Rearrangements within human spliceosomes captured after exon ligation (original) (raw)
In spliceosomes, dynamic RNA/RNA and RNA/protein interactions position the pre-mRNA substrate for the two chemical steps of splicing. Not all of these interactions have been characterized, in part because it has not been possible to arrest the complex at clearly defined states relative to chemistry. Previously, it was shown in yeast that the DEAD/H-box protein Prp22 requires an extended 3′ exon to promote mRNA release from the spliceosome following second-step chemistry. In line with that observation, the authors find that shortening the 3′ exon blocks cleaved lariat intron and mRNA release in human splicing extracts, which allows stalling human spliceosomes in a new post-catalytic complex (P complex). In comparison to C complex, which is blocked at a point following first-step chemistry, they detect specific differences in RNA substrate interactions near the splice sites. These differences include extended protection across the exon junction and changes in protein crosslinks to specific sites in the 5′ and 3′ exons.
Keywords: pre-mRNA splicing, spliceosome, mass spectrometry, crosslinking
Abstract
In spliceosomes, dynamic RNA/RNA and RNA/protein interactions position the pre-mRNA substrate for the two chemical steps of splicing. Not all of these interactions have been characterized, in part because it has not been possible to arrest the complex at clearly defined states relative to chemistry. Previously, it was shown in yeast that the DEAD/H-box protein Prp22 requires an extended 3′ exon to promote mRNA release from the spliceosome following second-step chemistry. In line with that observation, we find that shortening the 3′ exon blocks cleaved lariat intron and mRNA release in human splicing extracts, which allowed us to stall human spliceosomes in a new post-catalytic complex (P complex). In comparison to C complex, which is blocked at a point following first-step chemistry, we detect specific differences in RNA substrate interactions near the splice sites. These differences include extended protection across the exon junction and changes in protein crosslinks to specific sites in the 5′ and 3′ exons. Using selective reaction monitoring (SRM) mass spectrometry, we quantitatively compared P and C complex proteins and observed enrichment of SF3b components and loss of the putative RNA-dependent ATPase DHX35. Electron microscopy revealed similar structural features for both complexes. Notably, additional density is present when complexes are chemically fixed, which reconciles our results with previously reported C complex structures. Our ability to compare human spliceosomes before and after second-step chemistry has opened a new window to rearrangements near the active site of spliceosomes, which may play roles in exon ligation and mRNA release.
INTRODUCTION
Pre-mRNA splicing is catalyzed by a macromolecular ribonucleoprotein complex known as the spliceosome. Spliceosomes are composed of over 100 distinct components, including five small nuclear ribonucleoproteins (snRNPs). Each of the five snRNPs (U1, U2, U4, U5, U6) contains a small uridine rich RNA and associated proteins. During the process of splicing, the U snRNPs are sequentially recruited to a pre-mRNA substrate to assemble spliceosomes, which undergo ATP-dependent rearrangements to form intermediates known as E, A, B, Bact, B*, and C complexes (Will and Luhrmann 2011).
The association and interactions of components in the spliceosome is highly dynamic. Proteomic studies have shown that many proteins join and/or leave the complex at each assembly stage (Will and Luhrmann 2011). In yeast, eight members of the DExD/H box RNA-dependent ATPase family have been shown to promote several spliceosome rearrangements. For example, in the activated spliceosome (B*), Prp2 activity destabilizes the association of the U2 snRNP subcomplexes SF3a and SF3b and promotes first-step chemistry that results in cleavage at the 5′ splice site and formation of the lariat intron intermediate (Kim and Lin 1996; Silverman et al. 2004; Lardelli et al. 2010). In later stages, Prp16 promotes a conformational rearrangement required for second-step chemistry, which cleaves the 3′ splice and ligate the exons (Schwer and Guthrie 1991, 1992). After exon ligation, Prp22 promotes mRNA release in an ATP-dependent manner (Schwer and Gross 1998), followed by the displacement of the cleaved intron from U2, U5, and U6 snRNPs by Prp43 (Tsai et al. 2005). In the human spliceosome, two additional predicted DExD/H box ATPases, DDX41 and DHX35, are also present in catalytic spliceosomes (Jurica et al. 2002). These proteins do not have clear Saccharomyces cerevisiae orthologs, and their potential roles in splicing are not yet known.
During catalytic activation of spliceosomes, structural rearrangements must occur to position the pre-mRNA at the active sites for the two steps of splicing chemistry. In activation of the spliceosome for first-step chemistry, the complex must bring the 2′-OH of the branch point adenosine to attack the 5′ splice site (5′ ss). To promote this pre-mRNA conformation, U2 snRNA base-pairs with the branch point sequence (Parker et al. 1987; Wu and Manley 1989), U6 snRNA base-pairs near the 5′ ss (Wu and Manley 1991; Sawa and Abelson 1992), and both of these snRNAs base-pair with each other (Datta and Weiner 1991; Madhani and Guthrie 1992; Wassarman and Steitz 1993; Sun and Manley 1995). Additional contacts to the region upstream of the 5′ ss by U5 snRNA (Newman et al. 1995), as well as by the U5 snRNP protein Prp8, are proposed to also stabilize the first-step active site conformation (Wyatt et al. 1992; Teigelkamp et al. 1995a,b; Chiara et al. 1996; Reyes et al. 1996, 1999).
In preparing for the second step of splicing, spliceosomes must next bring the 3′-OH of the 5′ exon to attack at the 3′ splice site (3′ ss) for exon ligation. In spliceosomes blocked for second-step chemistry by either mutation of the 3′ ss or inactivating Prp16, Prp8 has been shown to crosslink just downstream from the 3′ ss (Teigelkamp et al. 1995a,b; Umen and Guthrie 1995; Chiara et al. 1996; McPheeters et al. 2000; McPheeters and Muhlenkamp 2003). U5 snRNA also crosslinks with the 3′ exon in the lariat intermediate (Wassarman and Steitz 1992; Newman et al. 1995). How formation of these contacts relates to second-step chemistry is unknown. When the second step is blocked by these different methods, it is not clear whether the spliceosome remains in first-step active site conformation, has transitioned to second-step active site conformation, or exists in an uncharacterized intermediate conformation. In order to clarify active site interactions related to second-step chemistry, a snapshot of the spliceosome following exon ligation, but prior to mRNA release, is required.
In this study, we have captured the human spliceosome in a post-catalytic state that we term P complex. Biochemical and EM analysis confirms that purified P complex is a large stable splicing complex that, along with C complex, which is stalled after the first step of splicing, now allows us to further examine the spliceosome before and after exon ligation. Comparing the protein composition and pre-mRNA interactions of P complex to C complex, we find distinct differences that speak to the changes in active site structure that occur in the process of exon ligation. Our studies provide insight into how spliceosomes rearrange pre-mRNA in the active site to facilitate the second step of splicing chemistry.
RESULTS
Shortening the 3′ exon stalls the human spliceosome after second-step chemistry but prior to mRNA release
Previous work in yeast showed that, after the second step of splicing, Prp22 contacts the 3′ exon and uses its helicase activity to promote mRNA release (Schwer 2008). In that study, the yeast spliceosome was also demonstrated to retain the mRNA product following second-step chemistry if the 3′ exon is shortened to 13 nt to make the binding site for Prp22 unavailable. We wondered whether shortening the 3′ exon of the pre-mRNA substrate would also inhibit mRNA release with the human splicing machinery. To test this hypothesis, we created a series of pre-mRNA substrates with 3′ exons ranging in length from 6 to 40 nt for in vitro splicing in a HeLa nuclear extract. After denaturing polyacrylamide gel electrophoresis (PAGE), we examined the radiolabeled RNA splicing products by phosphorimaging. With all of the substrates, mRNA appears over time, which indicates that neither step of splicing chemistry was blocked by shortening the 3′ exon (Fig. 1A, lanes 5–9; Supplemental Fig. S1).
FIGURE 1.

A short 3′ exon pre-mRNA substrate stalls human spliceosomes between the second step of splicing and mRNA release. (A) Denaturing PAGE analysis of in vitro splicing reactions with radiolabeled pre-mRNA substrates containing a mutated 3′ splice site and a 51-nt 3′ exon (mut 3′ ss), wild-type 3′ splice site and a 51-nt 3′ exon (wt 3′ ss), or wild type 3′ splice site and a 19-nt 3′ exon (short 3′ exon). Reaction time points, in minutes, are noted at the top of the gel, and splicing intermediates are indicated on the left as, from top to bottom, lariat-intron intermediate, lariat intron, pre-mRNA, mRNA, and 5′ exon. The different positions of pre-mRNA, lariat-intron intermediate, and mRNA with 51- vs. 19-nt 3′ exons are indicated by brackets. (B) Glycerol gradient (10%–30%) sedimentation of splicing reactions using the same pre-mRNA substrates as in A. All three panels show denaturing PAGE analysis of fractions collected from top to bottom of the gradient (1_–_16, respectively). Quantification of indicated band intensities vs. fraction number is graphed at the right. (C) Native gel analysis of splicing reactions correlating to those shown in A. The position of H/E, A, B, C/C-like, and mRNP complexes are indicated on the left.
We next used glycerol gradient centrifugation to determine whether the spliceosome releases mRNA with a short 3′ exon. We separated the splicing reactions on 10%–30% glycerol gradients and examined RNA from different fractions by denaturing PAGE. As a control for the migration of the catalytic spliceosome (C complex) in the gradient, we used a pre-mRNA substrate with a mutant 3′ ss that blocks splicing after first-step chemistry. As expected with this substrate, the free 5′ exon and lariat intermediate comigrate deep in the gradient, consistent with them associating in a large complex (Fig. 1B, top panel, lanes 10,11). When we used a pre-mRNA substrate with a wild-type 3′ ss and the normal 51-nt 3′ exon, both the lariat intron and mRNA peak earlier in the gradient, which indicates that they have been released from the spliceosome (Fig. 1B, middle panel, lanes 7,8). However, when we truncated the 3′ exon of the pre-mRNA to 19 nt, the mRNA peak is deeper in the gradient and nearly overlaps with the small amounts of remaining first-step intermediates (Fig. 1B, bottom panel, lanes 10,11). This result is consistent with retention of the mRNA with the short 3′ exon in a large complex.
We tested additional substrates with 3′ exons of different lengths (Supplemental Fig. S1). With shorter 3′ exons, more mRNA was retained in the spliceosome. However, as the 3′ exons are shortened, there is a decrease in second-step chemistry. In contrast, with longer 3′ exons, second-step chemistry increases but with more mRNA release. For the studies of splicing complexes trapped after second-step chemistry described below, we chose to use a pre-mRNA with a 19-nt 3′ exon because it exhibited the highest accumulation of post-catalytic splicing complexes that retain the mRNA and cleaved intron, which we now refer to as P complex.
Native gel analysis of splicing reactions using pre-mRNA substrate with the short 3′ exon confirms normal spliceosome assembly (E→A→B→C). However, as the reaction progresses, a large complex that migrates at the same position as C complex accumulates (Fig. 1C, lanes 2,5–9). In contrast, full-length WT pre-mRNA substrate that completes the second step and releases mRNP does not show the same accumulation of the C-like complex (Fig. 1C, lane 4). From this and the glycerol gradient data, we conclude that P complex is a large stable splicing complex stalled between exon ligation and mRNA release.
Protection of the mRNA splice junction in P complex
Because P complex maintains its hold on mRNA, regions of that mRNA that are stably associated with spliceosome components will likely not be available to hybridize to complementary DNA probes. To identify these regions, we performed protection assays against RNase H digestion. In these assays, we assembled P complex on pre-mRNA substrate with the short 3′ exon in a HeLa nuclear extract and then added a 100-fold excess of a series of 12- to 16-nt DNA oligos complementary to sequences along the entire mRNA (Fig. 2A). If a region of the mRNA is not protected by spliceosome interactions, then the oligo will form a DNA/RNA hybrid that makes the mRNA sensitive to digestion at that site by endogenous RNase H in the nuclear extract. In contrast, if the targeted mRNA region is protected by a component of the spliceosome, no DNA/RNA hybrid will form, and the mRNA will not be cleaved. We visualized mRNA cleavage by phosphorimaging after denaturing PAGE. In the 5′ exon, regions >30 nt upstream of the exon junction are largely unprotected, as evidenced by a decrease of mRNA band intensity after digestion (Fig. 2B, lanes 1–6, compared to control lane U). In contrast, the entire 30-nt region upstream of the exon junction is protected from digestion (lanes 7–9). The exon junction through 9 nt of the 3′ exon is also protected (lanes 10,11). The extent of protection in the 5′ exon in P complex is similar to that previously shown for C complex (Ilagan et al. 2009). However, in C complex, the entire 3′ exon is unprotected (Fig. 2C). The additional protection in the 3′ exon in P complex could indicate a conformational rearrangement of spliceosome components upon the second step of chemistry. For released mRNA in mRNP, Le Hir et al. showed protection in an 8-nt region ∼24 nt upstream of the exon junction by the exon junction complex (EJC) but reported no protection across the exon junction and 3′ exon (Newman et al. 1995; Le Hir et al. 2000). These results suggest that the exon junction may remain situated in the second-step active site in P complex.
FIGURE 2.

RNase H protection mapping of mRNA in the post-catalytic spliceosome complex. (A) Sequence of mRNA with short 3′ exon (gray shaded) and DNA oligos used for RNase H digestion shown in B. (B) Denaturing PAGE analysis of splicing reactions using a pre-mRNA substrate with a short 3′ exon to which DNA oligos were added after 60 min to initiate RNase H digestion of complementary RNA sequences. Oligos targeted the mRNA diagrammed at the top in the 5′ exon (lanes 1–9), exon junction of the mRNA (lane 10), and 3′ exon (lane 11). Lane U is a splicing reaction in the absence of added DNA oligo. Splicing intermediates are indicated on the left as, from top to bottom, lariat-intron intermediate, lariat intron, pre-mRNA, mRNA, and 5′ exon, and the positions of RNase H cleavage products are shown. Because digestion products also arise from unspliced pre-mRNA, RNase H cleavage was determined by comparing mRNA band intensities with the no-oligo control (U) and reported as % cleaved at the bottom of each lane. (C) Same as B but with spliceosomes assembled on a pre-mRNA substrate with a 3′ ss mutation.
Changes in protein-exon contacts in P complex
To further examine interactions with pre-mRNA in the spliceosome before and after second-step chemistry, we performed site-specific UV crosslinking experiments targeting the protected regions that are likely close to the active site(s) in both C and P complexes. We incorporated a single 32P-label at positions −10 or −2 nt upstream of the 5′ ss and +1 nt downstream from the 3′ ss in pre-mRNA substrates with either a mutant 3′ ss (to form C complex) or short 3′ exon (to form P complex) (Fig. 3A). For the +1 crosslink, both complexes were assembled on a substrate with the identical truncated 3′ exon. After UV-crosslinking of the complexes assembled on these substrates, we digested with RNase A/T1 and separated proteins in the splicing reaction by denaturing PAGE. We then detected proteins crosslinked to the radiolabeled nucleotides by phosphorimaging (Fig. 3B). To control for protein binding not specific to splicing, we also crosslinked to single-labeled pre-mRNA incubated at 4°C in HeLa extract in the absence of ATP to form so-called H complex (Fig. 3B, lanes 1,4,7).
FIGURE 3.

UV crosslinking of proteins to exons in P complex. (A) Schematic of mRNA produced by a short 3′ exon (dark gray) pre-mRNA containing a single 32P (marked by an asterisk) at indicated nucleotide positions relative to the eventual exon junction (marked by the black bar). The labels at the same positions were also included in a pre-mRNA with a longer 3′ exon (light gray) and mutant 3′ ss. For the +1 label, a 4-thio-uridine residue (marked by a black dot) was also included, and the mutant 3′ ss substrate also had a short 3′ exon. (B) SDS-PAGE of proteins crosslinked near the indicated single 32P label by UV irradiation following in vitro splicing with the short 3′ exon pre-mRNA (P) or mutant 3′ ss pre-mRNA (C). As a control for proteins that bind in the absence of splicing, the labeled pre-mRNA was also incubated in splicing reactions lacking ATP at 4°C, followed by UV crosslinking (H). Positions of molecular weight marker proteins are indicated. Crosslinked protein bands detected only with the short 3′ exon pre-mRNA that accumulates P complex are marked by an asterisk (*).
With the label at −10 nt upstream of the 5′ ss, a radiolabeled protein of ∼70 kD is present in both C and P that is not present in H (Fig. 3B, lanes 2,3). Because there is a small amount of C complex (10%–20%) remaining in the P complex preparation, we cannot absolutely state that a protein crosslink identified in both complexes represents a stable interaction resulting from second-step chemistry. Nevertheless, because the −10 nt position has not been directly examined before for any splicing complex, this is an interesting observation. Five proteins in this size range have been identified in C complex by mass spectrometry: SLU7, CDC40 (Prp17 ortholog), PPWD1, BUD13, and DDX41. Unfortunately, we have not been able to identify the 70-kD protein because of the lack of antibodies with good immunoprecipitation efficiency. We also do not know at what point in spliceosome assembly this protein first contacts the 5′ exon, although it is notable that these proteins first stably associate in Bact (CDC40, BUD13) or C (SLU7, PPWD1, DDX41) complexes.
At the −2 nt position, an ∼250-kD protein weakly crosslinks in both C and P complexes and is not detected in H. This protein is likely PRPF8, which has been shown to crosslink at this site in splicing complexes before first-step chemistry (Wyatt et al. 1992; Teigelkamp et al. 1995a,b; Chiara et al. 1996; Reyes et al. 1996, 1999). Notably, an ∼30-kD protein strongly crosslinks at −2 nt in P, which is not detected in C or H complexes (Fig. 3B, lanes 5,6). Because this crosslink is found only with P complex, it very likely reflects a conformational change at the 5′ ss after exon ligation. A protein of this size has never been reported to crosslink at the 5′ splice site. Our mass spectrometry analysis of P complex (see below) identified several proteins in the size range of 26–34 kD that join through spliceosome assembly: PPIE, SYF2, BCAS2, ISY1, CWC15, C9orf78, SNRPA1 (U2 A′), LENG1, and PQBBP1. To narrow the field, we blotted the crosslinking gel and performed Western analysis for four of these proteins: PPIE, SYF2, BCAS2, and ISY1 (Supplemental Fig. S3). The BCAS2 antibody recognized two bands, both of which migrate lower than the crosslinked protein, and the band detected by the ISY1 antibody migrated higher than the crosslinked protein, indicating that, likely, neither of these proteins is the ∼30-kD protein. Western blots of NE with a SNRPA1 antibody identify a band of ∼25 kD (data not shown), which suggests that this is also not the crosslinked protein. In contrast, PPIE and SYF2 both migrated to the same position as the crosslinked protein, making them candidates for the 30-kD crosslink. Unfortunately, the antibodies used for these Westerns are also not efficient for immunoprecipitation, and so with the low efficiency of UV crosslinking and limited amounts of material, we have not been able to conclusively identify the protein.
To probe the +1 position in the 3′ exon, we incorporated a 4-thioU adjacent to the radiolabel to increase crosslinking efficiency. In C complex, we detect relatively weak crosslinks to proteins of ∼75 and 130 kD and a very weak crosslink to a 250-kD protein (Fig. 3B, lane 9). In P complex, there are similar weak crosslinks to ∼75- and 130-kD proteins, but the ∼250-kD protein crosslink was much stronger (Fig. 3B, lane 8). Again, the 250-kD protein is likely PRPF8, which was shown to crosslink in this region in complexes blocked for second-step chemistry by inactivation of Prp16 (Teigelkamp et al. 1995a,b; Umen and Guthrie 1995; Chiara et al. 1996; McPheeters et al. 2000; McPheeters and Muhlenkamp 2003). We cannot confidently attribute the difference in crosslinking efficiency of the protein between C and P as a conformational change in the spliceosome. Instead, the difference may reflect the 3′ ss mutation used to trap C complex. Our previous studies indicated that a mutated 3′ ss does not stably occupy the second-step active site (Ilagan et al. 2009). Therefore, the difference in crosslinking to the 250-kD protein between P and C may be due to the modified substrate, although second-step chemistry may still have an impact.
Affinity purification of P complex
To isolate P complex, we assembled spliceosomes on a short 3′ exon pre-mRNA and, as previously described, carried out affinity purification via an MS2-tag in the intron (Jurica et al. 2002). We hypothesized that, as suggested by our glycerol gradient analysis, mRNA would copurify along with the affinity-tagged lariat intron in P complex. Denaturing PAGE of radiolabeled RNA isolated from the purified complex confirmed that the mRNA is indeed present after affinity purification (Fig. 4A, short 3′ exon elution), showing that we have isolated a post-catalytic splicing complex.
FIGURE 4.

Affinity purification of P complex. Affinity purification was carried out using MS2-MBP tagged pre-mRNA substrates labeled as in Figure 1. (A) Denaturing PAGE analysis of RNA from in vitro splicing reaction time points (0′, 60′), following RNase H-mediated cleavage of substrate that did not incorporate into splicing complexes (indicated by *) as detailed in Jurica et al. (2002) (80′), the material loaded onto amylose affinity column (load), or eluted from the affinity column with maltose (elution). (B) Denaturing PAGE analysis of RNA extracted from purified spliceosome complexes, which are visualized by SYBRGold staining. RNA from nuclear extract (NE) contains U2, U1, U4, U5, and U6 snRNAs as labeled. (• indicates tRNA in the extract and * indicates pre-mRNA digestion product.) RNA from purified C complex (C), mRNP complex (M), and P complex (P) assembled on the three pre-mRNA substrates shown in panel A, respectively. The positions of cleaved 5′ exon and short mRNA that copurify are noted.
To compare the RNA composition of P complex, we also isolated C complex and mRNP by the same MS2-affinity tag in the intron or 3′ exon, respectively (Fig. 4A, right two panels). We analyzed RNA extracted from the three complexes by SYBRGold staining after denaturing PAGE (Fig. 4B). As expected, we observed U2, U5, and U6 snRNAs in C complex (Jurica et al. 2002) and no snRNA association with mRNP (Merz et al. 2007). P complex also contained the U2, U5, and U6 snRNAs, indicating that these components are maintained in the spliceosome after exon ligation.
Loss and stabilization of spliceosome proteins after second-step chemistry
To determine the protein composition of P complex, we affinity-purified spliceosomes and identified components by tandem mass spectrometry (MS/MS). Like the U snRNAs, many proteins associated with C complex are present in P complex (Table 1). These included U5 snRNP proteins, the Prp19 complex, and second-step factors. We did not identify any proteins that were not previously found in C complex and thus appear to be specifically recruited to the spliceosome following second-step chemistry.
TABLE 1.
Mass spectrometry analysis of purified C and P complexes
In order to compare C and P complex components quantitatively, we used selective reaction monitoring (SRM) mass spectrometry, which measures the amount of specific peptides in a complex mixture and allows relative amounts of individual proteins between two samples to be determined. For 38 spliceosome-associated proteins, we identified two peptides consistently sequenced in previous MS/MS analysis of spliceosomes and determined their liquid chromatography elution timing. We then used SRM to quantify these peptides in C and P complexes, which resulted in a list of peak areas for each peptide. We compared these areas for individual peptides between C and P as a log2 ratio. The log2 ratio values of 87% of protein peptide pairs were within one standard deviation of the mean, indicating that the data are, by and large, robust, and these are shown in Table 1 (peptide sequences are shown in Supplemental Table S2). The five peptide pairs that did not track closely together were not used for further analysis. Previously identified post-translational modifications (PTM) or protein degradation, which would result in differential loss of a peptide signal, are plausible explanations for the difference between them.
For the remaining proteins, the average P/C log2 ratio was 0.6 with a standard deviation of 1.2, and the distribution is shown in Supplemental Figure S4. Seventy-six percent of peptides have a log2 ratio within one standard deviation of the mean, suggesting their parent proteins are equally represented in both samples. These include Sm proteins, some U2 snRNP proteins, U5 snRNP, and Prp19 complex proteins, as well as second-step factors. DHX8 (the human Prp22 ortholog) also is equally represented in both samples. MS/MS peptide counts correlate with these results in most cases.
Six proteins showed an average P/C log2 ratio outside one standard deviation of the mean. Five of these predicted enrichment in P complex relative to C complex: RNF113A, SF3B2, SF3B3, PRP38A, and CACTIN. In contrast, DHX35 peptide ratios predicted high enrichment in C complex relative to P complex. Notably, MS/MS peptide counts also followed the same trend of enrichment for these proteins.
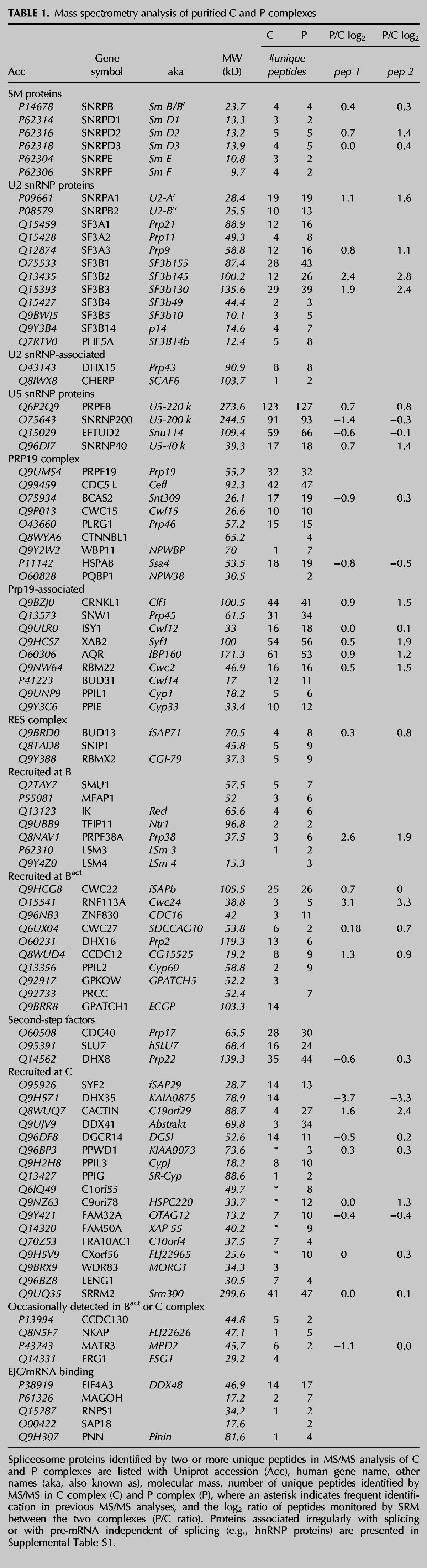
In order to confirm the SRM results, we performed Western analysis for six proteins that showed differential enrichment or no enrichment between C and P complexes (Fig. 5). We loaded the gel with 0.5 pmol of each complex (based on the amount of radiolabeled RNA splicing products) and then probed the same blot with different antibodies. We confirmed that there is some enrichment of SF3B3 in P complex and that DHX35 association is significantly reduced. The Western blot showed a very slight increase in the amount of PRPF8 in P complex, but the amounts of PRPF19, EIF4A3, and ISY1 were essentially equal, as predicted by the SRM and/or MS/MS analyses. One possible explanation for the loss of DHX35 is the difference in 3′ exon length of the substrate that we used to assemble the complex (55 vs. 20 nt). However, previous MS/MS analysis of C complex from which the 3′ exon is short or has been removed yielded a large number of DHX35 peptides (Ilagan et al. 2009; Bessonov et al. 2010; Agafonov et al. 2011). The difference in exon length only affected the amount of hnRNP and general RNA binding components and not the proteins that join through spliceosome assembly.
FIGURE 5.
Western analysis confirms enrichment of SF3B proteins and decrease of DHX35 in P complex. For Western analysis, equal amounts of purified C and P complexes were separated by denaturing PAGE, transferred to nitrocellulose, and the same blot was probed with the antibodies indicated on the left.
P and C complex have similar global structures that are stabilized by chemical crosslinking
We visualized negatively stained P complex by transmission electron microscopy (EM), which revealed monodispersed particles (Fig. 6A). Class-averaged views of these particles display similar dimensions (∼30 nm) and overall conformations as previously seen with C complex (Fig. 6B; Jurica et al. 2002).
FIGURE 6.
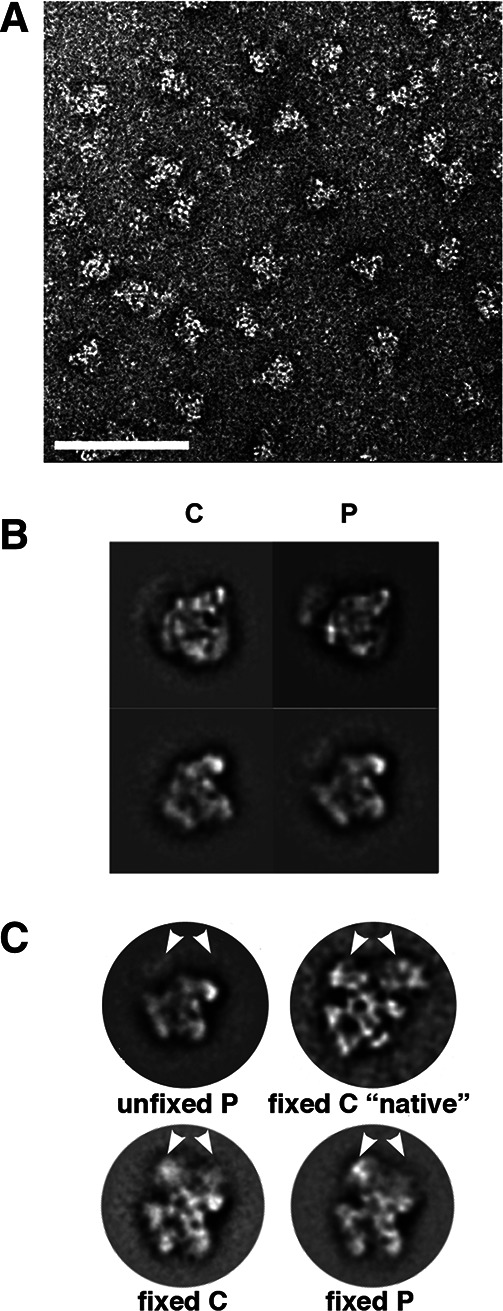
P and C complex have similar global structures that are stabilized by chemical crosslinking. (A) Transmission EM micrograph of negatively stained P complexes after affinity purification. White scale bar is 100 nm. (B) Two class-averaged views of C and P complex preferred orientations. (C) Comparisons of fixed and unfixed splicing complexes. “C native” is from Golas et al. (2010). White arrows point out the location of a density that is present only in fixed samples.
There has been a discrepancy between our structure of human C complex spliceosomes and that reported by the Lührmann and Stark groups, even though MS analyses by both groups suggest similar composition (Golas et al. 2010). Our complexes are distinctly smaller, and there is more “stuff” in the background of our EM images. In addition to slightly different purification methods, the Stark group chemically fixes samples prior to preparing EM grids. To see if fixation might account for the structural differences, we briefly treated samples of both P and C complex with glutaraldehyde prior to preparing EM grids. Images of these complexes show larger particles and a cleaner background, consistent with partial dissociation of spliceosome components during grid preparation of unfixed samples. Class-averaged views of these particles show that C and P complex still have very similar structures and are also similar to “native” C complex images from the Lührmann and Stark labs (Fig. 6C; Golas et al. 2010). Closer examination of the images of our unfixed complex images shows that they are similar to the 35S U5 snRNP complex but with some additional density, which may comprise U2 snRNP components and/or pre-mRNA. Part of the density that is missing from the unfixed C complex core is also missing from the Lührmann and Stark labs’ fixed salt-washed core of C complex (Golas et al. 2010). This suggests that the unfixed C complex core structure does not include proteins that join the spliceosome at Bact and C complex.
DISCUSSION
Understanding the RNA/RNA and RNA/protein interactions that position splice sites in the spliceosome for the two chemical steps of splicing is key to determining the molecular mechanisms of this large and dynamic complex. In this work, we establish a method to stall human spliceosomes following the second step of chemistry but before mRNA release (P complex). By comparing this post-catalytic complex to one stalled before the second step (C complex), we have characterized changes in spliceosome conformation that give insight into how the complex rearranges during splicing chemistry.
We accumulated P complex using a pre-mRNA substrate with a shortened 3′ exon. Work in yeast showed that the RNA-dependent ATPase Prp22 must contact the 3′ exon in an ATP-independent manner prior to promoting release of mRNA and that shortening the exon to <13 nt prevented this interaction (Schwer 2008). We find that human spliceosomes also require an extended 3′ exon in order to couple second-step chemistry with mRNA release. In this case however, at least 20 nt are required for some mRNA release to occur. This finding suggests that the human ortholog of Prp22, DHX8, uses a similar mechanism to promote mRNA release after exon ligation.
Comparing affinity-purified spliceosomes arrested before and after exon ligation (C and P complexes, respectively), we found both similarities and differences in the two states. Both complexes maintain the same snRNA components and nearly all the same proteins. In particular, MS/MS analysis did not identify any new proteins that stably associate with P complex spliceosomes.
The similarity between C and P complexes is also evident in the global structure that we observe by EM, with both complexes exhibiting roughly the same size and shape. Identifying specific differences between the EM structures will be challenging, given the limited resolution that is currently attainable for spliceosome complexes (Jurica et al. 2004; Behzadnia et al. 2007; Cohen-Krausz et al. 2007; Wolf et al. 2009; Bessonov et al. 2010; Golas et al. 2010). We were able to determine the basis of a puzzling difference in the structures of C complex spliceosomes reported by our lab and the Lürhmann/Stark group. Although we assemble and purify splicing complexes under similar conditions, our EM images of negatively stained complexes show somewhat smaller particles. We can now attribute this difference to chemical fixation of the complexes prior to EM grid preparation. A comparison of unfixed and fixed complexes reveals a significant region of density that appears to be stabilized by crosslinking. Comparisons of both EM images and MS/MS analysis of components between 35S U5 snRNP, C complex washed with high salt (Bessonov et al. 2008) and the U2/U5/U6 complex from Schizosaccharomyces pombe (Ohi et al. 2002, 2007) suggest that our unfixed core C complex (and P complex) particles contain U5 snRNP, U2 snRNP components, U6 snRNA, and PRP19 complex and related components. We previously showed that both exons are also present in the unfixed C complex core structure and that removing unprotected exon regions does not significantly alter its structure (Alcid and Jurica 2008; Ilagan et al. 2009). This comparison also suggests that a large part of the density that is stabilized by crosslinking comprises non-snRNP proteins that join at Bact and C complex. These results will be important for future interpretation and comparison of EM structures of splicing complexes.
Using an SRM mass spectrometry strategy for a more quantitative comparison of the protein components, we again see that, between C and P complexes, many proteins do not appear to change in their relative abundance, such as U5 snRNP and PRP19 complex proteins. However, there are distinct differences in the relative amounts of some spliceosome proteins. Two SF3b complex components, RNF113A, PRPF38A, and CACTIN were the most highly enriched in P complex, and Western analysis confirmed this observation for SF3B3. We also monitored single peptides for some additional spliceosome components (data not shown), including SF3B1, SF3B5, and PHF5A. High P/C log2 ratios for these SF3b proteins also indicated enrichment in P relative to C complex.
The difference for SF3b proteins suggests that its association with the spliceosome may be even more dynamic than previously thought. There is a potential relationship between SF3b stability in the spliceosome and toggling between U2 snRNA stem IIa and IIc structures. Formation of stem IIa in the U2 snRNP allows stable association of SF3b (Yan and Ares 1996). Some biochemical evidence suggests that SF3B2 may directly contact stem IIa (Kramer et al. 1999), and genetic interactions point to a functional relationship (Wells et al. 1996; Yan and Ares 1996). During prespliceosome assembly, the stem IIa conformation is required for stable U2 snRNP association with pre-mRNA and branch point recognition (Zavanelli and Ares 1991; Perriman and Ares 2007). Both of these rearrangements require the RNA-dependent ATPase Prp5. In a striking parallel, splicing inhibitors that target SF3b work by interfering with its role in stabilizing U2 snRNP association with pre-mRNA and promoting branch point recognition (Roybal and Jurica 2010; Corrionero et al. 2011; Folco et al. 2011).
Activation of the spliceosome for first chemistry requires stem IIc to form, although it is not clear if this occurs before, with, or after Prp2 ATPase activity that results in loss/destabilization of SF3b and Cwc24 (Hilliker et al. 2007; Perriman and Ares 2007; Warkocki et al. 2009; Lardelli et al. 2010; Ohrt et al. 2012). Following first-step chemistry, genetic suppression of cold-sensitive prp16 alleles suggests reformation of U2 stem IIa (Hilliker et al. 2007; Perriman and Ares 2007), while loss of Prp16 function correlates with destabilization of SF3b and Cwc24 association with spliceosomes (Lardelli et al. 2010). It is not clear what U2 snRNA conformation is present in spliceosomes arrested after second-step chemistry by loss of Prp22 activity, but both SF3b and Cwc24 are not present in yeast spliceosome isolated at this stage (Lardelli et al. 2010).
The conformation of U2 snRNA in yeast and human spliceosomes trapped by changes to the pre-mRNA substrate is also not known and may differ from spliceosomes trapped by inactivation of the ATPase activity of DEAD/H box proteins. Both Bact complex that accumulates on a substrate with a short polypyrimidine tract and no 3′ splice site and C complex that accumulates on a substrate with a longer polypyrimidine tract and missing or mutated 3′ splice site retain SF3b proteins, Cwc24/RNF113A and PRPF38A, but with less affinity than before catalytic activation (Jurica et al. 2002; Bessonov et al. 2008; Fabrizio et al. 2009; Ohrt et al. 2012). We previously found that, when C complex is challenged with mild denaturant, U2 snRNA and SNRPA1 (U2A′) and SNRPB2 (U2B′′) disassociated from the lariat intron intermediate, along with most U5 snRNP and PRP19 complex proteins (Coltri et al. 2011). In contrast, SF3b proteins remained with the lariat intron intermediate, along with DHX8 (Prp22) and a few other proteins. We interpret this to mean that SF3b switches affinity from U2 snRNA to the lariat intron. It would be interesting to know if this switch correlates with a toggle to the U2 snRNA stem IIc conformation.
The enrichment we observe for SF3b proteins RNF113A and PRPF38A in P complex indicates that rearrangements during or after exon ligation may restabilize their interactions with the spliceosome. The increase in these proteins is in contrast to yeast spliceosomes trapped in the post-catalytic state by mutations in Prp22, which lack these proteins (Lardelli et al. 2010). However, Beate Schwer showed that mutant Prp22 was still able to engage the 3′ exon, which is not the case for a truncated 3′ exon (Schwer 2008). It may be that other rearrangements associated with Prp22 engagement with the 3′ exon, such as destabilization of SF3b, have not yet have occurred in P complex. It will be interesting to challenge P complex with mild denaturant and use SRM to see if SF3b again shows higher affinity for the lariat intron than U2 snRNA. Knowing the conformation of U2 snRNA in C and P complexes would also be informative.
The role of RNF113A, which joins the spliceosome at Bact, is not clear, but there are hints of a link with U2 snRNP. RNF113A protein was recently shown to interact with SF3B2 by both yeast two-hybrid and coprecipitation (Hegele et al. 2012) and, in yeast, to play a role in stable association of U2 snRNP with pre-mRNA (Coltri and Oliveira 2012). Little is known about PRPF38A as well. It joins spliceosomes at B complex (Bessonov et al. 2008; Fabrizio et al. 2009; Herold et al. 2009; Agafonov et al. 2011), and the yeast ortholog, Prp38, has a role in spliceosome activation before U4 snRNP release (Xie et al. 1998). The protein CACTIN is even more of an enigma. It joins spliceosomes at C complex. CACTIN has been linked to regulation of the expression of Toll-like receptors and MHC class II gene by interacting with I-κB-like protein (Lin et al. 2000; Atzei et al. 2010), which is also found in some purifications of C complex.
DHX35, which joins the spliceosome at C complex (Jurica et al. 2002; Bessonov et al. 2010), is currently the only component that departs the complex upon exon ligation. DHX35 is a putative RNA-dependent ATPase, which does not have a S. cerevisiae ortholog. While additional studies are needed to define its specific role in splicing, our data now further limit DHX35 to a stage centered around second-step chemistry. Because this protein is not found in S. cerevisiae, its function may be coupled to splicing of pre-mRNAs with multiple introns or regulation of alternative splicing.
In addition to similarities and differences in protein association between C and P complex, we also found similarities and difference in interactions with the substrate RNA. One similarity is that the 5′ exon is protected to the same extent in both complexes. This result suggests that the spliceosome maintains a grip on the 5′ exon even after second-step chemistry, which is presumably disrupted by the ATPase activity of DHX8 (Prp22) for mRNA release. In both complexes, the same ∼70-kD protein crosslinks to RNA 10 nt upstream of the 5′ ss in a splicing-dependent manner. We do not know its identity, but of the proteins close to 70 kD in both complexes, SLU7 and CDC40 are known to play a role in the second step of splicing, and SLU7 has been shown to contact the 3′ ss (Chua and Reed 1999).
A clear difference in protein/RNA interaction is a crosslink just upstream of the 5′ splice site to an ∼30-kD protein that appears only in P complex. This interaction may result from repositioning the substrate for the second step of splicing chemistry or from rearrangements after the second step. There are several P complex proteins in the 26–34 kD range, but only BCAS2, CWC15, SNRPA1, SYF2, ISY1, and PPIE are consistently well-represented in MS/MS analyses of C and P complex. Western analysis show that BCAS2, SNRPA1 (data not shown), CWC15 (data not shown), and ISY1 in nuclear extract migrate differently from the 30-kD protein, where both PPIE and SYF2 do migrate at a similar position. Recently, Vijayraghavan and coworkers found that an ∼25-kD protein crosslinks to the −3 position of the 5¢ exon in yeast during yeast splicing (Saha et al. 2012). A temperature-sensitive mutation in Bud31 that affects second-step splicing chemistry inhibits formation of the crosslink to the 25-kD protein, which suggests that its interaction with the pre-mRNA might occur after exon ligation (Saha et al. 2012). SYF2 has a yeast ortholog of 25 kD that shows genetic interactions with Cdc40 (Prp17) (Ben-Yehuda et al. 2000), whereas PPIE has no yeast ortholog. This may increase the possibility that SYF2 is responsible for the crosslink in both species.
In summary, we have succeeded in capturing the human spliceosome in a new conformational state relative to splicing chemistry, namely after exon ligation but before mRNA release. By comparing this post-catalytic complex with that of C complex, which is trapped after first-step chemistry, we find several rearrangements in the interactions that position the substrate RNA near the active site. Further characterization of P complex will likely yield more insight into the structural transitions that occur in the human spliceosome after the second step of splicing that have not previously been accessible to biochemical study.
MATERIALS AND METHODS
In vitro splicing and RNase H protection assays
All substrate pre-mRNAs were derivatives of the AdML transcript with three MS2 sites incorporated at the XhoI site in the intron. We used PCR to amplify DNA templates with varying lengths of 3′ exon. To generate C complex, the template contained an AG ≥ GG mutation at the 3′ splice site. T7 runoff transcription was used to generate G(5′)ppp(5′)G-capped radiolabeled substrate that was gel-purified. For in vitro splicing reactions, substrate was incubated at a 10 nM concentration in 80 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg/mL tRNA, and 40% HeLa cell (Biovest International) nuclear extract at 30°C. For RNase H protection assays, reactions were supplemented after 60 min with DNA oligos (10–16 nt) targeting consecutive regions of the pre-mRNA and incubated an additional 20 min. RNA was then extracted with phenol/chloroform, ethanol-precipitated, and electrophoresed through a 15% denaturing acrylamide gel that was visualized with a phosphorimager (Molecular Dynamics) and quantified with ImageQuant software (Molecular Dynamics).
Glycerol gradients
Splicing reactions were centrifuged on a 600-µL linear 10%–30% glycerol gradient prepared in SCB (20 mM Tris, pH 7.9, 150 mM KCl, 0.5 mM EDTA) at 38,000 rpm for 2 h 30 min at 4°C using a Beckman SW 55 Ti rotor with tube adaptors. Sixteen 40-µL fractions were collected from the top to the bottom of the gradient. RNA from each fraction was extracted with phenol/chloroform, ethanol-precipitated, and separated on a 15% denaturing acrylamide gel that was visualized with a phosphorimager.
Native gel analysis
Splicing reactions were stopped at indicated time points by placing on ice and adding 5× native loading buffer (0.25 mg/mL heparin, 20 mM Tris, 20 mM glycine, 25% glycerol, 0.25% cyan blue, 0.25% bromophenol blue). After 5 min centrifugation at 4°C, samples were warmed to room temperature and loaded onto 2.1% native agarose gels. Gels were run in 20 mM Tris/glycine at 72 volts for 3 h and 50 min and immediately dried down on Whatman paper at 55°C for 30 min and visualized with a phosphorimager.
Synthesis of single 32P-labeled pre-mRNA and UV crosslinking
Site-specific 32P-labeled AdML pre-mRNA was generated by DNA splinted ligation following the protocol in Frilander and Turunen (2005). For the −10- and −2-nt labels, both 5′ and 3′ fragment templates were generated by PCR for in vitro T7 run-off transcription. For the +1-nt label, the 3′ fragment was chemically synthesized to include a 4′-thio-U at +2 position (Dharmacon). Ligation products were separated by denaturing PAGE and gel-purified. For UV crosslinking, spliceosomes were assembled in vitro on the labeled substrates and crosslinked using a 254-nm wavelength with ∼4 J. For pre-mRNA substrate containing 4-thioU, 365 nm UV light was used. The crosslinked complexes were then digested with RNase A/T1 and separated by 7% SDS PAGE, and radiolabeled proteins were visualized with a phosphorimager.
Splicing complex purification
Splicing complexes were assembled in vitro and purified using MS2:MBP protein to bind the MS2 sites in the pre-mRNA substrate as previously described (Jurica et al. 2002; Tange et al. 2005). For C and P complexes, remaining pre-mRNA substrate was removed after splicing using RNase H digestion (Jurica et al. 2002). No digestion was carried out during purification of mRNP. To examine the snRNA components, RNA was extracted from ∼150 fmol of purified complexes, separated by denaturing 8% polyacrylamide PAGE and visualized by SYBRGold direct staining (Molecular Probes).
Electron microscopy and image processing
Unfixed samples for electron microscopy were prepared by spotting 3 μL of purified complexes onto carbon-coated copper grids, followed by staining with 2% (w/v) uranyl acetate solution. For fixed samples, purified complexes were incubated in 0.1% EM grade glutaraldehyde, 10 mM Hepes-NaOH pH 8, 150 mM NaCl, 0.02% NP40, 1 mM MgAc, 1 mM imidazole, 2 mM EGTA, 10 mM mercaptoethanol for 30 min at 4°C. The remaining glutaraldehyde in the crosslinking reaction was quenched with 90 mM glycine-NaOH (pH 7.9) for an additional 30 min at 4°C. The samples were then immediately used to prepare grids in the same way as unfixed complexes.
Samples were analyzed using a JEOL 1230 microscope operating at 120 kV. Micrographs were exposed under low-dose conditions at 60,000× magnification on a 4 K × 4 K Gatan Ultrascan CCD camera, resulting in micrographs sampled at 4.9 Å per pixel. For each complex, approximately 3000 particles were selected manually from micrographs using the EMAN program Boxer (Ludtke et al. 1999). Following 2 × 2 pixel averaging using SPIDER (Frank et al. 1996), particle images were band pass-filtered, centered, and iteratively aligned, classified, and averaged using IMAGIC-5 (van Heel et al. 1996).
Protein identification using data-dependent MS/MS
For protein compositional analysis by mass spectrometry, ∼500 fmol of purified complexes were digested overnight with 150 ng Promega modified trypsin.
Samples were analyzed by LC-MS/MS using 1-h reverse-phase chromatography separations on a Waters NanoAcquity UPLC system interfaced to a Thermo LTQ-Orbitrap Velos mass spectrometer. Peptides were selected for CID fragmentation analysis as they eluted in a data-dependent fashion, and dynamic exclusion was employed to prevent repeated analysis of the same components.
Data were searched using Protein Prospector version 5.10 (Chalkley et al. 2008) against a database consisting of the UniprotKB protein database downloaded on March 21, 2012 with a randomized version of the database concatenated onto the end to allow for estimation of a false discovery rate (Elias and Gygi 2007). Only human entries were considered, so a total of 21,662 entries were included for searching. Precursor and fragment mass tolerances of 15 ppm and 0.6 Da, respectively, were allowed. Cysteines were assumed to be carbamidomethylated, and methionine oxidation, protein N-terminal acetylation, and pyroglutamate formation from N-terminal glutamine residues were all considered as possible modifications. A maximum expectation value of 0.005 was employed as a threshold for peptide identifications. At this threshold, there were no matches to the randomized portion of the database, suggesting that there may be no incorrect identifications among the reported results.
Peptide and protein relative quantification using SRM mass spectrometry
Purified C and PC spliceosome complexes (∼1 pmol) were analyzed by scheduled selective reaction monitoring mass spectrometry. LC-MS/MS separation using a Waters NanoAcquity HPLC system was interfaced to an AB-Sciex QTRAP 5500 mass spectrometer. The peptides chosen to monitor, fragments to measure, and retention times were determined based on data previously acquired in-house when analyzing these complexes. Additional peptides were also chosen from those that have been previously identified and stored in the PeptideAtlas database (Desiere et al. 2006). A total of 411 transitions (precursor forming a particular fragment) were monitored. Peak areas for quantification were extracted from the acquired data using the Skyline software (MacLean et al. 2010).
Western analysis
Antibodies for Western analysis were: DHX35 (Sigma-Aldrich), eIF4a3 (gift from Hervé Le Hir, Institut de Biologie de l’École Normale Supérieure, Paris, France), ISY1 (Sigma), PRPF8 (Proteintech), Prp19 (Abnova), and SF3B3 (Proteintech). Approximately 500 fmol of purified splicing complexes were separated by 10% SDS PAGE and blotted. The blot was stripped and reprobed with each antibody tested.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
Supplemental Material
ACKNOWLEDGMENTS
We are grateful to Drs. Alan Zahler, Manny Ares, and Rhonda Perriman, as well as to Jurica lab members for suggestions and discussion. This work was funded by National Institutes of Health grant R01GM72649 to M.S.J. Support for R.J.C. and A.L.B. was provided by National Center for Research Resources grants RR001614, RR015804, and RR019934 and National Institute of General Medical Sciences grant P41 GM103481. J.O.I. was supported by National Institutes of Health training grant T32GM08646, a Center for Biomolecular Science and Engineering Graduate Diversity Fellowship sponsored by the National Human Genome Research Institute, and National Institutes of Health Initiative for Maximizing Student Diversity Graduate Fellowship R25GM058903.
REFERENCES
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, Luhrmann R 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcid EA, Jurica MS 2008. A protein-based EM label for RNA identifies the location of exons in spliceosomes. Nat Struct Mol Biol 15: 213–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzei P, Gargan S, Curran N, Moynagh PN 2010. Cactin targets the MHC class III protein IκB-like (IκBL) and inhibits NF-κB and interferon-regulatory factor signaling pathways. J Biol Chem 285: 36804–36817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, et al. 2007. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J 26: 1737–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yehuda S, Dix I, Russell CS, McGarvey M, Beggs JD, Kupiec M 2000. Genetic and physical interactions between factors involved in both cell cycle progression and pre-mRNA splicing in Saccharomyces cerevisiae. Genetics 156: 1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452: 846–850 [DOI] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, Luhrmann R 2010. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16: 2384–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkley RJ, Baker PR, Medzihradszky KF, Lynn AJ, Burlingame AL 2008. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol Cell Proteomics 7: 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara MD, Gozani O, Bennett M, Champion-Arnaud P, Palandjian L, Reed R 1996. Identification of proteins that interact with exon sequences, splice sites, and the branchpoint sequence during each stage of spliceosome assembly. Mol Cell Biol 16: 3317–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K, Reed R 1999. The RNA splicing factor hSlu7 is required for correct 3′ splice-site choice. Nature 402: 207–210 [DOI] [PubMed] [Google Scholar]
- Cohen-Krausz S, Sperling R, Sperling J 2007. Exploring the architecture of the intact supraspliceosome using electron microscopy. J Mol Biol 368: 319–327 [DOI] [PubMed] [Google Scholar]
- Coltri PP, Oliveira CC 2012. Cwc24p is a general Saccharomyces cerevisiae splicing factor required for the stable U2 snRNP binding to primary transcripts. PLoS One 7: e45678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltri P, Effenberger K, Chalkley RJ, Burlingame AL, Jurica MS 2011. Breaking up the C complex spliceosome shows stable association of proteins with the lariat intron intermediate. PLoS One 6: e19061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Minana B, Valcarcel J 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25: 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta B, Weiner AM 1991. Genetic evidence for base pairing between U2 and U6 snRNA in mammalian mRNA splicing. Nature 352: 821–824 [DOI] [PubMed] [Google Scholar]
- Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R 2006. The PeptideAtlas project. Nucleic Acids Res 34: D655–D658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Luhrmann R 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell 36: 593–608 [DOI] [PubMed] [Google Scholar]
- Folco EG, Coil KE, Reed R 2011. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev 25: 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A 1996. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 116: 190–199 [DOI] [PubMed] [Google Scholar]
- Frilander JM, Turunen JJ 2005. RNA ligation using T4 DNA ligase. In Handbook of RNA biochemistry (ed. Hartmann RK et al. ). Wiley-VCH, Weinheim, Germany [Google Scholar]
- Golas MM, Sander B, Bessonov S, Grote M, Wolf E, Kastner B, Stark H, Luhrmann R 2010. 3D cryo-EM structure of an active step I spliceosome and localization of its catalytic core. Mol Cell 40: 927–938 [DOI] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Luhrmann R, Stelzl U 2012. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580 [DOI] [PubMed] [Google Scholar]
- Herold N, Will CL, Wolf E, Kastner B, Urlaub H, Luhrmann R 2009. Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol 29: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliker AK, Mefford MA, Staley JP 2007. U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev 21: 821–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan J, Yuh P, Chalkley RJ, Burlingame AL, Jurica MS 2009. The role of exon sequences in C complex spliceosome structure. J Mol Biol 394: 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ 2002. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8: 426–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Sousa D, Moore MJ, Grigorieff N 2004. Three-dimensional structure of C complex spliceosomes by electron microscopy. Nat Struct Mol Biol 11: 265–269 [DOI] [PubMed] [Google Scholar]
- Kim SH, Lin RJ 1996. Spliceosome activation by PRP2 ATPase prior to the first transesterification reaction of pre-mRNA splicing. Mol Cell Biol 16: 6810–6819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Gruter P, Groning K, Kastner B 1999. Combined biochemical and electron microscopic analyses reveal the architecture of the mammalian U2 snRNP. J Cell Biol 145: 1355–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli RM, Thompson JX, Yates JR III, Stevens SW 2010. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 16: 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ 2000. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J 19: 6860–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Huang LH, Steward R 2000. Cactin, a conserved protein that interacts with the Drosophila IκB protein cactus and modulates its function. Mech Dev 94: 57–65 [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W 1999. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128: 82–97 [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ 2010. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C 1992. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71: 803–817 [DOI] [PubMed] [Google Scholar]
- McPheeters DS, Muhlenkamp P 2003. Spatial organization of protein-RNA interactions in the branch site-3′ splice site region during pre-mRNA splicing in yeast. Mol Cell Biol 23: 4174–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPheeters DS, Schwer B, Muhlenkamp P 2000. Interaction of the yeast DExH-box RNA helicase prp22p with the 3′ splice site during the second step of nuclear pre-mRNA splicing. Nucleic Acids Res 28: 1313–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz C, Urlaub H, Will CL, Luhrmann R 2007. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA 13: 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AJ, Teigelkamp S, Beggs JD 1995. snRNA interactions at 5′ and 3′ splice sites monitored by photoactivated crosslinking in yeast spliceosomes. RNA 1: 968–980 [PMC free article] [PubMed] [Google Scholar]
- Ohi MD, Link AJ, Ren L, Jennings JL, McDonald WH, Gould KL 2002. Proteomics analysis reveals stable multiprotein complexes in both fission and budding yeasts containing Myb-related Cdc5p/Cef1p, novel pre-mRNA splicing factors, and snRNAs. Mol Cell Biol 22: 2011–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi MD, Ren L, Wall JS, Gould KL, Walz T 2007. Structural characterization of the fission yeast U5.U2/U6 spliceosome complex. Proc Natl Acad Sci 104: 3195–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrt T, Prior M, Dannenberg J, Odenwalder P, Dybkov O, Rasche N, Schmitzova J, Gregor I, Fabrizio P, Enderlein J, et al. 2012. Prp2-mediated protein rearrangements at the catalytic core of the spliceosome as revealed by dcFCCS. RNA 18: 1244–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Siliciano PG, Guthrie C 1987. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell 49: 229–239 [DOI] [PubMed] [Google Scholar]
- Perriman RJ, Ares M Jr 2007. Rearrangement of competing U2 RNA helices within the spliceosome promotes multiple steps in splicing. Genes Dev 21: 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JL, Kois P, Konforti BB, Konarska MM 1996. The canonical GU dinucleotide at the 5′ splice site is recognized by p220 of the U5 snRNP within the spliceosome. RNA 2: 213–225 [PMC free article] [PubMed] [Google Scholar]
- Reyes JL, Gustafson EH, Luo HR, Moore MJ, Konarska MM 1999. The C-terminal region of hPrp8 interacts with the conserved GU dinucleotide at the 5′ splice site. RNA 5: 167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal GA, Jurica MS 2010. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 38: 6664–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha D, Khandelia P, O’Keefe RT, Vijayraghavan U 2012. Saccharomyces cerevisiae NineTeen complex (NTC)-associated factor Bud31/Ycr063w assembles on precatalytic spliceosomes and improves first and second step pre-mRNA splicing efficiency. J Biol Chem 287: 5390–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa H, Abelson J 1992. Evidence for a base-pairing interaction between U6 small nuclear RNA and 5′ splice site during the splicing reaction in yeast. Proc Natl Acad Sci 89: 11269–11273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B 2008. A conformational rearrangement in the spliceosome sets the stage for Prp22-dependent mRNA release. Mol Cell 30: 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Gross CH 1998. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J 17: 2086–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Guthrie C 1991. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature 349: 494–499 [DOI] [PubMed] [Google Scholar]
- Schwer B, Guthrie C 1992. A conformational rearrangement in the spliceosome is dependent on PRP16 and ATP hydrolysis. EMBO J 11: 5033–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman EJ, Maeda A, Wei J, Smith P, Beggs JD, Lin RJ 2004. Interaction between a G-patch protein and a spliceosomal DEXD/H-box ATPase that is critical for splicing. Mol Cell Biol 24: 10101–10110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JS, Manley JL 1995. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev 9: 843–854 [DOI] [PubMed] [Google Scholar]
- Tange TO, Shibuya T, Jurica MS, Moore MJ 2005. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA 11: 1869–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teigelkamp S, Newman AJ, Beggs JD 1995a. Extensive interactions of PRP8 protein with the 5′ and 3′ splice sites during splicing suggest a role in stabilization of exon alignment by U5 snRNA. EMBO J 14: 2602–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teigelkamp S, Whittaker E, Beggs JD 1995b. Interaction of the yeast splicing factor PRP8 with substrate RNA during both steps of splicing. Nucleic Acids Res 23: 320–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RT, Fu RH, Yeh FL, Tseng CK, Lin YC, Huang YH, Cheng SC 2005. Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev 19: 2991–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C 1995. Prp16p, Slu7p, and Prp8p interact with the 3′ splice site in two distinct stages during the second catalytic step of pre-mRNA splicing. RNA 1: 584–597 [PMC free article] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M 1996. A new generation of the IMAGIC image processing system. J Struct Biol 116: 17–24 [DOI] [PubMed] [Google Scholar]
- Warkocki Z, Odenwalder P, Schmitzova J, Platzmann F, Stark H, Urlaub H, Ficner R, Fabrizio P, Luhrmann R 2009. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol 16: 1237–1243 [DOI] [PubMed] [Google Scholar]
- Wassarman DA, Steitz JA 1992. Interactions of small nuclear RNA’s with precursor messenger RNA during in vitro splicing. Science 257: 1918–1925 [DOI] [PubMed] [Google Scholar]
- Wassarman DA, Steitz JA 1993. A base-pairing interaction between U2 and U6 small nuclear RNAs occurs in >150S complexes in HeLa cell extracts: Implications for the spliceosome assembly pathway. Proc Natl Acad Sci 90: 7139–7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SE, Neville M, Haynes M, Wang J, Igel H, Ares M Jr 1996. CUS1, a suppressor of cold-sensitive U2 snRNA mutations, is a novel yeast splicing factor homologous to human SAP 145. Genes Dev 10: 220–232 [DOI] [PubMed] [Google Scholar]
- Will CL, Luhrmann R 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Kastner B, Deckert J, Merz C, Stark H, Luhrmann R 2009. Exon, intron and splice site locations in the spliceosomal B complex. EMBO J 28: 2283–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Manley JL 1989. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev 3: 1553–1561 [DOI] [PubMed] [Google Scholar]
- Wu JA, Manley JL 1991. Base pairing between U2 and U6 snRNAs is necessary for splicing of a mammalian pre-mRNA. Nature 352: 818–821 [DOI] [PubMed] [Google Scholar]
- Wyatt JR, Sontheimer EJ, Steitz JA 1992. Site-specific cross-linking of mammalian U5 snRNP to the 5′ splice site before the first step of pre-mRNA splicing. Genes Dev 6: 2542–2553 [DOI] [PubMed] [Google Scholar]
- Xie J, Beickman K, Otte E, Rymond BC 1998. Progression through the spliceosome cycle requires Prp38p function for U4/U6 snRNA dissociation. EMBO J 17: 2938–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Ares M Jr 1996. Invariant U2 RNA sequences bordering the branchpoint recognition region are essential for interaction with yeast SF3a and SF3b subunits. Mol Cell Biol 16: 818–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavanelli MI, Ares M Jr 1991. Efficient association of U2 snRNPs with pre-mRNA requires an essential U2 RNA structural element. Genes Dev 5: 2521–2533 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material