Identification of Salmonella Pathogenicity Island-2 Type III Secretion System Effectors Involved in Intramacrophage Replication of S. enterica Serovar Typhimurium: Implications for Rational Vaccine Design (original) (raw)
ABSTRACT
Salmonella enterica serovars cause severe diseases in humans, such as gastroenteritis and typhoid fever. The development of systemic disease is dependent on a type III secretion system (T3SS) encoded by Salmonella pathogenicity island-2 (SPI-2). Translocation of effector proteins across the _Salmonella_-containing vacuole, via the SPI-2 T3SS, enables bacterial replication within host cells, including macrophages. Here, we investigated the contribution of these effectors to intramacrophage replication of Salmonella enterica serovar Typhimurium using Fluorescence Dilution, a dual-fluorescence tool which allows direct measurement of bacterial replication. Of 32 strains, each carrying single mutations in genes encoding effectors, 10 (lacking sifA, sseJ, sopD2, sseG, sseF, srfH, sseL, spvD, cigR, or steD) were attenuated in replication in mouse bone marrow-derived macrophages. The replication profiles of strains combining deletions in effector genes were also investigated: a strain lacking the genes sseG, sopD2, and srfH showed an increased replication defect compared to single-mutation strains and was very similar to SPI-2 T3SS-deficient bacteria with respect to its replication defect. This strain was substantially attenuated in virulence in vivo and yet retained intracellular vacuole integrity and a functional SPI-2 T3SS. Moreover, this strain was capable of SPI-2 T3SS-mediated delivery of a model antigen for major histocompatibility complex (MHC) class I-dependent T-cell activation. This work establishes a basis for the use of a poly-effector mutant strain as an attenuated vaccine carrier for delivery of heterologous antigens directly into the cytoplasm of host cells.
IMPORTANCE
Live attenuated strains of Salmonella enterica serotype Typhi have generated much interest in the search for improved vaccines against typhoid fever and as vaccine vectors for the delivery of heterologous antigens. A promising vaccine candidate is the Δ_aroC_ Δ_ssaV_ S. Typhi strain, which owes its attenuation mainly to lack of a type III secretion system (SPI-2 T3SS). The SPI-2 T3SS is important for bacterial proliferation inside macrophages, but not all of the effectors involved in this process have been identified. Here, we show that 10 effectors of the related strain S. Typhimurium contribute to intracellular replication in macrophages. Moreover, we establish that a poly-effector mutant strain of S. Typhimurium can have a severe replication defect and maintain a functional SPI-2 T3SS, which can be exploited for delivery of heterologous antigens.
Introduction
Salmonella enterica serovars are Gram-negative bacteria that colonize a wide range of host species. In humans, Salmonella enterica serovars Typhimurium (S. Typhimurium) and Typhi (S. Typhi) usually cause self-limiting gastroenteritis and typhoid fever, respectively. In susceptible mouse strains, infection with S. Typhimurium leads instead to systemic disease, providing a commonly used model system for the study of typhoid fever. Salmonella has two functionally distinct type III secretion systems (T3SS) encoded by Salmonella pathogenicity islands 1 and 2 (SPI-1 and -2). The SPI-1 T3SS is central to the ability of Salmonella to invade nonphagocytic cells through the delivery of effector proteins which trigger extensive actin rearrangements on the surface of host cells (1). The SPI-2 T3SS, on the other hand, is activated intracellularly and is essential for bacterial replication inside host cells (2, 3). Furthermore, the ability of Salmonella to proliferate within phagocytic cells, including macrophages, is a hallmark of systemic disease (4). Bacterial replication occurs in a membrane-bound compartment called the _Salmonella_-containing vacuole (SCV). Effector proteins are translocated across the vacuolar membrane, via the SPI-2 T3SS, into the host endomembrane system and cytoplasm.
Intracellular bacterial growth is usually quantified by determination of net bacterial load within infected cells by counting CFU from cell or tissue lysates plated onto medium. However, CFU counts are the product of both replication and killing undergone by the bacteria. Work in our laboratory based on a dual-fluorescence reporter system, Fluorescence Dilution, permits direct assessment of bacterial replication (5). In both bone marrow-derived (BM) macrophages and the RAW 264.7 macrophage cell line, the SPI-2 T3SS was shown to enable bacterial proliferation rather than survival (5).
Despite the importance of the SPI-2 T3SS in S. Typhimurium replication, little is known about the translocated effectors that mediate this process. A total of 32 effectors were reported to be translocated via the SPI-2 T3SS (6–9), although only a small number have been confirmed to be directly involved in intracellular bacterial growth. The effectors SseF and SseG are essential for localizing SCVs to the Golgi network in epithelial cells. Strains lacking genes for SseF or SseG have a moderate growth defect in RAW macrophages (10, 11) and epithelial cells (12). Another three effectors functioning in SCV membrane dynamics, SifA, SseJ, and SopD2, are also important for bacterial growth. Several studies have reported a strong growth defect of a Δ_sifA_ mutant strain in macrophages (13–18). This can be explained by the finding that the majority of Δ_sifA_ mutant bacteria lose their vacuolar membranes (15) and are killed by the microbicidal environment of the macrophage cytosol (13). SseJ is a glycerophospholipid: cholesterol acyltransferase (GCAT) (19, 20). Since a strain with Δ_sifA_ and Δ_sseJ_ mutations retains its vacuolar membrane (21), SseJ is thought to counter the effects of SifA on the SCV membrane. A Δ_sseJ_ strain has a growth defect in RAW 264.7 cells (17) and peritoneal macrophages (21). SopD2 also regulates SCV membrane dynamics, although its exact function is unknown. A decrease in the bacterial load of a Δ_sopD2_ strain compared to a wild-type strain was shown in RAW 264.7 macrophages (22), but a second study in the same cell type did not detect a growth defect (18). In addition, our laboratory found recently that the effector SseL contributes to intracellular replication in macrophages (23). A recent study of the contribution of some SPI-2 T3SS effectors to replication found that, in RAW 264.7 macrophages, strains lacking the effector genes sifB, steC, or spvB or sseK1 sseK2 sseK3 had significantly reduced growth levels (24). In previous studies, however, intracellular growth defects were not detected in strains carrying deletions in these genes (17, 25, 26).
In this study, we determined the contributions of individual effectors of the SPI-2 T3SS to intramacrophage replication of S. Typhimurium. We found that approximately 30% of effectors contribute to intracellular replication. Based on these results, we combined mutations to generate a triple-mutation (Δ_sseG_ Δ_sopD2_ Δ_srfH_) strain with a replication defect similar to that of an SPI-2 null strain. This strain retains a functional T3SS and was used to deliver a heterologous antigen for MHC class I (MHC-I)-dependent T-cell activation.
RESULTS
Contribution of SPI-2 T3SS effectors to replication in mouse bone marrow-derived macrophages.
To determine the contribution of effectors of the SPI-2 T3SS to intracellular replication in macrophages, replication of wild-type and mutant strains was assessed by Fluorescence Dilution using a second-generation plasmid, pFCcGi, from which expression of mCherry is constitutive and that of green fluorescent protein (GFP) is under the control of an arabinose-inducible promoter (Fig. 1A). Bacteria carrying pFCcGi were initially grown in vitro in the presence of arabinose to drive production of GFP. Upon infection of macrophages with these bacteria, in the absence of arabinose, the preformed pool of GFP becomes halved with each bacterial cell division event. Therefore, bacterial replication is measured by dilution of the green fluorescence, with bacterial cells being detected on the basis of mCherry fluorescence (Fig. 1B). Use of pFCcGi-based Fluorescence Dilution as a reporter of bacterial replication was validated by measuring in vitro growth of bacteria. Dilution of GFP enabled accurate measurement of bacterial replication over 6 generations (data not shown). Moreover, there was no detectable effect on bacterial cell division time in macrophages, in contrast to the minor increase observed for pDiGc in a previous study (5).
FIG 1 .
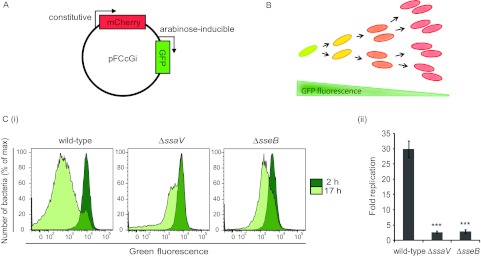
Use of Fluorescence Dilution to measure the impact of the SPI-2 T3SS on S. Typhimurium replication in bone marrow-derived macrophages. (A) Expression of fluorescent proteins from the pFCcGi plasmid is constitutive for mCherry and under the control of an arabinose-inducible promoter for gfp. (B) In the absence of arabinose, levels of GFP fluorescence in preinduced bacteria become diluted as successive rounds of cell division occur. (Ci and Cii) Macrophages were infected with S. Typhimurium strains carrying pFCcGi that were grown with arabinose prior to infection. Infections were carried out in the absence of arabinose, and the cells were lysed at 2 h and 17 h postinoculation. Replication of recovered intracellular bacteria was measured by flow cytometry analysis of GFP Fluorescence Dilution (n = 10,000 events analyzed). (Ci) Representative histograms of GFP fluorescence in intracellular populations of wild-type, Δ_ssaV_, and Δ_sseB_ mutant strains. (Cii) Fold replication values were calculated from the geometric means of GFP fluorescence as the ratio of replication at 17 h to replication at 2 h. Data are expressed as means ± standard errors of the means (SEM) of the results of five independent experiments. P values were obtained by Student’s t test (***, P < 0.001) relative to the wild-type strain results.
The replication rate of two S. Typhimurium SPI-2-T3SS null strains, Δ_ssaV_ and Δ_sseB_ (encoding a secretion apparatus component and translocon protein, respectively), was compared to that of a wild-type strain in bone marrow-derived macrophages extracted from C57 BL/6 mice. At 2 h and 17 h postuptake, dilution of GFP fluorescence in bacteria recovered from macrophages was analyzed by flow cytometry (Fig. 1Ci). Replication was quantified from the geometric mean of GFP fluorescence by calculating the fold change between the results determined at the two time points (Fig. 1Cii). Extensive replication of wild-type S. Typhimurium occurred in these macrophages over 17 h of infection, as illustrated by the shift in GFP fluorescence intensity, while replication of SPI-2-T3SS-deficient strains was much reduced. The number of generations undergone by the bacterial populations was deduced by Fluorescence Dilution and was higher than that reported in our previous paper, which can be attributed to the presence of pFCcGi in place of pDiGc (5). The mean rate of division of the wild-type strain (approximately 0.3 generations/h) was 3-fold higher than that of the two strains lacking a functional SPI-2 T3SS (approximately 0.1 generations/h). The large difference between the control strains (wild-type and SPI-2 T3SS null strains) in GFP fluorescence enabled relatively subtle contributions to replication to be detected in effector mutant strains.
Using the λ Red recombinase method (27), we generated 24 single-gene deletions to obtain, together with previously constructed strains, a total collection of 32 SPI-2 T3SS mutant strains representing all known effectors. Three genes for SPI-1 T3SS effectors (sopD, sptP, and avrA) were included in the study, as they have been reported to be secreted via the SPI-2 T3SS (6, 8). The 32 mutant strains were transformed with pFCcGi, and their intramacrophage replication rate was analyzed (Fig. 2). In each experiment, values of mean fold change in GFP fluorescence between 2 h and 17 h were calculated and normalized to that of the wild-type strain to obtain a replication index (RI), which, for the wild-type strain, was 1.0. The RI of 10 strains was significantly reduced compared to that of the wild-type strain. Those 10 strains contained mutations in sifA (0.29), sseJ (0.41), sopD2 (0.48), sseG (0.40), sseF (0.50), srfH (0.48), sseL (0.72), spvD (0.61), cigR (0.68), or steD (0.69) (Fig. 2). Displaying the strongest replication defects among these (values below 0.5) were five mutant strains with mutations in sifA, sseJ, sopD2, sseG, and sseF that were previously described to contribute to intramacrophage growth as assessed by CFU counts (10, 11, 13, 15–17, 22).
FIG 2 .

Contribution of individual SPI-2 T3SS effectors to S. Typhimurium intracellular replication. Fluorescence Dilution analysis of the replication of S. Typhimurium strains carrying deletions in genes of effectors of the SPI-2 T3SS was done in bone marrow-derived macrophages over 17 h. The color code used to group SPI-2 T3SS effectors according to their function is as follows: SCV membrane dynamics, green; SCV localization, yellow; cell migration, red; ubiquitin pathways, purple; unknown function, dark gray; actin cytoskeleton, brown; host immune signaling, pink; SPI-1 translocated effectors, turquoise. Data are expressed as the means ± SEM of the results of 3 to 30 independent experiments (N) and were normalized to wild-type values. Statistical analysis was performed with one-way analysis of variance (ANOVA) and a post hoc Dunnett test (*, P < 0.05; **, P < 0.01) relative to the wild-type strain results.
Effect of cumulative mutations in genes of SPI-2 T3SS effectors on replication.
Next we determined if combining deletions in certain genes could result in stronger replication defects. Some effectors of the SPI-2 T3SS have similar functions or are known to contribute to the same SPI-2-mediated process inside host cells. Therefore, as a first approach, mutations were combined in two or more genes related by function (Fig. 3A). Three groups were selected: (i) effectors involved in SCV membrane dynamics (SifA, SseJ, SopD2, and PipB2), based on the observed impact that the majority of these effectors have on replication; (ii) effectors sharing E3 ubiquitin ligase activity (SspH1, SspH2, and SlrP), to account for possible redundancy in their function; and (iii) effectors SseK1, SseK2, and SseK3, because, although their function is unknown, their amino acid sequences bear striking similarities (26, 28), suggesting they might have similar functions. We did not measure the replication rate of an sseF sseG double mutant, as this has been shown previously to be indistinguishable from single mutants in terms of virulence attenuation and intracellular proliferation (29).
FIG 3 .
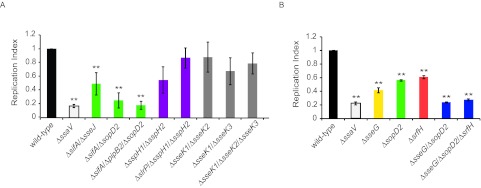
Replication of S. Typhimurium poly-effector mutant strains. Fluorescence Dilution analysis of intracellular replication was carried out over 17 h in bone marrow-derived macrophages. (A) Fold replication of strains combining deletions in genes involved in similar functions. Data are expressed as the means ± SEM of the results of 3 to 8 independent experiments and were normalized to wild-type values. (B) Fold replication of strains combining deletions in the genes associated with different activities within host cells. Data are expressed as the means ± SEM of the results of four independent experiments and were normalized to wild-type values. Statistical analysis was performed with a one-way ANOVA and post hoc Dunnett test (**, P < 0.01) relative to the wild-type strain results. Statistical differences (**, P < 0.01) between Δ_sseG_ Δ_sopD2_ or Δ_sseG_ Δ_sopD2_ Δ_srfH_ and each of the corresponding single-mutation strains (Δ_sseG_, Δ_sopD2_, and Δ_srfH_) were also confirmed. The color code used is as follows: SPI-2 T3SS effectors involved in SCV membrane dynamics, green; SCV localization, yellow; cell migration, red; ubiquitin pathways, purple; unknown function, dark gray.
Three strains were constructed that carry a sifA mutation but for which the vacuolar membrane is maintained through the counteracting and stabilizing effect of additional mutations: strains Δ_sifA_ Δ_sseJ_, Δ_sifA_ Δ_sopD2_, and Δ_sifA_ Δ_sopD2_ Δ_pipB2_ (18, 21). The three strains showed significantly less replication than the wild-type strain (RI = 0.49, 0.25, and 0.17, respectively) (Fig. 3A). Despite the presence of a vacuolar membrane, normal replication levels were not restored in these strains. On the other hand, none of these strains had a greater replication defect than the strain with the single sifA mutation (Fig. 2). Genes encoding effectors with E3 ubiquitin ligase activity were also investigated using strains carrying deletions in two genes (Δ_sspH1_ Δ_sspH2_) or three genes (Δ_slrP_ Δ_sspH1_ Δ_sspH2_). There was no additive effect on the replication defect caused by additional mutations, suggesting that this family of effectors does not contribute to intracellular replication (Fig. 2 and 3A). Likewise, combining mutations in genes of the sseK family (Δ_sseK1_ Δ_sseK2_, Δ_sseK1_ Δ_sseK3_, or Δ_sseK1_ Δ_sseK2_ Δ_sseK3_) had no significant effect on replication (Fig. 2 and 3A). This suggests that the SseK effectors have functions other than in bacterial replication, in agreement with previous findings (26).
As a second approach, mutations in genes associated with different functions were combined. A strain containing mutations in sseG and sopD2 replicated significantly less (RI = 0.24) than the corresponding single mutants (Fig. 3B) and similarly to the SPI-2-deficient Δ_ssaV_ strain (RI = 0.23). Introduction of a third mutation to this strain resulted in strain Δ_sseG_ Δ_sopD2_ Δ_srfH_, which had a replication defect similar to that of the double-mutation strain (RI = 0.27). The replication defect of the Δ_sseG_ Δ_sopD2_ mutant was complemented by introduction of a plasmid carrying wild-type alleles of both genes, followed by measurement of bacterial growth in bone marrow-derived macrophages at between 2 h and 17 h by CFU enumeration (see Fig. S1 in the supplemental material).
Competitive index analysis of Δ_sseG_ Δ_sopD2_ and Δ_sseG_ Δ_sopD2_ Δ_srfH_ mutant strains.
A competitive index (CI) analysis (30) was carried out in C57 BL/6 mice following intraperitoneal (i.p.) inoculation to measure the virulence attenuation of the mutant strains compared to wild-type S. Typhimurium in splenic macrophages (14). Mice were given a 1:1 mixture of the wild-type strain and either of the mutant strains. After 3 days, bacteria were recovered from the spleens of mice and the bacterial loads were determined for each strain by CFU enumeration. As expected, the Δ_ssaV_ mutant strain was strongly attenuated in virulence compared to the wild-type strain (Fig. 4). The Δ_sseG_ Δ_sopD2_ double-mutation and Δ_sseG_ Δ_sopD2_ Δ_srfH_ triple-mutation strains were also attenuated in virulence, although not to the extent seen with the Δ_ssaV_ mutant strain (Fig. 4).
FIG 4 .

Competitive index (CI) analysis of Δ_sseG_ Δ_sopD2_ and Δ_sseG_ Δ_sopD2_ Δ_srfH_ strains. C57 BL/6 mice were inoculated by intraperitoneal (i.p.) injection (5 × 105 CFU) or intragastric (i.g.) inoculation (3 × 108 CFU) with equal amounts of wild-type and Δ_ssaV_, Δ_sseG_ Δ_sopD2_, or Δ_sseG_ Δ_sopD2_ Δ_srfH_ mutant strains. Bacteria were recovered from infected spleens 3 days (i.p.) or 5 days (i.g.) postinoculation. CI values were calculated as the ratio of wild-type strain to mutant strain recovered (Output) divided by the ratio of wild-type strain to mutant strain present in the inoculum (Input). The scatter plot displays values obtained for individual mice, and the means are indicated (long horizontal lines). P values obtained by Student’s t test confirmed that all CI values are statistically different from 1.0 (**, P < 0.01).
To measure virulence attenuation following the natural route of infection, a CI analysis was also done over 5 days following intragastric (i.g.) inoculation. The mutant strains had lower mean CI values after oral challenge than after inoculation by the i.p. route (Fig. 4). Moreover, although the introduction of a third mutation (Δ_srfH_) had no measurable effect on replication in macrophages, it resulted in CI values lower than those seen with the double-mutation strain.
The availability of a poly-effector mutant strain of Salmonella with a replication defect similar to that of an SPI-2 T3SS null strain, but with a functional secretion apparatus, might provide a means for delivery of heterologous antigens directly into the cytosol of host antigen-presenting cells against the background of a safe delivery system. Therefore, we next investigated the functionality of the SPI-2 T3SS in the triple-effector mutant strain.
Δ_sseG_ Δ_sopD2_ Δ_srfH_ mutant bacteria reside in vacuoles and retain a functional SPI-2 T3SS.
SCVs acquire some characteristics of late endosomes, including the presence of lysosomal glycoproteins such as LAMP-1 in the SCV membrane (31). LAMP-1 labeling of vacuoles containing Δ_sseG_ Δ_sopD2_ Δ_srfH_ bacteria was indistinguishable from the labeling seen with those containing wild-type bacteria, suggesting the presence of a vacuolar membrane around enclosing bacteria of the triple-mutation strain (Fig. 5Ai). To confirm the integrity of vacuoles enclosing the triple mutant, HeLa cells were infected with GFP-expressing wild-type, Δ_sifA_, or Δ_sseG_ Δ_sopD2_ Δ_srfH_ bacteria for 10 h and were subsequently exposed to digitonin at a concentration which permeabilizes the plasma membrane but not the vacuolar membrane (12), followed by labeling with an anti-Salmonella antibody. Thus, only Salmonella bacteria that were not enclosed in a vacuole were labeled with the antibody, allowing a distinction between intravacuolar and cytosolic bacteria to be made. The Δ_sifA_ mutant strain provides a positive control for loss of the vacuolar membrane, as the majority of the bacteria become cytosolic by 8 h postuptake (15). The majority (65.6%) of wild-type bacteria maintained vacuolar membrane integrity, while only a small proportion (28.8%) of Δ_sifA_ mutant bacteria remained enclosed within intact vacuoles (Fig. 5Aii), in accordance with previously published data (25). The proportion of the triple-mutation strain within vacuoles was very similar to that of the wild type (66.8%), confirming that the mutations present in this strain do not destabilize the SCV (Fig. 5Aii).
FIG 5 .
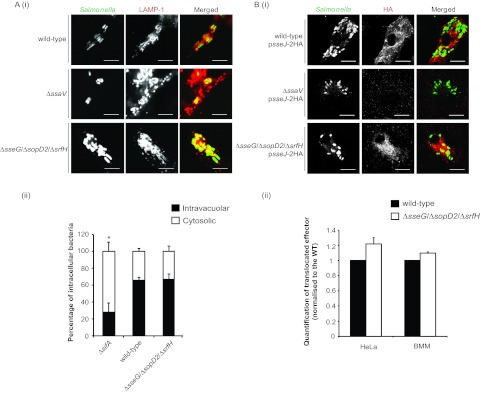
Vacuolar integrity and SPI-2 T3SS functionality in intracellular S. Typhimurium Δ_sseG_ Δ_sopD2_ Δ_srfH_. (Ai and Aii) Intravacuolar localization of Δ_sseG_ Δ_sopD2_ Δ_srfH_ in epithelial cells. (Ai) Confocal microscopy images of LAMP-1 labeling of HeLa cells infected with S. Typhimurium wild-type, Δ_ssaV_, or Δ_sseG_ Δ_sopD2_ Δ_srfH_ strains for 14 h. Cells were fixed and immunolabeled with anti-Salmonella (green in merged images) and anti-LAMP-1 (red in merged images) antibodies. Scale bars represent 5 µm. (Aii) HeLa cells infected for 10 h with GFP-expressing S. Typhimurium wild-type, Δ_sifA_, or Δ_sseG_ Δ_sopD2_ Δ_srfH_ strains were treated with digitonin to selectively permeabilize the plasma membrane and labeled with anti-Salmonella antibody. Percentages of intravacuolar and cytosolic bacteria were calculated based on differential labeling. (Bi and Bii) Translocation of an SPI-2 T3SS effector from S. Typhimurium Δ_sseG_ Δ_sopD2_ Δ_srfH_. Bone marrow-derived macrophages or HeLa cells were infected for 17 h or 14 h, respectively, with the indicated S. Typhimurium strains expressing HA-tagged SseJ from the plasmid pWSK29. (Bi) Confocal microscopy images of SseJ-2HA translocation in macrophages. Cells were fixed and immunolabeled with anti-Salmonella (green in merged images) and anti-HA (red in merged images) antibodies. Scale bars represent 5 µm. (Bii) Quantification of translocated SseJ-2HA by flow cytometry analysis of infected cells immunolabeled with anti-Salmonella and anti-HA antibodies. Data were calculated from the geometric means of fluorescence associated with anti-HA labeling and are expressed as means ± SEM of the results of three independent experiments. The Δ_ssaV_ strain was included as a negative control for effector translocation in each experiment (P < 0.001). P values were obtained by Student’s t test (*, P < 0.05) relative to the wild-type (WT) strain results.
The ability of the Δ_sseG_ Δ_sopD2_ Δ_srfH_ strain to translocate effectors via the SPI-2 T3SS into the cytosol of host cells was investigated with strains expressing hemagglutinin (HA)-tagged SseJ. The Δ_ssaV_ strain, which does not translocate effectors, was used as a negative control. As seen by immunofluorescence microscopy, levels of SPI-2 T3SS-mediated delivery of SseJ from the triple-mutation strain were similar to those of the wild-type strain in bone marrow-derived macrophages infected for 17 h (Fig. 5Bi). Translocation of a second SPI-2 T3SS effector, SseF, was also confirmed in macrophages (see Fig. S2 in the supplemental material). Flow cytometry analysis of infected cells was also done to quantify the levels of translocated SseJ. Bone marrow-derived macrophages or HeLa cells were infected for 17 h or 14 h, respectively, and labeled with an antibody against the translocated HA epitope. Similar amounts of SseJ were translocated by the Δ_sseG_ Δ_sopD2_ Δ_srfH_ and the wild-type strains in both cell types (Fig. 5Bii).
T-cell activation following SPI-2 T3SS-mediated antigen delivery.
We next tested whether a heterologous antigen produced in the Δ_sseG_ Δ_sopD2_ Δ_srfH_ strain could be delivered into the cytosol of dendritic cells (DC) through the SPI-2 T3SS and presented by MHC-I molecules (Fig. 6). Bone marrow-derived dendritic cells extracted from C57 BL/6 mice were infected with wild-type, Δ_ssaV_, or Δ_sseG_ Δ_sopD2_ Δ_srfH_ strains harboring a plasmid expressing the T-cell ovalbumin-derived SIINFEKL antigen fused to SseJ under the control of the sseA promoter (p3631) (32). Wild-type S. Typhimurium that did not carry the plasmid was used as a negative control. Purified SIINFEKL peptide added to cells infected with the same strain was included as a positive control. Following the killing of extracellular bacteria with gentamicin, infected cells were incubated for 22 h with the murine B3Z T-cell hybridoma, which is specific for SIINFEKL in the context of H-2Kb and which, upon activation, expresses the lacZ reporter gene (33). β-Galactosidase activity was used as a measure of T-cell stimulation in the assay. Cells infected with the wild-type or Δ_sseG_ Δ_sopD2_ Δ_srfH_ strain expressing the antigen showed a significant increase in β-galactosidase activity (Fig. 6) similar to that obtained with the positive control. On the other hand, the level of β-galactosidase activity following infection with the Δ_ssaV_ mutant carrying p3631 was similar to the background levels obtained with wild-type bacteria lacking p3631. This shows that T-cell stimulation is dependent on SPI-2 T3SS-mediated translocation of the SseJ-SIINFEKL fusion. Therefore, we conclude that despite the strong virulence attenuation caused by loss of sseG, sopD2, and srfH, this strain retains the ability to deliver a modified effector into the cytosol of dendritic cells and to stimulate T-cell activity following MHC-I antigen presentation.
FIG 6 .

T-cell stimulation by SPI-2-mediated delivery of a model antigen from S. Typhimurium Δ_sseG_ Δ_sopD2_ Δ_srfH_. Bone marrow-derived dendritic cells (BM-DC) were infected with indicated S. Typhimurium strains harboring the plasmid pWSK29 P sseA sseJ::OVA::HA (p3631). Cells infected with wild-type S. Typhimurium, in the absence of the plasmid, were used as a negative control. Stimulation with the SIINFEKL peptide was used as a positive control. Infected cells were cocultured for 22 h with B3Z T-cell hybridoma reporter cells (ratio of BM-DC to T cells of 1:4) followed by a 6-h incubation in the presence of the β-galactosidase substrate chlorophenyl red β-galactopyranoside. β-Galactosidase activity was measured colormetrically by the amount of substrate converted at 595 nm. Results were normalized to values obtained with the negative control and are expressed as means ± SEM of the results of four independent experiments, each performed in triplicate. P values were obtained by Student’s t test relative to the wild-type negative-control results (*, P < 0.05; **, P < 0.01).
DISCUSSION
In this work, we have compared the intracellular replication characteristics of 32 mutant strains of S. Typhimurium lacking individual effectors of the SPI-2 T3SS. Three of these mutations were then combined to make a triple-mutation strain with strong intramacrophage replication and virulence defects. Despite this attenuated phenotype, the strain displayed SPI-2 T3SS-dependent delivery of a model epitope into the cytosol of dendritic cells, which stimulated CD8-T-cell responses. This study was influenced by the results of clinical trials using the S. Typhi strain M01ZH09 Δ_aroC_ Δ_ssaV_, which has shown considerable promise as a potential single-oral-dose vaccine against typhoid (34–39). This strain owes its attenuation mostly to SPI-2 T3SS deficiency, which results in the reduction of replication of the strain in host cells and systemic spread (36). It might be feasible to achieve a similar profile of attenuation and immunogenicity through mutation of key effector genes in which the T3SS remains intact and competent for delivery of modified effectors into host cells. Such a strain could be exploited for cytosolic delivery of heterologous antigens in antigen-presenting cells to generate immunity against both Salmonella and an unrelated pathogen.
Others have reported the use of the Salmonella SPI-2 T3SS for delivery of heterologous antigens (40–42) and anticancer agents (43) directly into the cytoplasm of host cells. Those studies were successful in showing good levels of MHC-I presentation and CD8+ T-cell recognition. Previous studies have also focused on comparing the efficiencies of various _in vivo_-activated promoters and effector fusions of the SPI-2 T3SS in driving heterologous antigen expression and delivery from an attenuated purD htrA double-mutation strain as an attenuated carrier (32, 42). Antigens fused to SseJ were identified as eliciting a potent T-cell response (32).
Our study represents a comprehensive analysis of replication defects associated with strains lacking individual SPI-2 T3SS effectors in which mutant strains were simultaneously analyzed under the same conditions. A previous study tested the intracellular growth of a smaller group of effector mutant strains, comprising 11 single deletions (Δ_sifA_, Δ_sifB_, Δ_sseF_, Δ_sseG_, Δ_srfH_, Δ_sspH2_, Δ_slrP_, Δ_steA_, Δ_steC_, Δ_gogB_, and Δ_spvB_), as well as strains Δ_sseF_ Δ_sseG_ and Δ_sseK1_ Δ_sseK2_ Δ_sseK3_ (24). In RAW 264.7 macrophages, over 24 h, CFU counts showed a growth defect of Δ_sifA_, Δ_sifB_, Δ_steC_, Δ_spvB_, and Δ_sseK1_ Δ_sseK2_ Δ_sseK3_ mutant strains, although strains carrying deletions in sseF or sseG or both sseF and sseG were not significantly attenuated in those assays (24). With the exception of the Δ_sifA_ strain results, these findings are not in accord with our results or those of other studies (10, 11, 17, 25, 26, 44). It is possible that the discrepancies might be explained by differences in the types of macrophage that were used, as it was already reported that the extent and nature of replication of Salmonella are different in the macrophage-like cell line RAW264.7 or in primary bone marrow-derived macrophages (5). In our study, a significant replication defect was detected for 10 strains, indicating that sifA, sseJ, sopD2, sseF, sseG, and srfH are largely responsible for SPI-2 T3SS-mediated intracellular replication and that sseL, spvD, cigR, and steD also contribute to this process. The use of Fluorescence Dilution with the sifA mutant bacteria shows that its growth defect is not just the consequence of bacterial killing in the cytosol of macrophages, as a result of vacuolar loss (13), but that bacteria remaining inside SCVs undergo diminished replication. Moreover, the effect of deleting srfH, spvD, cigR, or steD on replication had not been previously demonstrated. SrfH is involved in migration of infected cells and is required for long-term systemic infection, but CFU analysis in macrophages had not previously revealed a function for SrfH in intracellular growth (45, 46). The three remaining effectors contributing to intracellular replication (SpvD, CigR, and SteD) were identified recently using mass spectrometry of secreted proteins (47), and their function is not yet known. Among the effectors with known function, those affecting replication appear to be involved mainly in maintenance of the SCV membrane and the localization of the SCV in the cell. This is not surprising, as fusion of vesicles with the SCV is likely to provide a source of nutrients for the dividing bacteria. Similarly, the spatial localization of vacuoles might facilitate their interactions with components of the host cell that contribute to bacterial replication.
The combined deletion of sseG, sopD2, and srfH in S. Typhimurium was sufficient to generate a strain that showed a strong replication defect in macrophages and virulence attenuation in mice. However, its level of virulence was still greater than that of the Δ_ssaV_ SPI-2 null mutant, which indicates that additional mutations may need to be made to further reduce its virulence. Such mutations could be selected on the basis of CI tests involving the remaining 22 effector mutants. Strains that do not have replication defects in macrophages, but which are attenuated for virulence, might carry mutations in genes whose products are involved in immune suppression, host cell cytotoxicity, and bacterial spread. Indeed, Salmonella has been reported to interfere with host MHC-I and MHC-II antigen presentation (48–50) as well as to inhibit T-cell proliferation (51, 52). Mutation of all effector genes would be predicted to generate a strain with levels of virulence attenuation comparable to that of the Δ_ssaV_ mutant while retaining a functional secretion apparatus. However, besides the technical challenge of producing a strain with 32 gene deletions, it is possible that not all mutations would contribute to the optimal efficacy of a live attenuated vaccine. Indeed, at least one effector (SteC) has been reported to attenuate virulence (53). Therefore, some effectors might have activities that could be useful to preserve in a vaccine. Further work on functional analysis of effectors is necessary to achieve the right balance between sufficient attenuation for safe administration and strong immunogenicity (54). Regarding the applicability of this knowledge to typhoid vaccine design, it is important to bear in mind that not all S. Typhimurium SPI-2 T3SS effectors are present in S. Typhi or S. Paratyphi (55). The present study nonetheless establishes proof of concept that a poly-effector mutant strain of Salmonella can have a severe replication defect and still be able to deliver modified heterologous antigen-containing effectors into the host cell and in sufficient amounts to generate an MHC-I-dependent T-cell response.
MATERIALS & METHODS
Bacterial strains and growth conditions.
S. Typhimurium strains used in this study are listed in Table S1 in the supplemental material. Bacteria were routinely grown in LB (Lennox L. Broth base), and, where appropriate, growth medium was supplemented with the following antibiotics at the indicated concentrations: carbenicillin (50 µg/ml), kanamycin (50 µg/ml), and chloramphenicol (35 µg/ml). For Fluorescence Dilution assays, bacterial strains carrying pFCcGi were grown in magnesium minimal medium (Mg-MES) containing 170 mM 2-(_N_-morpholino)ethanesulfonic acid (MES) at pH 5.0, 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 1 mM KH2PO4, 10 mM MgCl2, 38 mM glycerol, and 0.1% Casamino Acids (56), supplemented with 50 µg/ml carbenicillin and 0.2% l-arabinose to allow production of GFP.
Bacterial mutagenesis.
S. Typhimurium 12023 mutant strains were constructed using a one-step λ Red recombinase chromosomal inactivation system (27). Primer sequences designed for mutagenesis and for confirmation by PCR of the deletion genotype are listed in Table S2 in the supplemental material. Mutagenesis primers were designed to amplify an antibiotic resistance cassette and contained a 40-nucleotide sequence homologous to regions flanking the target gene on the chromosome.
Plasmids.
Plasmids used in this study are listed in Table S1 in the supplemental material. For bacterial mutagenesis, plasmid pKD3 or pKD4 was used as the template to amplify the chloramphenicol or kanamycin resistance gene, respectively, and amplification reaction products were electroporated into pKD46-containing bacteria expressing the λ Red recombinase enzyme (27). Clean deletions by excision of antibiotic resistance cassettes were generated with FLP recombinase expressed from pCP20 (27). For the construction of pFCcGi, mCherry was amplified from pmCherry using primers C1 and C2 (see Table S2 in the supplemental material) and introduced into the vector pFPV25.1, previously cleaved with XbaI and HindIII to release gfpmut3a. The vector was subsequently cleaved with EcoRV to allow insertion of a PBAD::gfpmut3a promoter fusion. This fragment was obtained from pGFP-arabinose (pGara), which was digested with ClaI and HindIII. pFCcGi was electroporated into S. Typhimurium 12023 wild-type and mutant strains. The plasmid pACYC_sseG_, carrying the sseG gene under the control of the tetracycline resistance (Tetr) promoter of pACYC184 (12), was used to construct pACYC_sseG_/sopD2. The primers pACYC-SopD2 FW and pACYC-SopD2 REV (see Table S2 in the supplemental material) were used to amplify sopD2 from S. Typhimurium 12023, introducing BamHI and SalI restriction sites flanking the gene. The fragment was introduced immediately downstream of sseG on the vector, also under the control of the Tetr promoter.
Cell culture.
RAW 264.7 murine macrophage-like cells and HeLa cells were grown in Dulbecco’s modified Eagle medium (DMEM; PAA Laboratories) supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (FCS; PAA Laboratories) and 2 mM glutamine at 37°C in 5% (vol/vol) CO2. The B3Z CD8+ T-cell hybridoma was grown in RPMI 1640 (Invitrogen) supplemented with 10% FCS, 2 mM glutamine, and 0.05 M β-mercaptoethanol at 37°C in 5% CO2. Primary bone marrow-derived macrophages or dendritic cells were extracted from C57 BL/6 mice (Charles River). Cells were recovered from mice tibias and femurs (57). Red blood cells were lysed with 0.83% NH4Cl, and the remaining undifferentiated bone marrow cells were cultured at 37°C in 5% CO2 in complete medium consisting of RPMI 1640 supplemented with 10% FCS, 2 mM glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 0.05 M β-mercaptoethanol, 100 U/ml penicillin-streptomycin, and 20% (vol/vol) L929-cell conditioned medium (National Institute of Medical Research) or 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Sigma-Aldrich) for macrophage or dendritic cell differentiation, respectively. After 3 days of culture, further fresh complete medium was added to the growing cells. On day 6, cells were washed and seeded in complete medium without antibiotic and incubated overnight before bacterial challenge.
Bacterial replication assay in macrophages by Fluorescence Dilution.
Bone marrow-derived macrophages were seeded at a density of 1 × 106 cells per well in 6-well plates 24 h prior to infection. Macrophages were infected at a multiplicity of infection (MOI) of approximately 10:1 with bacterial strains carrying pFCcGi that were grown to stationary phase and opsonized as previously described (15). Plates were centrifuged at 110 × g for 5 min and incubated at 37°C in 5% CO2 for 25 min. Cells were washed and incubated for a further 1 h with medium containing 100 µg/ml gentamicin to kill extracellular bacteria. This was replaced with medium containing 20 µg/ml gentamicin for the remainder of the experiment. No antibiotic selection was necessary for maintenance of the pFCcGi plasmid during infection assays. At specific time points, cells were washed and lysed with 0.1% (vol/vol) Triton X-100–phosphate-buffered saline (PBS) to release intracellular bacteria. Cell lysates were pelleted at 13,000 × g and resuspended in PBS solution. Fluorescence levels of the recovered bacteria were analyzed by flow cytometry. For each sample, 10,000 bacterial events were acquired on an LSR II flow cytometer (BD) or an LSR Fortessa flow cytometer (BD) using FACSDiva software. GFP and mCherry fluorescence intensities were detected at 525/50 nm and 610/20 nm, respectively. Data were analyzed with FlowJo Software version 8.1.1 (Treestar). To analyze GFP Fluorescence Dilution, bacteria were gated on an mCherry-positive signal. Replication was measured as the fold change in the geometric mean of GFP fluorescence intensity between two time points.
Bacterial growth assay of CFU.
Bone marrow-derived macrophages were seeded at a density of 2 × 105 in 24-well plates and infected with bacterial strains as described above. Upon cell lysis at selected time points, CFU numbers were determined by plating serial dilutions of the lysate on LB agar. Bacterial growth was measured as the fold change in CFU/ml between two time points.
Competitive infections.
Infection studies were carried out with 6-to-8-week-old female C57BL/6 mice (Charles River). Groups of 4 mice were inoculated intraperitoneally (i.p.) or intragastrically (i.g.) with 0.2 ml of bacteria suspended in PBS solution. The bacteria inoculum was 5 × 104 CFU (i.p.) or 3 × 108 CFU (i.g.) of combined S. Typhimurium wild-type and mutant strains (2.5 × 104 [i.p.] or 1.5 × 108 [i.g.] of each strain, at a ratio of 1:1). The CFU of each strain in the inoculum (input) was quantified by plating dilution series on LB agar and using antibiotic resistance to distinguish between strains. Mice were sacrificed after 3 days (i.p.) or 5 days (i.g.), and dilution series of spleen lysates were plated on LB agar for enumeration of CFU (output), using antibiotic resistance to differentiate strains. Values for competitive infections (CI) were calculated as the ratio of mutant to wild type in the output divided by that in the input.
Immunofluorescence microscopy and antibodies.
Bone marrow-derived macrophages or RAW 264.7 macrophages were seeded onto glass coverslips in 24-well plates at a density of 1 × 105 cells per well and infected as described above. HeLa cells were seeded onto glass coverslips in 24-well plates at a density of 5 × 104 cells per well at 24 h before infection. To induce bacterial invasion, bacterial cultures were diluted 1:33 in fresh LB medium and incubated at 37°C until an optical density at 600 nm (OD600) of 1.5 to 2.0 was reached. Bacteria were diluted in warm Earl’s balanced salt solution (EBSS; Invitrogen) and added to cell monolayers at an MOI of approximately 50. Invasion was allowed to proceed for 15 min at 37°C in 5% CO2 followed by incubation of the cells with 100 µg/ml gentamicin for 1 h under the same conditions. The concentration of gentamicin was subsequently reduced to 20 µg/ml for the remainder of the experiment. At selected times, cell monolayers were washed, fixed using 4% (wt/vol) paraformaldehyde (PFA)-PBS for 15 min at 25°C, and quenched with 10 mM NH4Cl. Cells were permeabilized with 0.1% (wt/vol) saponin and labeled with antibodies before observation using a confocal laser scanning microscope (Zeiss Axiovert LSM510). For quantification of intravacuolar bacteria, cells were permeabilized with 15 to 20 µg/ml digitonin for 1 min prior to antibody labeling.
Quantification of effector translocation.
HeLa cells or bone marrow-derived macrophages were seeded onto 6-well plates at a density of 2.5 × 105 or 1 × 106, respectively, and infected with strains expressing a HA epitope-tagged effector as described above. At specific time points, cells were washed and detached from the plate by using 0.05% (wt/vol) trypsin for HeLa cells or by scraping the macrophages. Recovered cells were pelleted at 200 × g for 5 min and resuspended in 3% (wt/vol) PFA–PBS for 15 min at 25°C. Cells in suspension were permeabilized in 0.1% (vol/vol) Triton X-100–PBS or 0.1% (wt/vol) saponin–PBS for HeLa cells or macrophages, respectively, for 10 min and labeled with primary antibody for 1 h and secondary antibody for 30 min in solutions containing 10% horse serum–PBS. Flow cytometry detection of the translocated epitope-tagged effector was done on a two-laser, four-color FACSCalibur flow cytometer (BD Biosciences). For each sample, at least 10,000 cells were analyzed. Collected data were analyzed with FlowJo software version 8.1.1 (Treestar).
Antibodies.
Anti-salmonella goat polyclonal antibody CSA-1 (Kirkegaard and Perry Laboratories) was diluted at 1:200 (immunofluorescence microscopy) or 1:400 (flow cytometry). Anti-HA mouse monoclonal antibody HA.11 (Covance) was diluted at 1:200 (immunofluorescence microscopy) or 1:400 (flow cytometry). Anti-LAMP-1 mouse monoclonal antibody H4A3 (Developmental Studies Hybridoma Bank) was diluted at 1:200 for immunofluorescence microscopy. Anti-HA rat monoclonal antibody 3F10 (Roche) was diluted at 1:400 for flow cytometry. Donkey monoclonal anti-goat Alexa 488- or Alexa 555- and anti-mouse Alexa 555- or Alexa 647-conjugated secondary antibodies (Invitrogen) were used at a dilution of 1:400 for immunofluorescence microscopy and flow cytometry.
T-cell stimulation.
Bone marrow-derived dendritic cells (BM-DC) were seeded at a density of 2 × 105 in 24-well plates and infected at an MOI of 25 or 100 for wild-type or mutant strains, respectively. Infections were carried out as described above for primary macrophages. Infection with the wild-type S. Typhimurium vector, with or without stimulation with 1 µM SIINFEKL peptide, was used as a positive or negative control, respectively. Following killing of extracellular bacteria with medium containing 100 µg/ml gentamicin, B3Z cells expressing the lacZ reporter gene under the control of the NFAT enhancer (33) were added to wells and cocultured for 22 h at a BM-DC/T-cell ratio of 1:8 in medium containing 20 µg/ml gentamicin. Cells were centrifuged at maximal speed for 5 min prior to the addition of 100 µl substrate solution containing a lysing agent (0.15 mM chlorophenyl red β-galactopyranoside–0.5% [vol/vol] Nonidet P-40–PBS). Following incubation for 6 h at 37°C, absorbance was determined at 595 nm.
SUPPLEMENTAL MATERIAL
Figure S1
Quantification of intracellular growth of S. Typhimurium multiple-mutation strains in CFU. Intracellular bacteria were recovered at 2 h and 17 h postinoculation. Values of fold increase in bacterial numbers were extracted from the ratio of CFU/ml at 17 h to CFU/ml at 2 h. Data are expressed as means ± SEM of the results of three independent experiments, each performed in triplicate. P values were obtained by Student’s t test (*, P < 0.05) relative to the wild-type strain results. Download
Figure S2
Translocation of SPI-2 T3SS effectors from S. Typhimurium Δ_sseG_ Δ_sopD2_ Δ_srfH_ in RAW 264.7 macrophages. Confocal microscopy images of RAW 264.7 macrophages infected with indicated S. Typhimurium strains expressing 2HA-tagged SseJ or SseF from the low-copy-number plasmid pWSK29 are shown. Cells were fixed at 8 h postinoculation and immunolabeled with anti-Salmonella and anti-HA antibodies (green and red, respectively, in merged images). Scale bars represent 5 µm. Download
Table S1
List of strains and plasmids used in this study. Abbreviations: Carb, carbenicillin; Km, kanamycin; Cm, chloramphenicol.
Table S2
List of primers used in this study.
ACKNOWLEDGMENTS
The ΔsifA sopD2 and ΔsifA ΔpipB2 ΔsopD2 strains were a gift from S. Méresse (CIML, Marseille, France), and the Δ_slrP_ strain was a gift from A. Bäumler (UC, Davis). The plasmid pWSK29 P sseA sseJ::OVA::HA was a gift from M. Hensel (Osnabrueck University, Germany). The B3Z T-cell hybridoma was kindly provided by K. Gould (Imperial College London, United Kingdom). We are grateful to Thomas Sutherland, Jaime Mota, Jayna Mistry, Jessica Thompson, Julie Guignot, and Junkal Garmendia for construction of mutant strains and to the Holden group for constructive reading of the manuscript.
This work was supported by grants from the Medical Research Council and Wellcome Trust (United Kingdom) to D.W.H. and from the Fundação para a Ciência e a Tecnologia, Portugal (FCT), to R.F.
Footnotes
CitationFigueira R, Watson KG, Holden DW, Helaine S. 2013. Identification of Salmonella pathogenicity island-2 type III secretion system effectors involved in intramacrophage replication of S. enterica serovar Typhimurium: implications for rational vaccine design. mBio 4(2):e00065-13. doi:10.1128/mBio.00065-13.
REFERENCES
- 1.van der Heijden J, Finlay BB. 2012. Type III effector-mediated processes in salmonella infection. Future Microbiol. 7:685–703 [DOI] [PubMed] [Google Scholar]
- 2.Ochman H, Soncini FC, Solomon F, Groisman EA. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U. S. A. 93:7800–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science 269:400–403 [DOI] [PubMed] [Google Scholar]
- 4.Fields PI, Swanson RV, Haidaris CG, Heffron F. 1986. Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc. Natl. Acad. Sci. U. S. A. 83:5189–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helaine S, Thompson JA, Watson KG, Liu M, Boyle C, Holden DW. 2010. Dynamics of intracellular bacterial replication at the single cell level. Proc. Natl. Acad. Sci. U. S. A. 107:3746–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geddes K, Worley M, Niemann G, Heffron F. 2005. Identification of new secreted effectors in Salmonella enterica serovar typhimurium. Infect. Immun. 73:6260–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figueira R, Holden DW. 2012. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158:1147–1161 [DOI] [PubMed] [Google Scholar]
- 8.Brumell JH, Kujat-Choy S, Brown NF, Vallance BA, Knodler LA, Finlay BB. 2003. SopD2 is a novel type III secreted effector of Salmonella typhimurium that targets late endocytic compartments upon delivery into host cells. Traffic 4:36–48 [DOI] [PubMed] [Google Scholar]
- 9.Cordero-Alba M, Bernal-Bayard J, Ramos-Morales F. 2012. SrfJ, a Salmonella type III secretion system effector regulated by PhoP, RcsB, and IolR. J. Bacteriol. 194:4226–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, Vazquez-Torres A, Gleeson C, Fang FC, Holden DW. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174 [DOI] [PubMed] [Google Scholar]
- 11.Kuhle V, Hensel M. 2002. SseF and SseG are translocated effectors of the type III secretion system of Salmonella pathogenicity island 2 that modulate aggregation of endosomal compartments. Cell. Microbiol. 4:813–824 [DOI] [PubMed] [Google Scholar]
- 12.Salcedo SP, Holden DW. 2003. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J. 22:5003–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beuzón CR, Salcedo SP, Holden DW. 2002. Growth and killing of a Salmonella enterica serovar typhimurium sifA mutant strain in the cytosol of different host cell lines. Microbiology 148:2705–2715 [DOI] [PubMed] [Google Scholar]
- 14.Salcedo SP, Noursadeghi M, Cohen J, Holden DW. 2001. Intracellular replication of Salmonella typhimurium strains in specific subsets of splenic macrophages in vivo. Cell. Microbiol. 3:587–597 [DOI] [PubMed] [Google Scholar]
- 15.Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19:3235–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brumell JH, Rosenberger CM, Gotto GT, Marcus SL, Finlay BB. 2001. SifA permits survival and replication of Salmonella typhimurium in murine macrophages. Cell. Microbiol. 3:75–84 [DOI] [PubMed] [Google Scholar]
- 17.Freeman JA, Ohl ME, Miller SI. 2003. The Salmonella enterica serovar typhimurium translocated effectors SseJ and SifB are targeted to the _Salmonella_-containing vacuole. Infect. Immun. 71:418–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroeder N, Henry T, de Chastellier C, Zhao W, Guilhon AA, Gorvel JP, Méresse S. 2010. The virulence protein SopD2 regulates membrane dynamics of _Salmonella_-containing vacuoles. PLoS Pathog. 6:e1001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lossi NS, Rolhion N, Magee AI, Boyle C, Holden DW. 2008. The Salmonella SPI-2 effector SseJ exhibits eukaryotic activator-dependent phospholipase A and glycerophospholipid: cholesterol acyltransferase activity. Microbiology 154:2680–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohlson MB, Fluhr K, Birmingham CL, Brumell JH, Miller SI. 2005. SseJ deacylase activity by Salmonella enterica serovar typhimurium promotes virulence in mice. Infect. Immun. 73:6249–6259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruiz-Albert J, Yu XJ, Beuzón CR, Blakey AN, Galyov EE, Holden DW. 2002. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 44:645–661 [DOI] [PubMed] [Google Scholar]
- 22.Jiang X, Rossanese OW, Brown NF, Kujat-Choy S, Galán JE, Finlay BB, Brumell JH. 2004. The related effector proteins SopD and SopD2 from Salmonella enterica serovar typhimurium contribute to virulence during systemic infection of mice. Mol. Microbiol. 54:1186–1198 [DOI] [PubMed] [Google Scholar]
- 23.Mesquita FS, Thomas M, Sachse M, Santos AJ, Figueira R, Holden DW. 2012. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 8:e1002743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckner MM, Croxen MA, Arena ET, Finlay BB. 2011. A comprehensive study of the contribution of Salmonella enterica serovar typhimurium SPI2 effectors to bacterial colonization, survival, and replication in typhoid fever, macrophage, and epithelial cell infection models. Virulence 2:208–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poh J, Odendall C, Spanos A, Boyle C, Liu M, Freemont P, Holden DW. 2008. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell. Microbiol. 10:20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown NF, Coombes BK, Bishop JL, Wickham ME, Lowden MJ, Gal-Mor O, Goode DL, Boyle EC, Sanderson KL, Finlay BB. 2011. Salmonella phage ST64B encodes a member of the SseK/NleB effector family. PLoS One 6:e17824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kujat Choy SL, Boyle EC, Gal-Mor O, Goode DL, Valdez Y, Vallance BA, Finlay BB. 2004. SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar typhimurium. Infect. Immun. 72:5115–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deiwick J, Salcedo SP, Boucrot E, Gilliland SM, Henry T, Petermann N, Waterman SR, Gorvel JP, Holden DW, Méresse S. 2006. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 74:6965–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beuzón CR, Holden DW. 2001. Use of mixed infections with Salmonella strains to study virulence genes and their interactions in vivo. Microbes Infect. 3:1345–1352 [DOI] [PubMed] [Google Scholar]
- 31.Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. 1993. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 90:10544–10548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hegazy WA, Xu X, Metelitsa L, Hensel M. 2012. Evaluation of Salmonella enterica type III secretion system effector proteins as carriers for heterologous vaccine antigens. Infect. Immun. 80:1193–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karttunen J, Sanderson S, Shastri N. 1992. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. U. S. A. 89:6020–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hindle Z, Chatfield SN, Phillimore J, Bentley M, Johnson J, Cosgrove CA, Ghaem-Maghami M, Sexton A, Khan M, Brennan FR, Everest P, Wu T, Pickard D, Holden DW, Dougan G, Griffin GE, House D, Santangelo JD, Khan SA, Shea JE, Feldman RG, Lewis DJ. 2002. Characterization of Salmonella enterica derivatives harboring defined aroC and Salmonella pathogenicity island 2 type III secretion system (ssaV) mutations by immunization of healthy volunteers. Infect. Immun. 70:3457–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyon CE, Sadigh KS, Carmolli MP, Harro C, Sheldon E, Lindow JC, Larsson CJ, Martinez T, Feller A, Ventrone CH, Sack DA, DeNearing B, Fingar A, Pierce K, Dill EA, Schwartz HI, Beardsworth EE, Kilonzo B, May JP, Lam W, Upton A, Budhram R, Kirkpatrick BD. 2010. In a randomized, double-blinded, placebo-controlled trial, the single oral dose typhoid vaccine, M01ZH09, is safe and immunogenic at doses up to 1.7 × 10(10) colony-forming units. Vaccine 28:3602–3608 [DOI] [PubMed] [Google Scholar]
- 36.Khan SA, Stratford R, Wu T, McKelvie N, Bellaby T, Hindle Z, Sinha KA, Eltze S, Mastroeni P, Pickard D, Dougan G, Chatfield SN, Brennan FR. 2003. Salmonella typhi and S. typhimurium derivatives harbouring deletions in aromatic biosynthesis and Salmonella pathogenicity island-2 (SPI-2) genes as vaccines and vectors. Vaccine 21:538–548 [DOI] [PubMed] [Google Scholar]
- 37.Kirkpatrick BD, Tenney KM, Larsson CJ, O’Neill JP, Ventrone C, Bentley M, Upton A, Hindle Z, Fidler C, Kutzko D, Holdridge R, LaPointe C, Hamlet S, Chatfield SN. 2005. The novel oral typhoid vaccine M01ZH09 is well tolerated and highly immunogenic in 2 vaccine presentations. J. Infect. Dis. 192:360–366 [DOI] [PubMed] [Google Scholar]
- 38.Kirkpatrick BD, McKenzie R, O’Neill JP, Larsson CJ, Bourgeois AL, Shimko J, Bentley M, Makin J, Chatfield S, Hindle Z, Fidler C, Robinson BE, Ventrone CH, Bansal N, Carpenter CM, Kutzko D, Hamlet S, LaPointe C, Taylor DN. 2006. Evaluation of Salmonella enterica serovar Typhi (Ty2 aroC-ssaV-) M01ZH09, with a defined mutation in the Salmonella pathogenicity island 2, as a live, oral typhoid vaccine in human volunteers. Vaccine 24:116–123 [DOI] [PubMed] [Google Scholar]
- 39.Tran TH, Dung NT, Truong NT, Van NTT, Bich Chau TN, Hoang NVM, Nga TTT, Thuy CT, Minh PV, Binh NTC, Ha TTD, Toi PV, Song Diep T, Campbell JI, Stockwell E, Schultsz C, Simmons CP, Glover C, Lam W, Marques F, May JP, Upton A, Budhram R, Dougan G, Farrar J, Vinh Chau NV, Dolecek C. 2010. A randomised trial evaluating the safety and immunogenicity of the novel single oral dose typhoid vaccine M01ZH09 in healthy Vietnamese children. PLoS One 5:e11778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Husseiny MI, Wartha F, Hensel M. 2007. Recombinant vaccines based on translocated effector proteins of Salmonella pathogenicity island 2. Vaccine 25:185–193 [DOI] [PubMed] [Google Scholar]
- 41.Panthel K, Meinel KM, Domènech VE, Retzbach H, Igwe EI, Hardt WD, Rüssmann H. 2005. Salmonella pathogenicity island 2-mediated overexpression of chimeric SspH2 proteins for simultaneous induction of antigen-specific CD4 and CD8 T cells. Infect. Immun. 73:334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X, Husseiny MI, Goldwich A, Hensel M. 2010. Efficacy of intracellular activated promoters for generation of _Salmonella_-based vaccines. Infect. Immun. 78:4828–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong G, Husseiny MI, Song L, Erdreich-Epstein A, Shackleford GM, Seeger RC, Jäckel D, Hensel M, Metelitsa LS. 2010. Novel cancer vaccine based on genes of Salmonella pathogenicity island 2. Int. J. Cancer 126:2622–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Browne SH, Hasegawa P, Okamoto S, Fierer J, Guiney DG. 2008. Identification of Salmonella SPI-2 secretion system components required for SpvB-mediated cytotoxicity in macrophages and virulence in mice. FEMS Immunol. Med. Microbiol. 52:194–201 [DOI] [PubMed] [Google Scholar]
- 45.McLaughlin LM, Govoni GR, Gerke C, Gopinath S, Peng K, Laidlaw G, Chien YH, Jeong HW, Li Z, Brown MD, Sacks DB, Monack D. 2009. The Salmonella SPI2 effector SseI mediates long-term systemic infection by modulating host cell migration. PLoS Pathog. 5:e1000671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Worley MJ, Nieman GS, Geddes K, Heffron F. 2006. Salmonella typhimurium disseminates within its host by manipulating the motility of infected cells. Proc. Natl. Acad. Sci. U. S. A. 103:17915–17920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niemann GS, Brown RN, Gustin JK, Stufkens A, Shaikh-Kidwai AS, Li J, McDermott JE, Brewer HM, Schepmoes A, Smith RD, Adkins JN, Heffron F. 2011. Discovery of novel secreted virulence factors from Salmonella enterica serovar typhimurium by proteomic analysis of culture supernatants. Infect. Immun. 79:33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheminay C, Möhlenbrink A, Hensel M. 2005. Intracellular salmonella inhibit antigen presentation by dendritic cells. J. Immunol. 174:2892–2899 [DOI] [PubMed] [Google Scholar]
- 49.Halici S, Zenk SF, Jantsch J, Hensel M. 2008. Functional analysis of the Salmonella pathogenicity island 2-mediated inhibition of antigen presentation in dendritic cells. Infect. Immun. 76:4924–4933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tobar JA, Carreño LJ, Bueno SM, González PA, Mora JE, Quezada SA, Kalergis AM. 2006. Virulent Salmonella enterica serovar typhimurium evades adaptive immunity by preventing dendritic cells from activating T cells. Infect. Immun. 74:6438–6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsui K, Nagano K, Arai T, Hirono I, Aoki T. 1998. DNA sequencing of the gene encoding _Salmonella typhimurium_-derived T-cell inhibitor (STI) and characterization of the gene product, cloned STI. FEMS Immunol. Med. Microbiol. 22:341–349 [DOI] [PubMed] [Google Scholar]
- 52.van der Velden AW, Copass MK, Starnbach MN. 2005. Salmonella inhibit T cell proliferation by a direct, contact-dependent immunosuppressive effect. Proc. Natl. Acad. Sci. U. S. A. 102:17769–17774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Odendall C, Rolhion N, Förster A, Poh J, Lamont DJ, Liu M, Freemont PS, Catling AD, Holden DW. 2012. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12:657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galen JE, Pasetti MF, Tennant S, Ruiz-Olvera P, Sztein MB, Levine MM. 2009. Salmonella enterica serovar Typhi live vector vaccines finally come of age. Immunol. Cell Biol. 87:400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forest CG, Ferraro E, Sabbagh SC, Daigle F. 2010. Intracellular survival of Salmonella enterica serovar Typhi in human macrophages is independent of Salmonella pathogenicity island (SPI). Microbiology 156:3689–3698 [DOI] [PubMed] [Google Scholar]
- 56.Beuzón CR, Banks G, Deiwick J, Hensel M, Holden DW. 1999. pH-dependent secretion of SseB, a product of the SPI-2 type III secretion system of Salmonella typhimurium. Mol. Microbiol. 33:806–816 [DOI] [PubMed] [Google Scholar]
- 57.Racoosin EL, Swanson JA. 1989. Macrophage colony-stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J. Exp. Med. 170:1635–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Quantification of intracellular growth of S. Typhimurium multiple-mutation strains in CFU. Intracellular bacteria were recovered at 2 h and 17 h postinoculation. Values of fold increase in bacterial numbers were extracted from the ratio of CFU/ml at 17 h to CFU/ml at 2 h. Data are expressed as means ± SEM of the results of three independent experiments, each performed in triplicate. P values were obtained by Student’s t test (*, P < 0.05) relative to the wild-type strain results. Download
Figure S2
Translocation of SPI-2 T3SS effectors from S. Typhimurium Δ_sseG_ Δ_sopD2_ Δ_srfH_ in RAW 264.7 macrophages. Confocal microscopy images of RAW 264.7 macrophages infected with indicated S. Typhimurium strains expressing 2HA-tagged SseJ or SseF from the low-copy-number plasmid pWSK29 are shown. Cells were fixed at 8 h postinoculation and immunolabeled with anti-Salmonella and anti-HA antibodies (green and red, respectively, in merged images). Scale bars represent 5 µm. Download
Table S1
List of strains and plasmids used in this study. Abbreviations: Carb, carbenicillin; Km, kanamycin; Cm, chloramphenicol.
Table S2
List of primers used in this study.