Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury (original) (raw)
Abstract
We investigated whether permeability transition-mediated release of mitochondrial cytochrome c is a potential therapeutic target for treating acute spinal cord injury (SCI). Based on previous reports, minocycline, a second-generation tetracycline, exerts neuroprotection partially by inhibiting mitochondrial cytochrome c release and reactive microgliosis. We first evaluated cytochrome c release at the injury epicenter after a T10 contusive SCI in rats. Cytochrome c release peaked at ≈4-8 h postinjury. A dose-response study generated a safe pharmacological regimen that enabled i.p. minocycline to significantly lower cytosolic cytochrome c at the epicenter 4 h after SCI. In the long-term study, i.p. minocycline (90 mg/kg administered 1 h after SCI followed by 45 mg/kg administered every 12 h for 5 days) markedly enhanced long-term hind limb locomotion relative to that of controls. Coordinated motor function and hind limb reflex recoveries also were improved significantly. Histopathology suggested that minocycline treatment alleviated later-phase tissue loss, with significant sparing of white matter and ventral horn motoneurons at levels adjacent to the epicenter. Furthermore, glial fibrillary acidic protein and 2′,3′ cyclic nucleotide 3′ phosphodiesterase immunocytochemistry showed an evident reduction in astrogliosis and enhanced survival of oligodendrocytes. Therefore, release of mitochondrial cytochrome c is an important secondary injury mechanism in SCI. Drugs with multifaceted effects in antagonizing this process and microgliosis may protect a proportion of spinal cord tissue that is clinically significant for functional recovery. Minocycline, with its proven clinical safety, capability to cross the blood-brain barrier, and demonstrated efficacy during a clinically relevant therapeutic window, may become an effective therapy for acute SCI.
Traumatic spinal cord injury (SCI) causes permanent neurological deficits because of the loss of spinal cord neurons and axons. Clinically, SCI most frequently results from vertebral fracture or dislocation that produces spinal cord bruising or contusion (1, 2). Experimental studies typically focus on incomplete contusive or compression injuries (3). Those experiments have indicated that the pathophysiology after SCI results from the combination of a primary mechanical trauma and secondary injury events. Unlike the primary mechanical trauma, which is immediate and beyond therapeutic management, the secondary injury occurs over the hours and days after SCI, further exacerbating tissue loss and functional impairments (4). However, research over the past two decades has demonstrated that therapeutic interventions attenuating common secondary injury mechanisms (e.g., glutamate toxicity, free radical cellular damage, Na+ and Ca2+ influx-related neuronal death, etc.) required either pre- or immediate postinjury (p.i.) application for demonstration of therapeutic effectiveness in experimental SCI and thus did not lead to successful clinical trials (5-7). Although i.v. administration of methylprednisolone has been recommended as the standard clinical treatment for SCI, its application is largely based on the beneficial results reported in a large-scale clinical trial (8). However, side effects such as trends toward higher infection rates, gastrointestinal hemorrhage, and respiratory complications, as well as questions about the validity of the conclusions drawn from the clinical trial, have generated controversy regarding the standardized use of mega-dose i.v. methylprednisolone for treating acute SCI (6). Because ≈50% of patients initially have incomplete lesions (8), it is crucial to identify therapeutic strategies that can promote functional recovery after SCI when they are implemented systemically within a pragmatic therapeutic window. A key to developing therapies is the identification at the molecular level of pathophysiological events underlying delayed tissue loss (4, 7). Notably, the caspase cell death pathway and one of its upstream triggers, mitochondrial cytochrome c release, have been elucidated in SCI. They are clearly targets for the development of therapeutic interventions (9-11).
Minocycline has been shown to have robust neuroprotective effects in rodent models of neurodegeneration, cerebral ischemia, and traumatic brain injury (12-17) through blocking the permeability transition-mediated release of cytochrome c that activates the caspase cell death pathway, directly inhibiting activated caspases, and through mitigating reactive microgliosis (18). Because delayed cell death and reactive microgliosis play critical roles in secondary injury after SCI, we hypothesized that blocking mitochondrial cytochrome c release might reduce the spinal cord tissue loss after SCI that is functionally significant. We therefore evaluated the potential therapeutic effects of minocycline in a standardized contusion model of SCI in rats.
Methods
SCI. Female Sprague-Dawley rats (280-330 g) were anesthetized with i.p. ketamine (75 mg/kg) and xylazine (20 mg/kg). A T10 contusion injury (10 g × 12.5 mm) was produced [by using the New York University (NYU) impactor] as previously described (19).
Minocycline Administration. Minocycline hydrochloride powder (Sigma) was prepared in normal saline (NS) and heated briefly in a water bath until completely dissolved into a clear solution. For the dose-response study, minocycline was administered i.p. at three different doses (18, 90, and 180 mg/kg) beginning 1 h p.i. The spinal cord tissues were collected 3 h later (i.e., 4 h p.i.) followed by exploration of the abdominal cavity for pathological signs of inflammation or necrosis. For the behavioral and histological experiments, rats were initially given 90 mg/kg minocycline i.p. in 10 ml of NS (pH ≈5.7) starting 1 h after SCI, followed by minocycline (45 mg/kg in 5 ml of NS) every 12 h for 5 consecutive days. Control groups received equivolumetric i.p. NS vehicle injections at the corresponding time points.
Experimental Protocol. The experiments were conducted according to a randomized block design. The size of the experimental groups was determined on the basis of power analyses as previously reported (20-22). Behavioral tests were performed at days 1, 7, 14, 21, and 28 p.i. All animals survived the study. All values are presented as mean ± SEM. Application of the term “significant” in the tests implies P < 0.05. All experimental procedures were reviewed and approved by the Animal Care and Use Committee of the VA Boston Healthcare System.
Time Course of Cytochrome c Release. Rats were divided into six groups (n = 3 rats per group) that were defined as preinjury, immediate p.i., and 4, 8, 24, and 48 h p.i. groups, respectively, based on the time points when spinal cords were collected. For each group, spinal tissue from T9-T11 segments [with the injured group receiving a T10 contusive SCI (10 g × 12.5 mm)] was collected for permeability transition-mediated mitochondrial cytochrome c release analysis with methods reported previously (12). Cytochrome c release in spinal cord segments further distal to the lesion epicenter (i.e., C1-C3 and L2-L4) also was analyzed. All blotting was repeated at least 4 times with consistent results.
Behavioral Evaluation of Functional Deficits. Examination of functional deficits 1, 7, 14, 21, and 28 days p.i. was conducted as previously described (23). At each time point a battery of tests of hind limb reflexes, as well as coordinated use of hind limbs, was performed by using the BBB scale for overall hind limb function, as previously described (23-25).
Histopathology. After the 4-week behavioral evaluation, animals were deeply anesthetized and perfused intracardially with 0.1 M phosphate buffer (Sigma) followed by 4% paraformaldehyde in phosphate buffer (pH 7.4). The abdominal cavity of each rat was examined for pathological signs of inflammation, necrosis, and/or fibrosis. Spinal cord tissue was dissected from the vertebral canal and processed as previously reported (26). Serial 20-μm cross sections were mounted sequentially on 10 slides (Superfrost*/Plus microscope slides, Fisher Scientific) per series, a process repeated until each slide carried five sections (1,000 μm of tissue), at which point a new series began. Serial spinal cord sections from the treated and control groups were stained in pairs to allow comparison of identically processed tissue. All morphological analyses were done by evaluators blinded to the treatment groups.
Tissue sections were examined for lesion cells and cavities as previously described (26), and the injury epicenters (regions of maximal damage) were identified. Solvent blue- and hematoxylin/eosin-stained sections at the lesion epicenter and 1, 2, 3, and 4 mm rostral and caudal to the epicenter were digitized by using the spot version 3.5.9.1 for mac os program (Diagnostic Instruments, Sterling Heights, MI) and analyzed with the mcid elite program (Imaging Research, St. Catherine's, ON, Canada). Lesion areas and areas of white matter sparing were defined as previously described (21, 24, 26).
For assessment of neuroprotection, slides representing the epicenter and specific locations rostral and caudal to it were stained with cresyl violet and analyzed for the presence of surviving ventral horn neurons that exhibited Nissl substance, a euchromatic nucleus, and a distinct nucleolus (20, 21).
Immunocytochemistry of Glial Fibrillary Acidic Protein (GFAP) and 2′**,3**′ Cyclic Nucleotide 3′ Phosphodiesterase (CNPase). The second or the third sets of slides of the first series from 3 and 4 mm caudal to the epicenter were used for immunocytochemistry of the GFAP of reactive astroglial cells (27) and the CNPase of oligodendroglial cells (28), respectively. Briefly, an anti-GFAP monoclonal antibody (Sigma) was used with a FITC-conjugated secondary antibody. The analysis was performed on slides with tissue 3 mm caudal to the epicenter from minocycline- and vehicle-treated animals (n = 8 rats per group). GFAP-positive astroglial cells of the ventral funiculi were captured by using the spot program. GFAP-positive astroglia with a clearly identifiable soma and at least one cell process (Fig. 4_c_) in the bilateral ventral funiculi of two transverse sections per rat that were 200 μm apart were counted at a magnification of ×40 and averaged to get the number of astroglia per section (i.e., 20 μm of tissue) for statistics. Oligodendrocytes were examined with similar procedures by using an anti-CNPase monoclonal antibody (Sternberger Monoclonals, Lutherville, MD) that was detected by its secondary antibody and Texas red as a chromagen. Areas of ≈50,000 μm2 from the ventromedial portion of the ventral funiculi 4 mm caudal to the epicenter (one section per rat, n = 8 rats per group) were measured for mean luminosity by using photoshop version 7.0 (Adobe Systems, Mountain View, CA). The mean luminosities for each group were compared by using an unpaired Student's t test.
Fig. 4.

(a and b) GFAP immunocytochemistry demonstrating reduced astrogliosis in rat spinal cord white matter receiving minocycline (a) vs. equivolumetric saline vehicle (b). (Scale bar = 250 μm.) (c) Hypertrophic astrocyte of a rat receiving saline vehicle treatment. (Scale bar = 20 μm.) (d) The average number of reactive astrocytes (y axis, number of astroglia per 20 μm of tissue) was significantly lower in the treated group (hatched bar) vs. the controls (open bar) (n = 8 rats per group; P = 0.017, unpaired Student's t test).
Statistical Analyses. BBB scores, behavior on inclined planes, white matter sparing, and ventral horn motor counts were analyzed by using repeated-measures ANOVA followed by unpaired Student's t tests (25). Fisher's exact test was used for the statistical evaluation of reflex recovery.
Results
Time Course of Cytochrome c Release from the Injury Epicenter. Western blot analyses revealed a negligible cytochrome c signal in the cytosolic compartment of noninjured spinal cord tissues (i.e., from sham-operated rats) sampled from equivalent spinal segments (i.e., T9-T11). In SCI rats, cytochrome c release from the mitochondria into the cytoplasm was not detected immediately after SCI but peaked 4-8 h p.i. (Fig. 1_a_). Conversely, cytochrome c levels in mitochondrial pellets were reduced proportionally (Fig. 1_b_). We therefore decided to administer minocycline 1 h p.i. and examine its effect on blocking cytochrome c release at the SCI epicenter 4 h p.i. In addition, we found that cytosolic cytochrome c levels in C1-C3 and L2-L4 spinal cord, segments more distal to the T10 injury epicenter, were still significantly elevated 48 and 24 h p.i., respectively (data not shown). Thus, a 5-day drug administration plan was formed for the treatment of minocycline.
Fig. 1.

(a and b) Cytochrome c Western blots of cytosolic (a) and mitochondrial (b) fractions of precontusion and postcontusion spinal cord tissue at the epicenter. (c) Dose-dependent effect of minocycline on cytochrome c release at the injury epicenter, with significant blocking at the 90 and 180 mg/kg doses. COX IV, cyclooxygenase IV.
Dose-Response Effect of Minocycline on Cytochrome c Release After SCI. Minocycline at doses of 18, 90, and 180 mg/kg was injected i.p. 1 h after SCI. We observed a dose-dependent effect of minocycline on cytochrome c release at the injury epicenter. Minocycline at 90 and 180 mg/kg i.p. showed significant blocking effects, essentially reducing cytochrome c release to the negligible pre-SCI level (Fig. 1_c_).
Effect of Minocycline on Hind Limb Paralysis After SCI. To avoid the potential significant side effects seen in drug preparation regimens reported previously (e.g., i.p. necrosis due to minocycline precipitates and, to a lesser degree, acidity of the minocycline preparation) (29), a more conservative dosing regimen was developed, beginning with 90 mg/kg i.p. minocycline (in 10 ml of NS per rat) 1 h after SCI, followed by 45 mg/kg minocycline (in 5 ml of NS per rat) every 12 h for 5 consecutive days. At this dose, rats did not demonstrate any signs of abdominal irritation immediately after injection, nor any pathological signs of inflammation, necrosis, or fibrosis after fixative perfusion. The minocycline-treated rats also did not show other general toxicities such as excessive loss of body weight (see below).
As described before in similar SCI models (19, 21, 24), all experimental rats demonstrated complete hind limb paralysis at 1 day p.i., including areflexia and lack of coordinated motor functions, such as locomotion. Thereafter, partial recovery of function was observed over the next few weeks. By 4 weeks p.i., the long-term chronic functional deficits became apparent. Greater return to a normal withdrawal reflex to an extension stimulus was noted in minocycline-treated rats vs. controls (70% vs. 25% of hind limbs; P = 0.010, Fisher's exact test) at 4 weeks p.i. A trend toward greater recovery to normal of the toe-spread reflex was noted in the minocycline-treated group vs. controls (65% vs. 35% of hind limbs; P = 0.370, Fisher's exact test) at 4 weeks p.i.
For recovery of coordinated motor function, animals were tested facing upward and downward on an inclined plane. Whereas performance in the upward-facing orientation reflected forelimb strength (which should not have been affected by a lower thoracic SCI in otherwise healthy animals), performance facing downward measured coordinated hind limb motor function (22, 23, 30). In the upward-facing orientation, the performances of the two groups were not statistically different (P > 0.05; Fig. 2_a_). However, the minocycline-treated group showed a statistically significant improvement compared with controls in the downward-facing orientation (P = 0.040; Fig. 2_a_) beginning on day 7 p.i. and persisting through the duration of the study. In addition, recovery of the capacity to use the hind limbs in open-field locomotion, as graded by the BBB scale, also significantly improved in an overall statistical comparison (P = 0.033, repeated-measures ANOVA) and at days 21 and 28 p.i. of the post hoc analysis (P < 0.05, unpaired Student's t test; Fig. 2_b_). Thus, the regimen of minocycline treatment significantly improved overall hind limb function compared with that of the controls beginning 7 days after SCI and throughout the 28-day study.
Fig. 2.

Effect of minocycline on the recovery of coordinated hind limb functions. (a) Comparison of groups (n = 10 rats per group) that received vehicle alone or minocycline in inclined plane performance. Data points represent the average ± SEM maximum angle at which rats can maintain position for 5 s. Data were analyzed with repeated-measures ANOVA, which showed an overall significant (P < 0.05) effect of treatment. Asterisks indicate that means are significantly different from the control group at the specified times after SCI (Student's t test). When facing upward, animals in the two groups (▴ and ▵) were statistically similar for all time points, indicating that they were similar in general body condition. When facing downward, the minocycline-treated group (▾) performed significantly better starting at day 7 p.i., compared with the vehicle control group (▿). (b) Locomotor function graded on the BBB scale that ranges from 21 in normal rats to 0 in rats with complete hind limb paralysis (25). Data points represent the group average ± SEM. Results were analyzed with repeated-measures ANOVA, which showed an overall significant (P < 0.05) effect of treatment. Asterisks indicate that means are significantly different from the vehicle-treated control group at the specified times after SCI (Student's t test).
Body Weight Changes After SCI. SCI in our model caused a small and statistically insignificant decrease in body weight at 24 h p.i. relative to body weight before injury (data not shown). Afterward, body weight gradually increased over the entire study period. There was no significant difference in body weight between the minocycline- and vehicle-treated groups at any time (P > 0.05, repeated-measures ANOVA).
Histopathological Examination of the Effect of Minocycline on SCI. In the contusion model of SCI (24, 26), the so-called lesion volume consists of an area of maximal tissue loss (i.e., lesion per injury epicenter) and the lesion rostral and caudal to the epicenter that tapers gradually until its most distal elements end in the dorsal funicular white matter.
To evaluate whether systemic minocycline treatment reduced lesion volume, we outlined the lesion area in transverse sections of the cord sampled from the injury epicenter and adjacent tissue up to 4 mm rostral and caudal to the epicenter in 1-mm increments. The epicenters were characterized by a peripheral, and in some cases incomplete, rim of residual, hypomyelinated white matter. This residual rim surrounded a central lesion that consisted of cavities and a loose network of nonneuronal cells. The overall cross-section profiles of the lesion epicenters from the vehicle- and minocycline-treated groups were comparable (Figs. 3 a and b). Additionally, there were no significant differences in lesion area size in tissue at other loci rostral and caudal to the epicenter (Fig. 3_a_). Measurement of longitudinal lesion lengths also did not indicate any significant effect of minocycline (9.01 ± 0.47 mm vs. 9.12 ± 0.59 mm; P > 0.05, unpaired Student's t test) (20, 21).
Fig. 3.

(a) Lack of significant effect of minocycline (total dose of 495 mg/kg per rat over 5 days) on lesion volume at the injury epicenter and adjacent tissue up to 4 mm rostral and caudal to the epicenter, in 1-mm increments (solvent blue and hematoxylin/eosin staining). (b) Representative transverse spinal cord sections after T10 contusion injury at the epicenter and in 1-mm increments rostral and caudal to the epicenter in a rat receiving minocycline (Upper) and saline vehicle (Lower). Epicenter sections are marked by arrows. (c) Overall significant difference (P = 0.014, repeated-measures ANOVA) between rats receiving minocycline or saline vehicle (n = 10 rats per group) in the average area of residual total white matter (i.e., white matter and hypomyelinated white matter; solvent blue and hematoxylin/eosin staining) (24), as well as 2, 3, and 4 mm rostral and 3 and 4 mm caudal to the lesion epicenter (P < 0.03, unpaired Student's t test). (d) Minocycline treatment significantly protected ventral horn neurons 3 and 4 mm rostral to the epicenter (P < 0.03, repeated-measures ANOVA), as well as 2, 3, and 4 mm caudal to the epicenter (P < 0.05, unpaired Student's t tests). (e and f) Representative transverse cresyl violet-stained sections 2 mm caudal to the injury epicenter in a rat receiving minocycline (e) or saline vehicle (f). (Scale bar = 50 μm.)
To determine whether minocycline treatment spared white matter, the cross-section profiles of spinal cord tissue at the epicenter and sections 1, 2, 3, and 4 mm rostral and caudal to the epicenter were examined for residual total white matter area (i.e., white matter and hypomyelinated white matter) (24, 26). There was an overall significant difference between the two groups in the average area of residual total white matter (P = 0.014, repeated-measures ANOVA) as well as at 2, 3, and 4 mm rostral and 3 and 4 mm caudal to the lesion epicenter on post hoc tests (P < 0.03, unpaired Student's t test; Fig. 3_c_). Additionally, the relationship between white matter sparing 4 mm caudal to the lesion epicenter and chronic hind limb function 4 weeks after SCI was examined. Linear regression analysis indicated a significant positive correlation between the area of preserved white matter 4 mm caudal to the injury epicenter and overall hind limb function, as measured by the BBB scale 4 weeks after SCI (n = 10 rats per group; P < 0.036, ANOVA).
To gain specific information about the neuronal protective effect of minocycline, we first examined the gray matter-sparing effect of minocycline and found no significant difference between the two experimental groups (data not shown). We then compared the numbers of healthy-appearing ventral horn neurons with euchromatic nuclei and prominent nucleoli in the vehicle- and minocycline-treated groups (20). Motoneurons of the ventral horns were counted in a series of cresyl violet-stained sections at the lesion epicenter and at 1-mm increments both rostral and caudal to the lesion epicenter (20). Motoneurons were not present at the injury epicenter or in tissue 1 mm rostral or caudal to the epicenter. Starting 2 mm rostral and caudal to the epicenter, motoneuron sparing was observed. Minocycline treatment significantly protected ventral horn neurons in the overall analysis (P < 0.03, repeated-measures ANOVA) and 3 and 4 mm rostral to the epicenter, as well as 2, 3, and 4 mm caudal to the epicenter in post hoc analyses (P < 0.05, unpaired Student's t tests; Fig. 3 _d_-f).
Effect of Minocycline on Reactive Astrocytes and Oligodendrocytes After SCI. To determine whether minocycline treatment inhibited posttraumatic astrogliosis, the numbers of GFAP-positive glia were compared in the two experimental groups. Spinal sections 3 mm caudal to the injury epicenter were quantitatively examined for GFAP-positive glial cells in the ventral funiculi, a white matter region with axonal tracts that has important roles in rat locomotion (31, 32). Minocycline treatment demonstrated an overall significant effect on reducing the number of reactive astroglia (Fig. 4_d_) in the ventral funiculi (18.0 ± 2.3 astroglia per 20 μm of tissue in the minocycline-treated group vs. 26.1 ± 1.9 astroglia per 20 μm of tissue in the control group; P = 0.017, unpaired Student's t test) (Fig. 4).
Systemic minocycline administration also protected oligodendrocytes. Because CNPase is a diffused signal linked with myelin sheets and cell soma, we compared the immunoreactivity in terms of difference in luminosity between the two groups in the ventral funiculi of the spinal cord sections 4 mm caudal to the epicenter (20). A significantly greater luminosity was detected in the treated group than in the controls (73.8 ± 3.5 luminosity units vs. 26.1 ± 1.9 luminosity units, respectively; P < 0.001, unpaired Student's t test; 1 section per rat, n = 8 rats per group) (Fig. 5_d_, where black = luminosity unit 0 and white = luminosity unit 256).
Fig. 5.
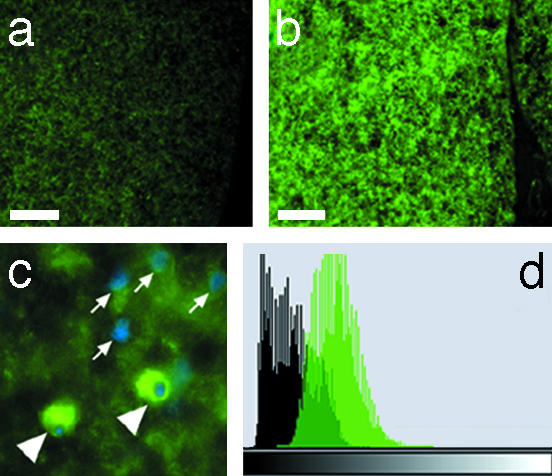
(a and b) CNPase immunocytochemistry demonstrating increased oligodendrocyte preservation in minocycline-treated rat spinal cord ventral funiculi 4 mm caudal to the epicenter (b) vs. that in the controls (a) (n = 8 rats per group). (Scale bar = 75 μm). (c) Anti-CNPase and 4′,6-diamidino-2-phenylindole (DAPI) double-staining, demonstrating oligodendroglia (arrowheads) as well as the nuclei of cells not reacting with the anti-CNPase antibody (arrows). (d) Composite histograms of total luminosities summarized from each experimental group showed a significant difference in anti-CNPase immunoreactivity (i.e., brightness intensity of the immunostain as measured by luminosity units) between the minocycline-treated (green) vs. the vehicle-treated (black) rats (n = 8 rats per group, with group average luminosity of 73.8 ± 3.5 luminosity units vs. 26.1 ± 1.9 luminosity units, respectively; P < 0.001, unpaired Student's t test). The histograms represent the distribution of total pixels (y axis) in the measured sections (480,000 pixels per section, 1 section per rat, and 8 sections per group for a total of 3.84 million pixels per group) of each group, plotted against luminosity units (x axis) where luminosity ranges from luminosity unit 0 (black) to 256 (white).
Discussion
Our results demonstrate that minocycline administered systemically 1 h after SCI significantly diminishes mitochondrial cytochrome c release resulting from SCI and reduces functional deficits starting 7 days p.i. The minocycline-treated group recovered hind limb reflexes more rapidly, and a higher percentage of these rats regained responses comparable with those of noninjured rats. The treated rats also achieved a faster and greater degree of recovery of coordinated hind limb use in maintaining position in a downward orientation on an inclined plane (Fig. 2_a_). The overall hind limb functional improvement, as measured by the BBB scale, was significantly enhanced in the minocycline-treated group relative to vehicle-treated controls 21 and 28 days p.i. (Fig. 2_b_). Additionally, there were no notable side effects at the 90 mg/kg i.p. dosing regimen. Our data show that the therapeutic outcome of minocycline most likely results from its comprehensive effects of blockade of cytochrome c release, resulting in protection of white matter, oligodendrocytes, and neurons and in inhibition of astrogliosis.
The hind limb deficits in this model of lower thoracic SCI are largely due to loss of white matter axonal tracts (24, 32). To a much lesser degree, the death of motoneurons related to trunk stability (33) is an additional contributing factor. The white matter degeneration is caused by the primary injury (i.e., mechanical lesion) and secondary injury events such as Na+ influx-mediated intraaxonal Ca2+ accumulation leading to proteinase activation, which destroys the cytoskeleton (24, 34), as well as the induction of oligodendroglial apoptosis with subsequent demyelination of the surviving axons (35, 36). Earlier work demonstrated that minocycline exerts remarkable neuroprotective effects in models of cerebral ischemia (13, 16), Huntington's disease, Parkinson's disease (14, 17), multiple sclerosis, and amyotrophic lateral sclerosis (12). Under these pathophysiological conditions, members of the caspase family of enzymes responsible for cell death, in particular caspase-1 and caspase-3, are activated and have important roles in the pathogenesis of these diseases (14, 16). Recently, we have demonstrated that mitochondria are a key target for minocycline's broad-spectrum neuroprotective action (12, 37). Minocycline works by inhibiting permeability transition-mediated release of cytochrome c from the mitochondria into the cytoplasm, where cytochrome c becomes a potent stimulus for caspase-9 and caspase-3 activation (38). This cascade of events was observed in a similar rat SCI model and was suggested to be potentially responsible for delayed cell death in white matter pathology (9). In addition to the caspases, inducible nitric oxide synthase (iNOS) is also up-regulated and activated under these circumstances (14, 16, 39). It has been shown that minocycline-mediated neuroprotection is associated with the inhibition of caspase-1, caspase-3, and iNOS transcriptional up-regulation and activation (14-17). Moreover, minocycline-mediated neuroprotection has been linked with iminocycline's inhibition of p38 mitogen-activated protein kinase and microglial activation (16, 40). Notably, caspase activation, iNOS up-regulation, and activated microglia have been identified as major secondary injury events responsible for a significant proportion of the neuronal and glial death resulting from experimental SCI (9, 35, 36, 41). In particular, post-SCI oligodendroglial apoptosis is a lengthy phenomenon that lasts for days or even weeks (35, 36). However, previous reports showed that pharmacological strategies aimed at countering apoptosis by directly inhibiting caspases or protein synthesis as well as by suppressing iNOS needed to be carried out immediately p.i. to achieve therapeutic effectiveness in experimental SCI. These reports left doubts as to whether such targets could be therapeutically practical, as seen in efforts to antagonize other commonly known secondary injury mechanisms such as glutamate toxicity, free radical damages, and ionic imbalances (see refs. 10, 36, and 41; also see reviews in refs. 5 and 6). Hence, it is essential to seek therapeutic targets that are further upstream in mediating caspase activation-related cell death.
Our treatment regimen consisted of an initial high dose of minocycline (90 mg/kg) followed by a maintenance dose of 45 mg/kg every 12 h for 5 days. The total dose of 495 mg/kg per rat is significantly higher than those used in previous studies (12-17, 42). This pharmacological design produces an effective plasma concentration immediately after the first dose, which was sustained by succeeding doses over 5 days, a critical period for apoptotic neuronal and glial death as well as microglial activation after SCI (35, 36, 43, 44). We observed a dramatic increase in the level of cytosolic cytochrome c that peaked 4-8 h p.i., a period that offers a pragmatic therapeutic window (Fig. 1 a and b). Our finding is consistent with an earlier report in which mitochondrial cytochrome c release peaked 4 h p.i., after a more severe SCI produced by the same type of impactor (9). We applied minocycline 1 h after a mild contusion injury, the time at which necrotic pathology in the gray matter (i.e., neurons) becomes plain at the injury epicenter (45). However, nonnecrotic and apoptotic cell death in tissues adjacent to the epicenter generally takes hours or even weeks to complete (26, 35, 36, 46). Thus, the outcome of this study suggests that a significant portion of the secondary tissue loss in SCI may result from cytosolic cytochrome _c_-mediated neuronal and glial death (9, 12). Minocycline, therefore, is a very promising therapeutic intervention for SCI, because of its multifaceted antisecondary injury properties: counteracting the spark-off mechanism of cytochrome c and, simultaneously, directly inhibiting downstream caspase activation and other key degenerative events such as inducible nitric oxide synthase (iNOS) up-regulation, microglial activation, and reactive astrogliosis.
Our histopathological data strongly support these notions. For example, our treatment regimen did not alter the average size of lesion at the injury epicenters, a pathological feature largely defined by the early necrotic process (26, 45). Minocycline, however, significantly spared white matter and increased ventral horn motoneuron survival at spinal cord levels adjacent to the injury site (Fig. 3 c and d), where neural cell apoptosis was found to be active during similar post-SCI time courses (35, 36). More importantly, the strong correlation between white matter preservation and enhanced chronic functional recovery further supports the hypothesis that minocycline treatment in this SCI model recovers hind limb function by reducing later-phase white matter loss that contributes significantly to the deleterious consequences of SCI. Additionally, there was a discernable reduction in the number of reactive astrocytes and an augmented survival of oligodendrocytes in the spared white matter (Figs. 4 and 5). Reactive astrocytes release inhibitory molecules such as neurocan and trabecular meshwork-inducible glucocorticoid response protein (TIGR) to further restrict neuronal repair and regeneration (27, 47), and astroglial scarring prevents axonal regeneration (27). There is also evidence that post-SCI demyelination caused by oligodendrocyte death/malfunction contributes significantly to chronic SCI functional deficits (48, 49).
Our rationale is further supported by behavioral results in the rats receiving minocycline after SCI. The overall hind limb functional improvement as assessed by the BBB locomotor score only became statistically significant 21 days p.i., a time frame suggesting that neither minimization of necrosis nor axonal regeneration is the likely underlying mechanism. Reduction in necrosis would have resulted in an earlier recovery pattern, whereas axonal regeneration would have resulted in a much more prolonged period before recovery. Hence, white matter sparing with less gliosis and greater oligodendrocyte and motoneuron survival may best explain the beneficial outcomes of minocycline treatment observed in this experimental model of SCI.
The significant functional hind limb improvement 4 weeks p.i., which was linked with significant neuronal and axonal protection and mitigation of astrogliosis and was not associated with any observable side effects of the treatment, is consistent with previously published data on other neurological disorders. It is also consistent with a recent report (42) in which systemic minocycline treatment starting 1 h p.i. significantly improved hind limb function in mice by decreasing lesion size and axonal loss. The histopathological discrepancy between the two studies can be largely explained by the different pathological responses between the two species (e.g., unlike in clinical SCI, in mice SCI essentially causes no cyst formation) (50). In a separate study, we found that methylprednisolone (30 mg/kg i.v., administered 1 h p.i., followed by 5.4 mg/kg i.v. for 23 h), the currently recommended pharmacological treatment for acute clinical SCI, did not improve functional recovery as evaluated by the BBB score in this model of SCI relative to the controls (n = nine rats per group). Therefore, the present finding supports our hypothesis that minocycline, by blocking an upstream event that triggers delayed neural cell death and the broad-spectrum neuroprotective effects of mitochondrial release of cytochrome c, can ameliorate functional deficits after traumatic SCI. Furthermore, the significant functional improvement after SCI was achieved by the systemic administration of minocycline at 1 h p.i., a time window that is practical for administering the drug in the field. Therefore, our results, together with the safety record of minocycline and its ability to penetrate the blood-brain barrier (51), suggest that it may become a clinical therapy for SCI.
Acknowledgments
Xiao Yu assisted in tissue cryostat processing. This work was supported by a Veterans Affairs Medical Research Grant, Eastern PVA VA B2958O, National Institutes of Health Grant 1-R21-NS41999, National Institute of Neurological Disorders and Stroke Grants RO1 NS39324 and NS41635, and by Project ALS.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SCI, spinal cord injury; p.i., postinjury; GFAP, glial fibrillary acidic protein; CNPase, 2′,3′ cyclic nucleotide 3′ phosphodiesterase.
References
- 1.Riggins, R. S. & Kraus, J. F. (1977) J. Trauma 17**,** 126-133. [DOI] [PubMed] [Google Scholar]
- 2.Kurtzke, J. F. (1977) Neurol. Neurocir. Psiquiatr. 18**,** 157-191. [PubMed] [Google Scholar]
- 3.Anderson, T. E. & Stokes, B. T. (1992) J. Neurotrauma 9**,** S135-S142. [PubMed] [Google Scholar]
- 4.Hulsebosch, C. E. (2002) Adv. Physiol. Educ. 26**,** 238-255. [DOI] [PubMed] [Google Scholar]
- 5.Blight, A. R. (2002) Nat. Neurosci. Suppl. 5, 1051-1054. [DOI] [PubMed]
- 6.Hurlbert, R. J. (2001) Spine 26**,** S39-S46. [DOI] [PubMed] [Google Scholar]
- 7.McTigue, D. M., Popovich, P. G., Jakeman, L. B. & Stokes, B. T. (2002) Prog. Brain Res. 128**,** 3-8. [DOI] [PubMed] [Google Scholar]
- 8.Bracken, M. B., Shepard, M. J., Collins, W. F., Holford, T. R., Young, W., Baskin, D. S., Eisenberg, H. M., Flamm, E., Leo-Summers, L., Maroon, P. H., et al. (1990) N. Engl. J. Med. 322**,** 1405-1411. [DOI] [PubMed] [Google Scholar]
- 9.Springer, J. E., Azbill, R. D. & Knapp, P. E. (1999) Nat. Med. 5**,** 943-946. [DOI] [PubMed] [Google Scholar]
- 10.Li, M., Ona, V. O., Chen, M., Kaul, M., Tenneti, L., Zhang, X., Stieg, P. E., Lipton, S. A. & Friedlander, R. M. (2000) Neuroscience 99**,** 333-342. [DOI] [PubMed] [Google Scholar]
- 11.Emery, E., Aldana, P., Bunge, M. B., Puckett, W., Srinivasan, A., Keane, R. W., Bethea, J. & Levi, A. D. (1998) J. Neurosurg. 89**,** 911-920. [DOI] [PubMed] [Google Scholar]
- 12.Zhu, S., Stavrovskaya, I. G., Drozda, M., Kim, B. Y., Ona, V., Li, M., Sarang, S., Liu, A. S., Hartley, D. M., Wu, C., et al. (2002) Nature 417**,** 74-78. [DOI] [PubMed] [Google Scholar]
- 13.Arvin, K. L., Han, B. H., Du, Y., Lin, S. Z., Paul, S. M. & Holtzman, D. M. (2002) Ann. Neurol. 52**,** 54-61. [DOI] [PubMed] [Google Scholar]
- 14.Chen, M., Ona, V. O., Li, M., Ferrante, R. J., Fink, K. B., Zhu, S., Bian, J., Guo, L., Farrell, L. A., Hersch, S. M., et al. (2000) Nat. Med. 6**,** 797-801. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez Mejia, R. O., Ona, V. O., Li, M. & Friedlander, R. M. (2001) Neurosurgery 48**,** 1393-1399. [DOI] [PubMed] [Google Scholar]
- 16.Yrjanheikki, J., Keinanen, R., Pellikka, M., Hokfelt, T. & Koistinaho, J. (1998) Proc. Natl. Acad. Sci. USA 95**,** 15769-15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu, D. C., Jackson-Lewis, V., Vila, M., Tieu, K., Teismann, P., Vadseth, C., Choi, D. K., Ischiropoulos, H. & Przedborski, S. (2002) J. Neurosci. 22**,** 1763-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tikka, T. M. & Koistinaho, J. E. (2001) J. Immunol. 166**,** 7527-7533. [DOI] [PubMed] [Google Scholar]
- 19.Basso, D. M., Beattie, M. S. & Bresnahan, J. C. (1996) Exp. Neurol. 139**,** 244-256. [DOI] [PubMed] [Google Scholar]
- 20.Teng, Y. D., Mocchetti, I. & Wrathall, J. R. (1998) Eur. J. Neurosci. 10**,** 798-802. [DOI] [PubMed] [Google Scholar]
- 21.Teng, Y. D., Mocchetti, I., Taveira-DaSilva, A. M., Gillis, R. A. & Wrathall, J. R. (1999) J. Neurosci. 19**,** 7037-7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gale, K., Kerasidis, H. & Wrathall, J. R. (1985) Exp. Neurol. 88**,** 123-134. [DOI] [PubMed] [Google Scholar]
- 23.Teng, Y. D., Lavik, E. B., Qu, X., Ourednik, J., Zurakowski, D., Langer, R. & Snyder, E. Y. (2002) Proc. Natl. Acad. Sci. USA 99**,** 3024-3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teng, Y. D. & Wrathall, J. R. (1997) J. Neurosci. 17**,** 4359-4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basso, D. M., Beattie, M. S. & Bresnahan, J. C. (1995) J. Neurotrauma 12**,** 1-21. [DOI] [PubMed] [Google Scholar]
- 26.Noble, L. J. & Wrathall, J. R. (1989) Exp. Neurol. 103**,** 34-40. [DOI] [PubMed] [Google Scholar]
- 27.Jones, L. L., Margolis, R. U. & Tuszynski, M. H. (2003) Exp. Neurol. 182**,** 399-411. [DOI] [PubMed] [Google Scholar]
- 28.Morin-Richaud, C., Feldblum, S. & Privat, A. (1998) Brain Res. 783**,** 85-101. [DOI] [PubMed] [Google Scholar]
- 29.Nessler, S., Dodel, R., Bittner, A., Reuss, S., Du, Y., Hemmer, B. & Sommer, N. (2002) Ann. Neurol. 52**,** 689-690. [DOI] [PubMed] [Google Scholar]
- 30.Rivlin, A. S. & Tator, C. H. (1977) J. Neurosurg. 47**,** 577-581. [DOI] [PubMed] [Google Scholar]
- 31.Guth, L., Albuquerque, E. X., Deshpande, S. S., Barrett, C. P., Donati, E. J. & Warnick, J. E. (1980) J. Neurosurg. 52**,** 73-86. [DOI] [PubMed] [Google Scholar]
- 32.Blight, A. R. (1983) Neuroscience 10**,** 521-543. [DOI] [PubMed] [Google Scholar]
- 33.Holstege, G. (1991) Prog. Brain Res. 87**,** 307-421. [DOI] [PubMed] [Google Scholar]
- 34.Stys, P. K., Waxman, S. G. & Ransom, B. R. (1992) J. Neurosci. 12**,** 430-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowe, M. J., Bresnahan, J. C., Shuman, S. L., Masters, J. N. & Beattie, M. S. (1997) Nat. Med. 3**,** 73-76. [DOI] [PubMed] [Google Scholar]
- 36.Liu, X. Z., Xu, X. M., Hu, R., Du, C., Zhang, S. X., McDonald, J. W., Dong, H. X., Wu, Y. J., Fan, G. S., Jacquin, M. F., et al. (1997) J. Neurosci. 15**,** 5395-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, X., Zhu, S., Drozda, M., Zhang, W., Stavrovskaya, I. G., Cattaneo, E., Ferrante, R. J., Kristal, B. S. & Friedlander, R. M. (2003) Proc. Natl. Acad. Sci. USA 100**,** 10483-10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green, D. R. & Reed, J. C. (1998) Science 281**,** 1309-1312. [DOI] [PubMed] [Google Scholar]
- 39.Almer, G., Vukosavic, S., Romero, N. & Przedborski, S. (1999) J. Neurochem. 72**,** 2415-2425. [DOI] [PubMed] [Google Scholar]
- 40.Tikka, T., Fiebich, B. L., Goldsteins, G., Keinanen, R. & Koistinaho, J. (2001) J. Neurosci. 21**,** 2580-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatzipanteli, K., Garcia, R., Marcillo, A. E., Loor, K. E., Kraydieh, S. & Dietrich, W. D. (2002) J. Neurotrauma 19**,** 639-651. [DOI] [PubMed] [Google Scholar]
- 42.Wells, J. E., Hurlbert, R. J., Fehlings, M. G. & Yong, V. W. (2003) Brain 126**,** 1628-1637. [DOI] [PubMed] [Google Scholar]
- 43.Cunha, B. A., Comer, J. B. & Jonas, M. (1982) Med. Clin. North Am. 66**,** 293-302. [DOI] [PubMed] [Google Scholar]
- 44.Chang, H. R., Comte, R., Piguet, P. F. & Pechere, J. C. (1991) J. Antimicrob. Chemother. 27**,** 639-645. [DOI] [PubMed] [Google Scholar]
- 45.Balentine, J. D. (1978) Lab. Invest. 39**,** 236-253. [PubMed] [Google Scholar]
- 46.Balentine, J. D. (1978) Lab. Invest. 39**,** 254-266. [PubMed] [Google Scholar]
- 47.Jurynec, M. J., Riley, C. P., Gupta, D. K., Nguyen, T. D., McKeon, R. J. & Buck, C. R. (2003) Mol. Cell. Neurosci. 23**,** 69-80. [DOI] [PubMed] [Google Scholar]
- 48.Beattie, M. S., Farooqui, A. A. & Bresnahan, J. C. (2000) J. Neurotrauma 7**,** 915-925. [DOI] [PubMed] [Google Scholar]
- 49.Kakulas, B. A. (1999) J. Spinal Cord Med. 22**,** 119-124. [DOI] [PubMed] [Google Scholar]
- 50.Steward, O., Schauwecker, P. E., Guth, L., Zhang, Z., Fujiki, M., Inman, D., Wrathall, J., Kempermann, G., Gage, F. H., Saatman, K. E., et al. (1999) Exp. Neurol. 157**,** 19-42. [DOI] [PubMed] [Google Scholar]
- 51.Saivin, S. & Houin, G. (1988) Clin. Pharmacokinet. 15**,** 355-366. [DOI] [PubMed] [Google Scholar]