC is able to regulate sarcoplasmic reticulum Ca2+ release into the cytoplasm.
FIGURE 5.

ARC reduces Ca2+ release from sarcoplasmic reticulum. A, analysis of ARC intracellular localizations. The left panel shows ARC in the subcellular fractions of cardiomyocytes as analyzed by immunoblotting. The cytochrome (Cyto) oxidase subunit IV (COX IV) is a mitochondria (Mito) marker. SERCA2 is a marker of sarcoplasmic reticulum. Endogenous ARC and SERCA2 were visualized by immunofluorescence (right panel). Bar, 20 μm. B, ARC inhibits the reduction of [Ca2+]SR induced by hydrogen peroxide. Neonatal rat cardiomyocytes were infected with the adenoviral ARC or β-gal at an m.o.i. of 80. 24 h after infection, cells were treated with hydrogen peroxide. *, p < 0.05. C, knockdown of ARC sensitizes [Ca2+]SR reduction upon treatment with hydrogen peroxide. Neonatal rat cardiomyocytes were infected with the adenoviral ARC RNAi or its scrambled form (ARC-S-RNAi) at an m.o.i. of 50. 24 h after infection, cells were treated with hydrogen peroxide. *, p < 0.05. Data are expressed as mean ± S.E. from three independent experiments.
ARC Attenuates Mitochondrial Ca2+ Uptake
It has been well documented that the imbalance in mitochondrial Ca2+ ([Ca2+]m) leads to apoptosis. We tested whether ARC was involved in the maintenance of [Ca2+]m homeostasis. Enforced expression of ARC reduced [Ca2+]m rises upon treatment with hydrogen peroxide (Fig. 6A). Knockdown of ARC sensitized the elevation of [Ca2+]m upon treatment with 10 μm hydrogen peroxide (Fig. 6B). We isolated cardiomyocytes from ARC transgenic mice, and they exhibited a lower level of [Ca2+]m than those from the wild type mice upon hydrogen peroxide treatment (Fig. 6C), suggesting that ARC serves to inhibit the elevation of [Ca2+]m.
FIGURE 6.

ARC attenuates the elevation of [Ca2+]m. A, enforced expression of ARC reduces [Ca2+]m rises induced by hydrogen peroxide. Neonatal rat cardiomyocytes were infected with the adenoviral ARC or β-gal at an m.o.i. of 80. 24 h after infection, cells were treated with hydrogen peroxide. The left panel shows the representative confocal image. The right upper panel shows ARC levels in mitochondrion-enriched heavy membranes analyzed by immunoblotting. The right lower panel shows [Ca2+]m. *, p < 0.05. B, knockdown of endogenous ARC sensitizes hydrogen peroxide-induced [Ca2+]m elevations. Neonatal rat cardiomyocytes were infected with the adenoviral ARC RNAi or its scrambled form (ARC-S-RNAi) at an m.o.i. of 50. 24 h after infection, cells were treated with hydrogen peroxide. The upper panel shows ARC levels in mitochondrion-enriched heavy membranes analyzed by immunoblotting. The lower panel shows [Ca2+]m. *, p < 0.05. C, cardiomyocytes from ARC transgenic mice show a lower level of [Ca2+]m in response to hydrogen peroxide stimulation. The upper panel shows ARC levels in ARC transgenic mice (Tg) and wild type mice (WT). [Ca2+]m levels are shown in the lower panel. *, p < 0.05. D, fluorescent analysis of M/GFP/ARC. Neonatal rat cardiomyocytes were transfected with the construct of M/GFP/ARC. Mitochondria were visualized by Mitotracker Red. Bar, 20 μm. E, ARC attenuates [Ca2+]m accumulation induced by hydrogen peroxide. Neonatal rat cardiomyocytes were transfected with the constructs of the empty vector (M/GFP) or M/GFP/ARC and subjected to hydrogen peroxide. *, p < 0.05. F, ARC attenuates mitochondrial membrane potential depolarization. Neonatal rat cardiomyocytes were treated as described for E. Mitochondrial membrane potential was analyzed by TMRM (red). GFP positive cells exhibit green color (left panel). Bar, 20 μm. The right panel shows the summary of TMRM fluorescence. G, subcellular distributions of cytochrome c. Neonatal rat cardiomyocytes were treated as described for E, then fixed, and stained for cytochrome c. Bar, 20 μm. Data are expressed as mean ± S.E. from three independent experiments.
To further understand the role of ARC in regulating [Ca2+]m, we made a construct in which ARC was fused to mitochondrion-targeted GFP, and we termed this construct as M/GFP/ARC, which was specifically localized in mitochondria (Fig. 6D). The empty mitochondrion-targeted GFP was termed as M/GFP. M/GFP/ARC was able to attenuate [Ca2+]m elevations induced by hydrogen peroxide (Fig. 6E). Also, it could inhibit the collapse of mitochondrial membrane potential (Fig. 6F). We analyzed cytochrome c distributions in cells expressing M/GFP/ARC. In the control cells without treatment, cytochrome c distribution pattern was coincident with that of M/GFP. Upon treatment with hydrogen peroxide, cytochrome c and M/GFP showed a differential pattern. However, in cells expressing M/GFP/ARC, the cytochrome c distribution pattern was not significantly altered (Fig. 6G).
Foxo3a Regulates Ca2+ through ARC
Foxo3a is a transcription factor. How can it influence Ca2+ machinery in cardiomyocytes? Because of the ability of ARC to regulate Ca2+, we tested whether ARC can be a downstream mediator of Foxo3a to target Ca2+. Knockdown of ARC could abolish the effect of Foxo3a on attenuating [Ca2+]c elevations (Fig. 7A) and apoptosis (Fig. 7B) induced by hydrogen peroxide. The effects of Foxo3a on attenuating caspase-3 activation (Fig. 7C), [Ca2+]m increases (Fig. 7D), and cytochrome c release (Fig. 7E) were also abolished upon ARC knockdown. These data indicate that ARC is a downstream mediator of Foxo3a in regulating calcium homeostasis.
FIGURE 7.
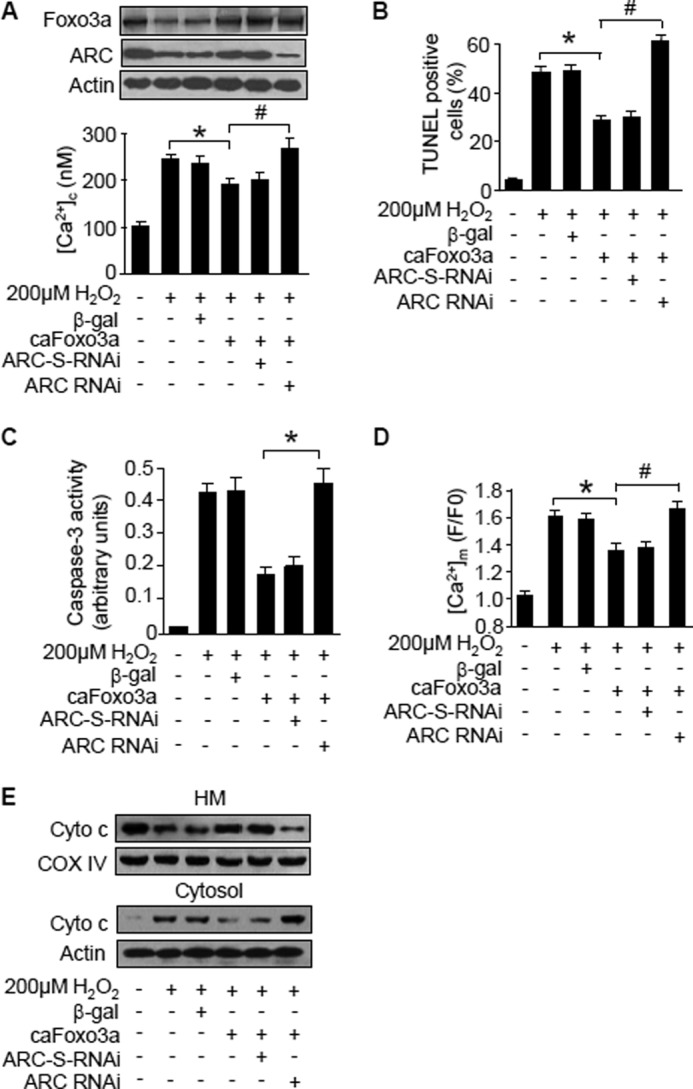
Foxo3a regulates Ca2+ through ARC. A–C, knockdown of ARC abolishes the effect of caFoxo3a on [Ca2+]c (A), apoptosis (B), and caspase-3 activation (C). Neonatal rat cardiomyocytes were co-infected with the adenoviral caFoxo3a at an m.o.i. of 80 and ARC RNAi or ARC-S-RNAi at an m.o.i. of 50. The cells were treated with hydrogen peroxide 24 h after adenoviral infection. The upper panel in A shows Foxo3a and ARC levels analyzed by immunoblotting. The lower panel in A shows [Ca2+]c. * and #, p < 0.05. D and E, knockdown of ARC abolishes the effect of caFoxo3a on [Ca2+]m (D) and cytochrome c distributions (E). Neonatal rat cardiomyocytes were treated as described for A. * and #, p < 0.05. Cytochrome c distributions in mitochondrion-enriched heavy membranes (HM) and cytosol were analyzed by immunoblotting. Data are expressed as mean ± S.E. from three independent experiments.
ARC Is a Transcriptional Target of Foxo3a
We explored the relationship between ARC and Foxo3a. An elevated level of ARC mRNA in caFoxo3a-expressed cardiomyocytes could be observed (Fig. 8A). Also, ARC protein levels were elevated upon caFoxo3a stimulation (Fig. 8B). We further tested whether endogenous Foxo3a participates in the regulation of ARC. Knockdown of endogenous Foxo3a by RNAi led to a reduction in mRNA (Fig. 8C) and protein (Fig. 8D) levels of ARC.
FIGURE 8.

ARC is a transcriptional target of Foxo3a. A, Foxo3a stimulates ARC mRNA expression. Neonatal rat cardiomyocytes were infected with the adenoviral caFoxo3a or β-gal at an m.o.i. of 80. Cells were harvested for the analysis of ARC mRNA by quantitative RT-PCR. The values were normalized to that of GAPDH. *, < 0.05 versus control. B, Foxo3a up-regulates ARC protein levels. Neonatal rat cardiomyocytes were infected as described for A. Cells were harvested for the analysis of ARC protein by immunoblotting. The blots shown here are the representative blots from three independent experiments. Numbers above immunoblots show the ratios of the band intensity of ARC to that of actin. C, knockdown of Foxo3a leads to a reduction in ARC mRNA levels. Neonatal rat cardiomyocytes were infected with adenoviral Foxo3a-RNAi or its scrambled form (Foxo3a-S-RNAi) at an m.o.i. of 100. Cells were harvested 48 h after infection for the analysis of ARC mRNA levels by quantitative RT-PCR, *, p < 0.05 versus control. D, knockdown of Foxo3a leads to a reduction in ARC protein levels. Neonatal rat cardiomyocytes were infected as described for C. Cells were harvested 48 h after infection for the analysis of ARC protein levels by immunoblotting. The blots shown here are the representative blots from three independent experiments. Numbers above immunoblots show the ratios of the band intensity of ARC to that of actin. E, ARC promoter region has three potential Foxo3a-binding sites. Rat ARC promoter contains three potential Foxo3a-binding sites (BS) indicated as BS1, BS2, and BS3. Three fragments of ARC promoter were synthesized and linked to luciferase reporter vector, respectively. F, Foxo3a stimulates the activity of ARC promoter containing BS1. HEK-293 cells were infected with adenoviral β-gal, wild type Foxo3a (wtFoxo3a), or caFoxo3a at an m.o.i. of 80. 24 h after infection, cells were transfected with the constructs of the empty vector (pGL-4) or ARC promoter constructs, respectively. Firefly luciferase activities were normalized to Renilla luciferase activities. G, inhibition of endogenous Foxo3a leads to a reduction of ARC promoter activity. Neonatal rat cardiomyocytes were infected with adenoviral Foxo3a-RNAi or its scrambled form (Foxo3a-S-RNAi) at an m.o.i. of 100. 24 h after infection, cells were transfected with the constructs of the empty vector (pGL-4) or the pGL-4-ARC promoter-1, respectively. Firefly luciferase activities were normalized to Renilla luciferase activities. *, p < 0.05 versus pGL-4-ARC promoter-1 alone. H, Foxo3a directly binds to BS1 of ARC promoter as analyzed by ChIP assay. Neonatal rat cardiomyocytes were infected with adenoviral β-gal, WTFoxo3a, or caFoxo3a at an m.o.i. of 80. Cells were harvested 24 h after infection for ChIP analysis. Chromatin-bound DNA was immunoprecipitated with the anti-Foxo3a antibody. The anti-actin antibody was used as a negative control (Neg). Immunoprecipitated DNA was analyzed by PCR using a primer combination that encompassed the three potential Foxo3a-binding sites in ARC promoter (BS1, BS2, and BS3), respectively. M, means marker/ladder. I, ARC protein levels in hearts from Foxo3a knock-out mice were less than that from wild type mice. The hearts from Foxo3a knock-out mice and their wild type littermates were harvested, and ARC levels were analyzed by immunoblotting. Data are expressed as mean ± S.E. from three independent experiments.
The influence of Foxo3a on ARC expression led us to consider whether ARC is a transcriptional target of Foxo3a. To address this consideration, we analyzed the rat ARC promoter region, and we found that the promoter region of ARC contains three potential Foxo3a-binding sites (Fig. 8E). We first tested whether Foxo3a can regulate ARC promoter activity. Luciferase assay revealed that although enforced expression of wild type Foxo3a (WTFoxo3a) and caFoxo3a could stimulate ARC promoter activity, caFoxo3a had a stronger effect. Furthermore, only the full-length ARC promoter could be activated by Foxo3a, indicating that the BS1 was responsible for the luciferase activity (Fig. 8F). Knockdown of endogenous Foxo3a by RNAi significantly reduced ARC promoter activity in cardiomyocytes (Fig. 8G). To determine Foxo3a-binding sites in the ARC promoter in vivo, we performed ChIP assays using primers directed against the BS1, BS2, and BS3, respectively. Foxo3a bound to the BS1 but not BS2 and BS3 in the ARC promoter (Fig. 8H). Finally, we analyzed ARC levels in Foxo3a knock-out mice and observed that these mice had a low level of ARC in comparison with the wild type mice (Fig. 8I). Taken together, we demonstrate that ARC is a transcriptional target of Foxo3a.
DISCUSSION
Although Foxo3a plays a role in regulation of apoptosis in a variety of cell types, its role in cardiac apoptosis remains to be fully understood. This work demonstrated that Foxo3a inhibits apoptosis in cardiomyocytes, and the cardiac anti-apoptotic protein ARC attenuates the release of calcium from the sarcoplasmic reticulum and inhibits calcium elevations in the cytoplasm and mitochondria under oxidative stress. Foxo3a is shown to maintain calcium homeostasis and inhibit apoptosis in cardiomyocytes through ARC. Furthermore, ARC is a direct transcriptional target of Foxo3a. Thus, our results provide novel evidence that Foxo3a regulates cardiac apoptosis by maintaining calcium homeostasis through ARC.
The role of Foxo3a in apoptosis is dependent on the cell types and the cellular contexts. It can provoke apoptosis in hematopoietic (4, 5) and neuronal cells (6) but can prevent apoptosis in neutrophils. This discrepancy is probably due to the multiple targets of Foxo3a, and its final transcriptional output is determined by the equilibrium of the pro- and anti-apoptotic factors. The identified downstream targets of Foxo3a include Fas ligand (FasL) (6), Bim (49), Mn-superoxide dismutase (50), and catalase (51). FasL activates the extrinsic apoptotic pathway by associating with Fas and consequently leading to the formation of the death-inducing signaling complex and caspase-8 activation. Bim is a member of the Bcl-2 family and counteracts Bcl-2 and Bcl-xL, thereby inducing apoptosis (52). Mn-superoxide dismutase and catalase can scavenge reactive oxygen species that are important apoptotic stimuli. This work revealed that Foxo3a is able to prevent apoptosis in cardiomyocytes. Notably, we have found that ARC is a transcriptional target of Foxo3a and necessary for Foxo3a to exert its anti-apoptotic function.
It has been shown that ARC is a calcium-binding protein and can suppress Ca2+-mediated apoptosis (53). This study demonstrated that ARC is present in the sarcoplasmic reticulum and participates in the regulation of mitochondrial Ca2+ homeostasis by attenuating the release of [Ca2+]SR induced by oxidative stress. ARC requires the C terminus to bind to the calcium, and the deletion of the C terminus leads to the inability of ARC to bind to calcium (53). Intriguingly, the subcellular localization of ARC is controlled by the C terminus. The mutation in threonine 149 in the C terminus results in the alteration of ARC subcellular localization and loss of ARC anti-apoptotic function (54). Thus, it appears that the C terminus is important for ARC function. Our data shed new light on understanding the novel molecular mechanism by which ARC inhibits apoptosis.
SR and mitochondria locate close to each other in cardiomyocytes (15). There is a tight coupling of Ca2+ signaling between SR release sites and nearby mitochondria, because the SR Ca2+ release results in Ca2+ microdomains sufficient to promote local mitochondrial Ca2+ uptake (16, 17). In this study, we have shown that the increase in [Ca2+]m may result from the reduction of [Ca2+]SR. The membrane-permeable Ca2+ chelator BAPTA-AM is highly selective for Ca2+ and can be used to control the level of intracellular Ca2+. The efficiency of mitochondrial Ca2+ uptake depends on the upstroke velocity of cytosolic Ca2+ transients (55). It would be interesting to test whether BAPTA-AM can affect the effects of ARC on regulating calcium homeostasis in future studies.
ARC is involved in the control of myocardial infarction. ARC transgenic mice exhibit less myocardial infarction sizes (56), whereas ARC knock-out mice demonstrate accelerated myocardial infarction (47). ARC levels are significantly decreased in cardiomyocytes upon treatment with hydrogen peroxide and hypoxia (20, 23). Furthermore, ARC levels are reduced upon heart failure (47) or ischemia (57). The expression reduction of a protein can be due to its decrease in synthesis and/or increase in degradation. ARC degradation is up-regulated upon apoptotic stimulation (57). This work has revealed that Foxo3a can transactivate ARC, and the reduced expression of Foxo3a contributes to ARC down-regulation. Thus, the dysregulation of ARC expression can be related to Foxo3a.
To keep the heart intact in both structure and function, it is necessary to prevent apoptosis so that the heart does not lose cardiomyocytes. Therefore, the development of anti-apoptotic strategies may prove useful as a means to prevent apoptosis-related cardiac diseases leading to heart failure. This work provides novel evidence that Foxo3a and ARC constitute an anti-apoptotic pathway that participates in the maintenance of calcium homeostasis. Our results can provide important information for exploring the beneficial effects of this pathway on apoptosis-related cardiac diseases.
*
This work was supported, in whole or in part, by National Institutes of Health Grants 1R01HL102202 and 5R21HL092315. This work was also supported by National Natural Science Foundation of China Grants 31010103911 and 81070119.
3
The abbreviations used are:
SR
sarcoplasmic reticulum
Z
benzyloxycarbonyl
fmk
fluoromethyl ketone
BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-_N,N,N_′,_N_′-tetraacetic acid-acetoxymethyl ester
m.o.i.
multiplicity of infection
ARC
apoptosis repressor with caspase recruitment domain
TMRM
tetramethylrhodamine methyl ester
LV
left ventricular
I/R
ischemia/reperfusion
LDH
lactate dehydrogenase
AAR
area at risk
INV
infarction
ca
constitutively active.
REFERENCES
- 1.Crow M. T., Mani K., Nam Y. J., Kitsis R. N. (2004) The mitochondrial death pathway and cardiac myocyte apoptosis. Circ. Res. 95, 957–970 [DOI] [PubMed] [Google Scholar]
- 2.Reeve J. L., Duffy A. M., O'Brien T., Samali A. (2005) Don't lose heart–therapeutic value of apoptosis prevention in the treatment of cardiovascular disease. J. Cell. Mol. Med. 9, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Accili D., Arden K. C. (2004) FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117, 421–426 [DOI] [PubMed] [Google Scholar]
- 4.Dijkers P. F., Medema R. H., Pals C., Banerji L., Thomas N. S., Lam E. W., Burgering B. M., Raaijmakers J. A., Lammers J. W., Koenderman L., Coffer P. J. (2000) Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 20, 9138–9148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dijkers P. F., Birkenkamp K. U., Lam E. W., Thomas N. S., Lammers J. W., Koenderman L., Coffer P. J. (2002) FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol. 156, 531–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 7.Furuyama T., Nakazawa T., Nakano I., Mori N. (2000) Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 349, 629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skurk C., Izumiya Y., Maatz H., Razeghi P., Shiojima I., Sandri M., Sato K., Zeng L., Schiekofer S., Pimentel D., Lecker S., Taegtmeyer H., Goldberg A. L., Walsh K. (2005) The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 280, 20814–20823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson M. J., Viars C. S., Czekay S., Cavenee W. K., Arden K. C. (1998) Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics 47, 187–199 [DOI] [PubMed] [Google Scholar]
- 10.Tan W. Q., Wang K., Lv D. Y., Li P. F. (2008) Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 283, 29730–29739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni Y. G., Berenji K., Wang N., Oh M., Sachan N., Dey A., Cheng J., Lu G., Morris D. J., Castrillon D. H., Gerard R. D., Rothermel B. A., Hill J. A. (2006) Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pacher P., Csordás G., Hajnóczky G. (2001) Mitochondrial Ca2+ signaling and cardiac apoptosis. Biol. Signals Recept. 10, 200–223 [DOI] [PubMed] [Google Scholar]
- 13.Long X., Goldenthal M. J., Wu G. M., Marín-García J. (2004) Mitochondrial Ca2+ flux and respiratory enzyme activity decline are early events in cardiomyocyte response to H2O2. J. Mol. Cell. Cardiol. 37, 63–70 [DOI] [PubMed] [Google Scholar]
- 14.Stamm C., Friehs I., Cowan D. B., Cao-Danh H., Choi Y. H., Duebener L. F., McGowan F. X., del Nido P. J. (2002) Dopamine treatment of postischemic contractile dysfunction rapidly induces calcium-dependent pro-apoptotic signaling. Circulation 106, I290–298 [PubMed] [Google Scholar]
- 15.Sharma V. K., Ramesh V., Franzini-Armstrong C., Sheu S. S. (2000) Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32, 97–104 [DOI] [PubMed] [Google Scholar]
- 16.Duchen M. R., Leyssens A., Crompton M. (1998) Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J. Cell Biol. 142, 975–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 [DOI] [PubMed] [Google Scholar]
- 18.Koseki T., Inohara N., Chen S., Núñez G. (1998) ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc. Natl. Acad. Sci. U.S.A. 95, 5156–5160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geertman R., McMahon A., Sabban E. L. (1996) Cloning and characterization of cDNAs for novel proteins with glutamic acid-proline dipeptide tandem repeats. Biochim. Biophys. Acta 1306, 147–152 [DOI] [PubMed] [Google Scholar]
- 20.Nam Y. J., Mani K., Ashton A. W., Peng C. F., Krishnamurthy B., Hayakawa Y., Lee P., Korsmeyer S. J., Kitsis R. N. (2004) Inhibition of both the extrinsic and intrinsic death pathways through nonhomotypic death-fold interactions. Mol. Cell 15, 901–912 [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson A. B., Tsai J. G., Logue S. E., Crow M. T., Gottlieb R. A. (2004) Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J. Biol. Chem. 279, 21233–21238 [DOI] [PubMed] [Google Scholar]
- 22.Ekhterae D., Lin Z., Lundberg M. S., Crow M. T., Brosius F. C., 3rd, Núñez G. (1999) ARC inhibits cytochrome c release from mitochondria and protects against hypoxia-induced apoptosis in heart-derived H9c2 cells. Circ. Res. 85, e70–e77 [DOI] [PubMed] [Google Scholar]
- 23.Neuss M., Monticone R., Lundberg M. S., Chesley A. T., Fleck E., Crow M. T. (2001) The apoptotic regulatory protein ARC (apoptosis repressor with caspase recruitment domain) prevents oxidant stress-mediated cell death by preserving mitochondrial function. J. Biol. Chem. 276, 33915–33922 [DOI] [PubMed] [Google Scholar]
- 24.Gustafsson A. B., Sayen M. R., Williams S. D., Crow M. T., Gottlieb R. A. (2002) TAT protein transduction into isolated perfused hearts: TAT-apoptosis repressor with caspase recruitment domain is cardioprotective. Circulation 106, 735–739 [DOI] [PubMed] [Google Scholar]
- 25.van Empel V. P., Bertrand A. T., van der Nagel R., Kostin S., Doevendans P. A., Crijns H. J., de Wit E., Sluiter W., Ackerman S. L., De Windt L. J. (2005) Down-regulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ. Res. 96, e92–e101 [DOI] [PubMed] [Google Scholar]
- 26.Tanaka M., Ito H., Adachi S., Akimoto H., Nishikawa T., Kasajima T., Marumo F., Hiroe M. (1994) Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ. Res. 75, 426–433 [DOI] [PubMed] [Google Scholar]
- 27.Long X., Boluyt M. O., Hipolito M. L., Lundberg M. S., Zheng J. S., O'Neill L., Cirielli C., Lakatta E. G., Crow M. T. (1997) p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J. Clin. Invest. 99, 2635–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webster K. A., Discher D. J., Kaiser S., Hernandez O., Sato B., Bishopric N. H. (1999) Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J. Clin. Invest. 104, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan W. Q., Wang J. X., Lin Z. Q., Li Y. R., Lin Y., Li P. F. (2008) Novel cardiac apoptotic pathway: the dephosphorylation of apoptosis repressor with caspase recruitment domain by calcineurin. Circulation 118, 2268–2276 [DOI] [PubMed] [Google Scholar]
- 30.Poolman R. A., Li J. M., Durand B., Brooks G. (1999) Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knock-out mice. Circ. Res. 85, 117–127 [DOI] [PubMed] [Google Scholar]
- 31.Song W., Lu X., Feng Q. (2000) Tumor necrosis factor-α induces apoptosis via inducible nitric-oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc. Res. 45, 595–602 [DOI] [PubMed] [Google Scholar]
- 32.von Harsdorf R., Li P. F., Dietz R. (1999) Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 99, 2934–2941 [DOI] [PubMed] [Google Scholar]
- 33.Chen H. P., Liao Z. P., Huang Q. R., He M. (2009) Sodium ferulate attenuates anoxia/reoxygenation-induced calcium overload in neonatal rat cardiomyocytes by NO/cGMP/PKG pathway. Eur. J. Pharmacol. 603, 86–92 [DOI] [PubMed] [Google Scholar]
- 34.Murtaza I., Wang H. X., Feng X., Alenina N., Bader M., Prabhakar B. S., Li P. F. (2008) Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 283, 5996–6004 [DOI] [PubMed] [Google Scholar]
- 35.Li P. F., Li J., Müller E. C., Otto A., Dietz R., von Harsdorf R. (2002) Phosphorylation by protein kinase CK2: a signaling switch for the caspase-inhibiting protein ARC. Mol. Cell 10, 247–258 [DOI] [PubMed] [Google Scholar]
- 36.Morris T. E., Sulakhe P. V. (1997) Sarcoplasmic reticulum Ca2+-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic. Biol. Med. 22, 37–47 [DOI] [PubMed] [Google Scholar]
- 37.Diwan A., Matkovich S. J., Yuan Q., Zhao W., Yatani A., Brown J. H., Molkentin J. D., Kranias E. G., Dorn G. W., 2nd (2009) Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J. Clin. Invest. 119, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li P. F., Maasch C., Haller H., Dietz R., von Harsdorf R. (1999) Requirement for protein kinase C in reactive oxygen species-induced apoptosis of vascular smooth muscle cells. Circulation 100, 967–973 [DOI] [PubMed] [Google Scholar]
- 39.Scorrano L., Oakes S. A., Opferman J. T., Cheng E. H., Sorcinelli M. D., Pozzan T., Korsmeyer S. J. (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135–139 [DOI] [PubMed] [Google Scholar]
- 40.Kao J. P., Harootunian A. T., Tsien R. Y. (1989) Photochemically generated cytosolic calcium pulses and their detection by fluo-3. J. Biol. Chem. 264, 8179–8184 [PubMed] [Google Scholar]
- 41.Yao A., Su Z., Nonaka A., Zubair I., Lu L., Philipson K. D., Bridge J. H., Barry W. H. (1998) Effects of overexpression of the Na+-Ca2+ exchanger on [Ca2+]i transients in murine ventricular myocytes. Circ. Res. 82, 657–665 [DOI] [PubMed] [Google Scholar]
- 42.Tajima M., Weinberg E. O., Bartunek J., Jin H., Yang R., Paoni N. F., Lorell B. H. (1999) Treatment with growth hormone enhances contractile reserve and intracellular calcium transients in myocytes from rats with postinfarction heart failure. Circulation 99, 127–134 [DOI] [PubMed] [Google Scholar]
- 43.Sato T., Saito T., Saegusa N., Nakaya H. (2005) Mitochondrial Ca2+-activated K+ channels in cardiac myocytes: a mechanism of the cardioprotective effect and modulation by protein kinase A. Circulation 111, 198–203 [DOI] [PubMed] [Google Scholar]
- 44.Jo H., Noma A., Matsuoka S. (2006) Calcium-mediated coupling between mitochondrial substrate dehydrogenation and cardiac workload in single guinea pig ventricular myocytes. J. Mol. Cell. Cardiol. 40, 394–404 [DOI] [PubMed] [Google Scholar]
- 45.Shannon T. R., Guo T., Bers D. M. (2003) Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 93, 40–45 [DOI] [PubMed] [Google Scholar]
- 46.Kabbara A. A., Allen D. G. (2001) The use of the indicator fluo-5N to measure sarcoplasmic reticulum calcium in single muscle fibres of the cane toad. J. Physiol. 534, 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donath S., Li P., Willenbockel C., Al-Saadi N., Gross V., Willnow T., Bader M., Martin U., Bauersachs J., Wollert K. C., Dietz R., von Harsdorf R., German Heart Failure Network (2006) Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation 113, 1203–1212 [DOI] [PubMed] [Google Scholar]
- 48.Mei Y., Zhang Y., Yamamoto K., Xie W., Mak T. W., You H. (2009) FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc. Natl. Acad. Sci. U.S.A. 106, 5153–5158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dijkers P. F., Medema R. H., Lammers J. W., Koenderman L., Coffer P. J. (2000) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10, 1201–1204 [DOI] [PubMed] [Google Scholar]
- 50.Kops G. J., Dansen T. B., Polderman P. E., Saarloos I., Wirtz K. W., Coffer P. J., Huang T. T., Bos J. L., Medema R. H., Burgering B. M. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 51.Nemoto S., Finkel T. (2002) Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 295, 2450–2452 [DOI] [PubMed] [Google Scholar]
- 52.O'Connor L., Strasser A., O'Reilly L. A., Hausmann G., Adams J. M., Cory S., Huang D. C. (1998) Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jo D. G., Jun J. I., Chang J. W., Hong Y. M., Song S., Cho D. H., Shim S. M., Lee H. J., Cho C., Kim D. H., Jung Y. K. (2004) Calcium binding of ARC mediates regulation of caspase 8 and cell death. Mol. Cell. Biol. 24, 9763–9770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Badorff C., Ruetten H., Mueller S., Stahmer M., Gehring D., Jung F., Ihling C., Zeiher A. M., Dimmeler S. (2002) Fas receptor signaling inhibits glycogen synthase kinase 3β and induces cardiac hypertrophy following pressure overload. J. Clin. Invest. 109, 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohlhaas M., Maack C. (2010) Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation 122, 2273–2280 [DOI] [PubMed] [Google Scholar]
- 56.Pyo J. O., Nah J., Kim H. J., Chang J. W., Song Y. W., Yang D. K., Jo D. G., Kim H. R., Chae H. J., Chae S. W., Hwang S. Y., Kim S. J., Kim H. J., Cho C., Oh C. G., Park W. J., Jung Y. K. (2008) Protection of cardiomyocytes from ischemic/hypoxic cell death via Drbp1 and pMe2GlyDH in cardio-specific ARC transgenic mice. J. Biol. Chem. 283, 30707–30714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nam Y. J., Mani K., Wu L., Peng C. F., Calvert J. W., Foo R. S., Krishnamurthy B., Miao W., Ashton A. W., Lefer D. J., Kitsis R. N. (2007) The apoptosis inhibitor ARC undergoes ubiquitin-proteasomal-mediated degradation in response to death stimuli: identification of a degradation-resistant mutant. J. Biol. Chem. 282, 5522–5528 [DOI] [PubMed] [Google Scholar]