Mapping the Local Organization of Cell Membranes Using Excitation-Polarization-Resolved Confocal Fluorescence Microscopy (original) (raw)
Abstract
Fluorescence anisotropy and linear dichroism imaging have been widely used for imaging biomolecular orientational distributions in protein aggregates, fibrillar structures of cells, and cell membranes. However, these techniques do not give access to complete orientational order information in a whole image, because their use is limited to parts of the sample where the average orientation of molecules is known a priori. Fluorescence anisotropy is also highly sensitive to depolarization mechanisms such as those induced by fluorescence energy transfer. A fully excitation-polarization-resolved fluorescence microscopy imaging that relies on the use of a tunable incident polarization and a nonpolarized detection is able to circumvent these limitations. We have developed such a technique in confocal epifluorescence microscopy, giving access to new regions of study in the complex and heterogeneous molecular organization of cell membranes. Using this technique, we demonstrate morphological changes at the subdiffraction scale in labeled COS-7 cell membranes whose cytoskeleton is perturbed. Molecular orientational order is also seen to be affected by cholesterol depletion, reflecting the strong interplay between lipid-packing regions and their nearby cytoskeleton. This noninvasive optical technique can reveal local organization in cell membranes when used as a complement to existing methods such as generalized polarization.
Introduction
Investigating biological processes using the polarization of light is a well developed approach for studying the structural organization of proteins and lipids. In cell biology, such organization is the direct signature of molecular interaction mechanisms that play a determining role in biological functions such as cell shape maintenance and motility (1), vesicular trafficking (2), or signaling (3). In fluorescence, the orientational organization of molecules comes into play, because they are preferentially excited when their absorption transition dipole is parallel to the incident polarization direction. This property has been exploited in fluorescence anisotropy for decades (4) by comparing the fluorescence signals recorded along analysis directions parallel and perpendicular to the incident polarization. Fluorescence anisotropy and its equivalent scheme, linear dichroism (which considers two orthogonal excitation polarizations), have been primarily developed for studies in solution, giving access to information on the orientational dynamics of proteins and, consequently, on their structure and conformation. The application of these methodologies to imaging, initiated in isotropic media such as for the mapping of cell cytoplasm viscosity (5), have been focused more recently on biomolecular assemblies exhibiting a degree of orientational order. Fluorescence anisotropy and linear dichroism have helped to solve important questions, giving evidence of glycoprotein’s confinement in cell membranes to nanometer-scale lipid domains (6), septin filament conformation in cell division (7), cytoskeleton actin organization (8,9), _α_-synuclein aggregation in fibrils involved in neurodegenerative diseases (10), membrane receptor conformational changes upon cell activation (11), and plasma membrane morphology specificities (2,12).
These techniques suffer from some limitations, however. First, they are not adapted to ordered samples, given that they rely on a preferential photoselection (or emission) direction imposed by the incident excitation polarization (or analyzer). They are consequently unable to provide complete information on how the molecular assembly is oriented (defined by its average orientation) and how it is angularly ordered (defined by the molecular orientational order). Since only one of these two parameters is accessible, a measure of molecular orientational order requires working with samples of controlled geometry, where the averaged orientation can be known a priori. This explains the wide range of studies in membranes of round shapes such as red blood cells (13), giant unilamellar vesicles (14), cell blebs (15), or round cells (16). Another way to adapt anisotropy or linear dichroism to oriented structures is to address only particular regions of the sample where the molecules lie on average along or perpendicular to the incident polarization direction, such as horizontal or vertical parts of a membrane in the cell contour image (11,12,17). Although both workarounds have allowed new findings, their limitations considerably reduce the range of exploration, making it impossible to grasp the full complexity of molecular organization in heterogeneous samples. Finally, fluorescence anisotropy can be easily biased by depolarizing mechanisms such as energy transfer between neighbor molecules or the existence of an angle between absorption and emission transition dipoles in the excited molecule. Although depolarization induced by fluorescence resonance energy transfer between molecules (homo-FRET) has been advantageously used to image changes in protein clustering (18,19) or actin polymerization in cells (20), it is detrimental when trying to quantify molecular structural organization. This mechanism can indeed introduce erroneous interpretations of the data if they are not studied separately (17,21).
Recently, new techniques, based on tuning of the excitation polarization, have been proposed to circumvent these limitations by exploiting the full potential of polarized fluorescence imaging, (21–24). Such techniques, which can be considered as more universal linear dichroism methods, allow retrieval of complete information on both molecular orientational order and average orientation in a nonambiguous manner in a whole image of the investigated samples, not just in particular regions. One method (21) was based on two-photon excitation-polarization tuning, allowing sample sectioning with polarized detection, which, because it is sensitive to depolarization mechanisms (such as homo-FRET), can suffer from a low level of signals. A second scheme (22,23) removed the polarized detection to circumvent these effects, and improved the time resolution using wide-field illumination. To avoid polarization distortion issues, however, this scheme was implemented in the forward-direction detection mode, which is not appropriate for many samples, leads to faster photobleaching, and does not yield depth sections (24). In this work, we implement an excitation-polarization-resolved technique in an epiconfocal sectioning mode, which is appropriate for the vast majority of biological systems. We discuss the temporal resolution limit of this method and its ability to work in 3D orientational directions. In addition, we develop a data analysis tool that provides considerable improvement in signal processing time and precision compared to previous approaches (21), allowing the monitoring of molecular orientational order and orientation dynamics over timescales of seconds. This method is appropriate for the measurement of phenomena averaged in space over the diffraction-limit size of an optical microscope and in time over a timescale of seconds.
We demonstrate the benefits of this configuration in molecular orientational order imaging along cell-membrane equatorial contours labeled with lipid probes. We show in particular that the subdiffraction-scale local morphological features of a cell membrane can be quantified in vivo, whereas until now these characteristics have been measured by indirect or invasive methods (25–27). Studies of cells affected by pharmacological treatments that induce cytoskeleton perturbation or cholesterol depletion show a significant change of their molecular orientational order. The observed effects corroborate previous findings based on membrane stiffness measurements (28) and generalized polarization (GP), which measures lipid packing by the spectral sensitivity of lipid probes to local polarity (29–31). The new piece of information on subdiffraction-scale morphological features can complement investigations of lipid packing and avoid biased interpretations in existing methods such as fluorescence correlation spectroscopy (FCS).
Materials and Methods
Cells preparation
All experiments were carried out on fibroblastlike COS-7 cells (American Type Culture Collection (ATCC) No. CRL-1657, Manassas, VA). COS-7 cells were grown in an incubator at 37°C in Lab-Tek chambers (Nunc, Rochester, NY) in DMEM medium (Gibco, Saint Aubin, France) completed by 10% FBS and 1% sodium pyruvate. Cells were washed and kept in Hanks buffered salt solution containing 10 mM HEPES (HBSS/HEPES), pH 7.4, at 37°C, for the duration of the imaging process. Cells were labeled with di-8-ANEPPQ directly before measurements and after the pharmacological treatments. For labeling, cells were washed and incubated in insertion buffer (NaCl 130 mM, KCl 5 mM, CaCl2 10 mM, glucose 5 mM, and HEPES 10 mM) with 1 _μ_M di-8-ANEPPQ (dissolved by absolute ethanol to 1 mM for preservation and usage) for 2 min at room temperature. After incubation, cells were washed again and kept in HBSS/HEPES at 37°C throughout the imaging process. Cells were labeled with DiIC18 directly before measurements and after the pharmacological treatments. For labeling, cells were washed and incubated in HBSS/HEPES with 1 _μ_M DiIC18 (dissolved by absolute ethanol to 1 mM for preservation and usage) for 2 min at room temperature. After incubation, cells were washed again and kept in HBSS/HEPES at 37°C for the whole imaging process. The concentration of the dyes used did not affect cell shape for the duration of the measurements. For pharmacological treatments that alter cytoskeleton fibers, cells were washed in HBSS/HEPES and incubated at 37°C with 1 _μ_M latrunculin A (5 min), 10 _μ_M cytochalasin D (30 min), or 0.5 _μ_M jasplakinolide (5 min). Cells were then washed with HBSS/HEPES and labeled and kept in HBSS/HEPES with 10-fold-diluted reagent for the duration of the imaging process. For pharmacological treatments that deplete cholesterol content, cells were washed in HBSS/HEPES and incubated at 37°C in serum-free medium with either 2.5 _μ_M or 100 _μ_M methyl-_β_-cyclodextrin (MBCD) (5 min). For treatment with cholesterol oxidase, cells were first starved with serum-free medium for 1 h at 37°C. Either 1 or 5 units of cholesterol oxidase stock solution was added per 1 ml serum-free medium, and the mixture was then incubated for 2 h. Cells were then washed with HBSS/HEPES and kept in HBSS/HEPES for the entire imaging process. Finally, to induce hypotonic shock, cells were swollen in a solution of 80% H2O and 20% HBSS/HEPES and imaged after 15 min.
Excitation-polarization-resolved fluorescence imaging set-up
Measurements were carried out on a custom confocal microscope system, adapted to allow full control of the polarization state of the excitation beam (Fig. S1 in the Supporting Material). Excitation is provided by a polarized continuous-wave laser (Calypso, Cobolt, Solona, Sweden) emitting at 491 nm that delivers linearly polarized light whose angle of polarization, α, is sequentially rotated by a half-wave plate (WPH05M-488, Thorlabs, Newton, NJ) mounted on a motorized rotation stage (PR50CC, Newport, Irvine, CA). After reflection on a dichroic mirror (XF2037-500DRLP, Omega Optical, Brattleboro, VT), the excitation light is focused onto the sample by a water-immersion objective lens (C-Apochromat 40× UV-VIS-NIR, NA 1.2, Carl Zeiss, Jena, Germany). Emitted fluorescence is collected by the same lens in an epigeometry, passes through the dichroic mirror, an emission filter (HQ540/80M-2P, Chroma, Bellows Falls, VT), and a 75-_μ_m-diameter (1.25 Airy unit) confocal pinhole. The fluorescence is detected by a single-photon counting module (SPCM-AQR-14, Perkin Elmer, Waltham, MA). Measurements are performed at 37°C. The typical excitation power at the entrance of the objective lens is 10 _μ_W. Imaging is performed by scanning the excitation/observation volume in the sample using a set of two galvanometric mirrors placed between the dichroic mirror and the objective lens. The transverse size of the observation volume, characterized previously (32), is ∼200 nm.
Polarization distortion effects in both excitation and collection arms have been characterized at the focal spot of the objective and in the detection path of the microscope. The polarization state of light was measured at different places in the optical set-up, both in the excitation and emission paths, for different incident polarization angles, α, between 0° and 180°. Indeed, several optical elements in the optical path can act as polarizing elements because of their possible optical anisotropy properties (diattenuation, γ, and retardance, δ). Using a polarization-state analysis based on the rotating quarter-wave plate method (33,34), we were able to model the excitation path as a series of effective polarizing elements whose optical axis orientation is denoted by θ. In particular, the reflection on the dichroic mirror introduces a notable retardance close to π/2 that could be partially compensated by inserting a quarter-wave plate (WPQ05M-488, Thorlabs) in the excitation path. The remaining effective polarizing elements were completely characterized and modeled (with parameters θ, γ, and δ, summarized in Table S1) to be accounted for in the data analysis. We dealt in the same way with the collection path where weak polarization distortions could be observed (Table S1). All measured polarization distortions were introduced in the theoretical model developed for polarization-resolved analysis (details are given in the Supporting Material). Note that these distortions are seen to be achromatic over the detection spectral range of the measurement, as ascertained by constant excitation-polarization-resolved data obtained in homogeneous samples (solution, giant unilamellar vesicles (GUVs)) in different detection spectral regions. Measurements consist of recording a stack of 90 images, obtained by varying the angle _α_k between _α_1 = 0° (with respect to the horizontal axis of the sample) and _α_90 = 178° by steps of 2°. Typically, 150 × 150-pixel images were recorded with 100-_μ_s pixel dwell time, yielding to an acquisition duration of ∼5 min, including instrument communication delays.
Theoretical model and data processing
The polarization-resolved steady-state fluorescence intensity is modeled using a formalism similar to that previously developed for two-photon fluorescence (21), except that here the signal is not analyzed and is therefore insensitive to depolarization processes at the emission. This model is based on the expression of the polarization dependence of the excitation photoselection of an ensemble of molecules, whose angular extent is defined by a molecular angular distribution function. The steady-state fluorescence intensity emitted by an ensemble of dipoles whose orientation is contained within an angular distribution function, fψ(θ,φ), is written as
| Iρ,ψ,η(α)=∫02πdφ∫0πdθsinθ|μ→(θ,φ,ρ,η)⋅E→(α)|2fψ(θ,φ)J(θ,φ,ρ,η), | (1) | | ----------------------------------------------------------------- | --- |
where the square modulus represents the excitation probability for an absorption transition dipole, μ→, to be excited by an electric field, E→, with a linear state of polarization along an angle α with respect to the x axis in the sample plane. In this model, a complete expression of E→ is introduced, including all polarization distortions measured in the set-up (see Supporting Material). (θ,φ) defines the orientation of μ→ in the local frame of the membrane (x, y, z) (Fig. S2 A). (ρ,η) defines the orientation of the distribution function in the macroscopic laboratory frame (X, Y, Z) (Fig. S2 C). The function J represents the detection probability of fluorescence and takes into account the numerical aperture of the objective lens (13). Following the methodology introduced by Axelrod (13), this function is recalculated to account for the polarization distortions occurring in the detection path of the microscope (see Supporting Material; note that we confirmed that the efficiency of the detector used in this study is not dependent on the detected polarization).
The normalized function, fψ(θ,φ), describes the dipole orientation distribution that is modeled by a full cone of aperture ψ whose symmetry axis is oriented at angle ρ with respect to the x axis, and an out-of-plane angle η. Depending on the lipid probe structure, the function fψ(θ,φ) can take different forms. In the case of a lipid probe whose dipoles lie along the membrane lipid molecules (such as di-8-ANEPPQ) (Fig. S2 A), the angular distribution in the frame of the membrane is written as
| fψ(θ,φ)={14πsin2(ψ/4)ifθ≤ψ/20otherwise. | (2) |
|---|
In the case of a lipid probe whose dipoles lie perpendicular to the membrane lipid molecules (such as DiIC18, 1,1'-dioctadecyl-3,3,3′,3′-tetramethyl indocarbocyanine perchlorate) (Fig. S2 B), the angular distribution in the frame of the membrane is written as
| fψ(θ,φ)={14πsin(ψ/2)if(π−ψ)/2≤θ≤(π+ψ)/20otherwise. | (3) |
|---|
Whatever the shape of the distribution function, one can show that the dependence on α in Eq. 1 can be written as a finite Fourier series
| Iρ,ψ,η(α)=I0[1+Aρ,ψ,ηcos(2α)+Bρ,ψ,ηsin(2α)], | (4) |
|---|
with
Aρ,ψ,η=2I0π∫0πdαIρ,ψ,η(α)cos(2α)
| Bρ,ψ,η=2I0π∫0πdαIρ,ψ,η(α)sin(2α), | (5) |
|---|
and I0=1/π∫0πdαIρ,ψ,η(α). Moreover, if η is assumed to be zero, any set (ρ,ψ) gives a unique set (Aρ,ψ,Bρ,ψ). Conversely, knowing A and B allows us to retrieve (ρ,ψ) unambiguously (as shown in simulations; Fig. S3). The assumption η=0° supposes that the information is measured close to the equatorial plane of the cell membranes in which the fluorophore orientation lies close to the sample plane. This treatment is valid until the cell curvature becomes large (n ∼ 45°) (see below).
In practice, the selection of the relevant pixels to be analyzed is performed on the basis of the total fluorescence image obtained by summing for each pixel photon counts obtained for all angles αk. First, to ensure good precision (see discussion below), only pixels with >5000 counts are considered. Pixels corresponding to the membrane signal are manually selected by the operator using a polygon selection tool. For each selected pixel, the photon counts, Iexp(αk), are added to those of the neighbor pixels, yielding a binning of 3 × 3 pixels. Then, for each pixel, experimental coefficients Aexp and Bexp are computed using
Aexp=2I0N∑k=1NIexp(αk)cos(2αk)
| Bexp=2I0N∑k=1NIexp(αk)sin(2αk), | (6) |
|---|
where I0=1/N∑k=1NIexp(αk). The corresponding (ρ,ψ) angles are retrieved by finding within a set (Aρ,ψ,Bρ,ψ) (where ρ and ψ have been sampled every degree) the angle closest to (Aexp,Bexp), i.e., the one that minimizes the quantity (Aρ,ψ−Aexp)2+(Bρ,ψ−Bexp)2. The validity of the obtained (ρ,ψ) angles is assessed by calculating the normalized chi-square value,
| χ2=1N∑k=1N[Iexp(αk)−Itheory(αk)]2σk2, | (7) |
|---|
where Itheory(αk)=I0[1+Aρ,ψcos(2αk)+Bρ,ψsin(2αk)] and the variance σk2 is assumed to be equal to the photon counts Itheory(αk), in agreement with Poisson statistics of low photon counts. Data corresponding to χ2>5 are discarded (Fig. S4). Thanks to this procedure, measurements that are affected by instabilities such as by sample drift, membrane local motion, photobleaching, etc., and therefore differ from theory by more than what is expected from photon shot noise, are systematically rejected.
The retrieved ψ and ρ data are shown as histograms for single cells, for ∼12 cells (∼4000 pixels), and by bar graphs with average values indicating the mean ± SE over all pixels. Statistical significance was determined and P values were calculated with an analysis of variance one-way Tukey HSD test. This representation specifically allows evaluation of heterogeneities from cell to cell as well as within the whole investigated population. Note that for all data analysis related to membrane behaviors, a careful inspection of the total fluorescence image of the cell ensures that statistics are made on cell membrane parts that are clearly isolated and not part of an accumulation of membranes of size above the diffraction limit.
Sensitivity to out-of plane orientations
Measurements and simulations have been performed to investigate the range of validity of the model and results developed below, in the case of excitation-polarization-resolved fluorescence data recorded at a different plane of the membrane than the equatorial plane for which η = 0°. Measurements of (ρ, ψ) were performed on a COS-7 cell membrane contour at different heights, Z, above the sample surface (cell adherence plane) (Fig. S5). As expected, ψ is overestimated when η increases, since the cone projection in the sample plane tends toward a more isotropic distribution function when this cone is tilted out of the sample plane. The bias on ψ is nevertheless seen to remain quite low for a few-micrometers range of variation of Z around the equatorial plane of the cell membrane, showing that the cell membrane 3D shape is well adapted to a robust measurement of ψ. In theory, the bias on ψ increases with respect to η in a nonlinear way (Fig. S5), which explains why increasing η leads to a low bias on ψ for relatively low values of η (<20°). For a case where ψ = 120°, the bias is seen to be within a 10% range when η reaches 45°. This shows that for experiments allowing such a bias, data far above the equatorial plane of the membrane can be measured. In a more constraining situation where only a few degrees of error are accepted for the measurements of ψ, out-of-plane tilt angles of η < 30° are still allowed.
Modeling the cell membrane spatial profile
The change of aperture of the cone distribution of di-8-ANEPPQ molecules in COS-7 cell membranes upon different cell treatments can be directly related to a modification of the spatial profile of the cell membrane contour. To model this effect in a simple way and retrieve an order of magnitude of the cell membrane shape modification, we assume that the value of ψ0 measured in cells under hypotonic shock is that of a flat membrane (ψ0 = 100°, averaged over 12 cells). We assume that nontreated cells exhibit a lateral 1D profile of sinusoidal shape with amplitude A: P(x)=A×cos(x×(n×π/d)), with d = 100 nm the half lateral size of the focal spot, n the number of period folds within this spot lateral size. The whole cone aperture induced by this curvature is Δψ=2atan(A×(n×π/d)). The measured cone aperture is therefore ψ=Δψ+ψ0, where ψ0=100° is the cone aperture value in a flat cell membrane. Finally, to deduce a cell membrane lateral 1D profile from a measured ψ value, we use the function
| Pdeduced(x,ψ)=[tan(ψ−ψ02)×dn×π]×cos(x×n×πd) | (8) |
|---|
The first term in brackets is the amplitude of the membrane profile, which depends on the number of periods of the sinusoid profile that is assumed within the focal spot size. Assuming one period of membrane profile sinusoid in the focal spot size (n = 1), its amplitude is seen to reach a maximum of 30 nm in the case of ψ=150°, which is the higher averaged value observed in cells treated with jasplakinolide. The minimum measurable amplitude is of the order of 2 nm, assuming a detection limit of ψ=5°. These values are upper limits, since they are decreased in situations where the profile contains more periods in the focal spot, according to Eq. 8.
Results
We quantify orientational order by the extent of angular freedom experienced by a molecule, using an angle called ψ (Fig. 1, A and B), which defines the rotational hindrance imposed by local molecular interactions in cell membranes or protein assemblies. The average orientation of this angular distribution is defined by the angles ρ (in the sample plane) (Fig. 1, A and B) and η (tilt angle out of the sample plane) (Fig. S2). Since fluorescence imaging primarily addresses processes that occur in the sample plane, we concentrate on orientational distributions in this plane where only ρ is relevant (η ∼ 0°). The most general case of 3D orientations is discussed below. Whereas the molecular orientational order, ψ, is intrinsic to the sample and contains biologically relevant information, the angle ρ depends on its global orientation and needs to be decoupled from ψ. The purpose of excitation-polarization-resolved fluorescence imaging is to provide simultaneously information on both ψ and ρ angles in each pixel of a fluorescence image.
Figure 1.
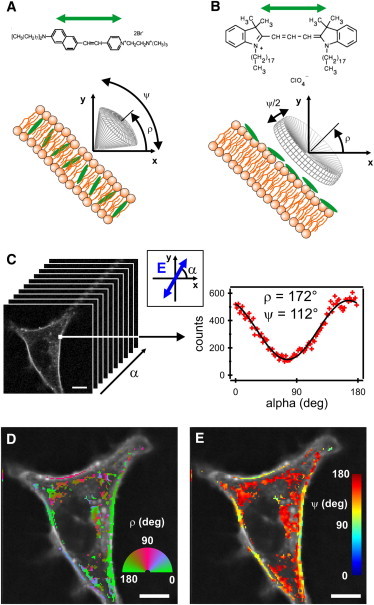
Schematics of the orientational order parameters of lipid probes in the cell-membrane equatorial plane. (A) Lipid probe whose dipole inserts into a cell membrane. (B) Lipid probe whose dipole lies along the cell-membrane contour. (C) Stack of polarization-resolved fluorescence data on a single COS-7 cell of arbitrary shape labeled with di-8-ANEPPQ. Polarization-resolved data recorded for one pixel in the membrane contour are shown with 90 input polarizations (shown schematically in the inset). (D) Retrieved image of the local orientation angle, ρ. (E) Corresponding image for ψ. Both images are superimposed with the fluorescence intensity image in greyscale. Scale bars, 10 _μ_m (C–E).
Principle of excitation-polarization-resolved fluorescence microscopy
The principle of the technique relies on the tuning of the excitation photoselection. Several images of a sample are recorded for a minimum of three linear, regularly spaced input polarization directions in the sample plane, ranging between 0° (along the horizontal axis of the image) and 180° (Fig. S1). The choice of number of input polarizations is purely governed by the precision chosen for the orientational parameter determination, which indeed depends on the total number of photons recorded for a given pixel (Fig. S6). In this study, we represent a static picture of the sample and chose 90 polarization steps in the measurement, which takes a few minutes of recording. This allows a precision of ∼2° on the retrieved angle (ρ) and order (ψ) parameters. Increasing the dynamics is possible by reducing the number of input polarizations, but with a cost of lower precision (typically four polarization steps leads to a 10° precision). Reaching a few degrees precision with a faster dynamics (typically a rate of 1/s) is nevertheless possible using confocal-like parallel imaging such as that obtained with Nipkow-type spinning disk devices (34).
The details of the set-up are described in the Materials and Methods section. Implementing several input polarization directions into a microscope requires careful control of the polarization state at the focal point of the objective, especially for intermediate polarizations that are neither horizontal nor vertical. We developed for this purpose a polarization diagnostic tool that can rapidly identify polarization distortions due to the optical set-up (see Materials and Methods). These distortions are mostly corrected for by an optical compensation, with the remaining corrections included in the data analysis process together with the collection polarization distortions (Table S1). The fluorescence is recorded without any analyzer; therefore, the data are only sensitive to the variation of the photoselection and not to the emission process. This makes the technique insensitive to depolarization processes occurring in the sample, which could arise, for instance, from homo-FRET between neighbor molecules. Finally, the recorded signal is analyzed in a simple and fast way to directly retrieve information on the local molecule’s averaged orientation (ρ) and order (ψ). The data processing relies on a Fourier series decomposition of the polarization-resolved signal, recorded in every pixel of the image (Fig. 1 C). From this analysis, the ρ and ψ parameters can be retrieved independently by solving a basic optimization problem for which the solution is unique (see Materials and Methods) (Figs. S3 and S4). This procedure leads finally to images of ρ (Fig. 1 D) and ψ (Fig. 1 E), with the latter including direct information on the spatial repartition of molecular orientational order in the sample. These images are obtained within a processing time of a few hundred milliseconds.
Sensitivity to local membrane morphology
A preliminary analysis was performed on model membranes to validate the method. Artificial lipid membranes made of DOPC (1,2-dioleoyl-_sn_-3-phosphatidylcholine) GUVs, labeled with the fluorescent lipid probe di-8-ANEPPQ (Fig. 1 A) exhibit a uniform molecular orientational order with ψ = 95° ± 4° (Fig. S7). An angle of ψ = 70° ± 6° is obtained for DiIC18, for which the positioning of the membrane is found to be along the membrane contour (Fig. S8). These numbers are very close to the order values obtained in similar systems using fluorescence anisotropy (14,21). The standard deviation of ψ is limited by the measurement noise, which confirms the remarkable homogeneity of molecular orientational order in such single-lipid vesicles.
The molecular orientational order of lipid probes in cell membranes exhibits a much different behavior (Fig. 1 E). Live (37°C) COS-7 fibroblastlike cell membranes labeled with di-8-ANEPPQ show a very heterogeneous molecular orientational order around the cell contour on its equatorial plane, with a visible isotropic behavior in the cell cytoplasm. Next to the membrane, a clear disorder is also visible, which we attribute to internalized lipid probes, internal membrane regions, or vesicles. The average angular aperture ψ around the cell membrane is quite large (ψ ∼ 120°), which confirms previous observations in two-photon microscopy obtained with a less direct analysis (21). The ψ image also shows that the distribution of ψ values is much larger than the expected precision of the measurement (a few degrees). Although the disordered parts of the cell membrane show high ψ values of ∼150° (Fig. 2 A), its most ordered parts reach ψ ∼ 100° (Fig. 2 B). This behavior can most likely be attributed to the characteristic membrane morphology, which is much more perturbed than in a GUV membrane, which is tense by nature. The cell membrane, in particular, undergoes strong perturbations due to the interaction of membrane proteins with the cytoskeleton and with the substrate, but also due to trafficking and its intrinsic molecular heterogeneity. We observed furthermore that regions of high molecular disorder are correlated with high intensity (Fig. 2, A and B), which is compatible with folded parts of the membrane where molecules accumulate at a scale below the diffraction limit of the microscope, or other types of lipid accumulation at the proximity of the plasma membrane, such as in internal membranes or submicrometer-scale vesicles. Since this technique is insensitive to potential homo-FRET between molecules, the retrieved information is relevant even in regions where molecules are highly concentrated, which is likely to occur in wrinkled parts of the cell membrane. The ψ image can ultimately be used to quantify the membrane fold sizes, which might be reached if wrinkles were the primary origin of molecular disorder. For this purpose, we modeled the membrane profile using a sinusoidal shape in one dimension (see Materials and Methods). Supposing that the membrane is locally flat when ψ is lowest (ψ = 100°), we deduce that its amplitude reaches 10 nm for ψ = 120°, and 30 nm for highly disordered regions (ψ ∼150°) (Fig. 2 C). In this model, one period of sinusoidal profile is assumed within the 200-nm-diameter length of the focal volume (see Materials and Methods); therefore, these values have to be considered as higher limits. The order of magnitude of the wrinkle amplitude obtained in this study is consistent with previous estimations of the membrane profile extent obtained using invasive or indirect methods (2,25–27). Note that this simplified curved-membrane model includes effects that cannot be discriminated in this measurement. First, the membrane shape fluctuations are highly dynamic; therefore, the retrieved membrane profile is an average over a time much larger than the timescale of the fluctuations. Second, the measurement reflects the behavior of molecules distributed within a whole focal volume of 200-nm lateral size, within which other phenomena are likely to occur: internalization, membrane trafficking, vesicle formation, etc. Nevertheless, this averaged quantity, similar to those obtained from other techniques dedicated to membranes (in particular, FCS and GP analyses) is representative of an overall morphological behavior that can be precisely measured when the cell nature or environment is affected.
Figure 2.
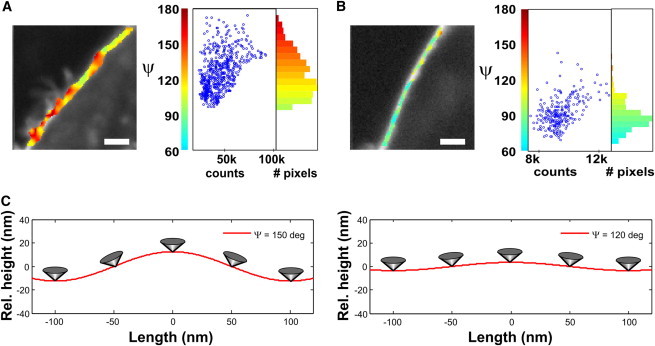
Zoomed areas of ψ images in COS-7 cell labeled with di-8-ANEPPQ. (A) Heterogeneous disordered region. (B) Ordered region. Scale bars, 2 _μ_m. The histograms of ψ are shown together with the correlation plot between intensity (total number of photons summed over all incident polarization angles) and ψ in the zoomed region. (C) Simulated 1D profile (cell-membrane relative height) of the lateral dimension of the cell membrane, assuming a sinusoidal shape, for measured cone apertures of ψ = 150° (left) and ψ = 120° (right) (see text). The change of orientation of the cone along the membrane is illustrated. The lateral extent of the profile is shown within the focal spot size of 200 nm.
To explore the efficiency of this technique in reporting membrane morphological changes, we investigated how molecular orientational order responded to different drugs affecting the actin network of the cells (Fig. 3). Single cells are illustrated in Fig. 3 A, and distributions of ψ over 12 cells are reported in Fig. 3 B. The ψ distributions of single cells visibly resemble those obtained from a population of cells, which shows that heterogeneities are intrinsic to a single sample. Mechanical disruption of the cytoskeleton by a hypotonic shock leads to an increase of molecular orientational order, an effect that is also observed using cytochalasin D, a drug that provokes actin depolymerization. Latrunculin A, an actin polymerization inhibitor, leads to a somewhat similar effect, with additional heterogeneities. jasplakinolide, which stabilizes actin filaments, provokes a new population of high disorder at ψ ∼ 150°, which seems to be correlated with the visible additional membrane wrinkles around its contour, but also with invisible features occurring at the subdiffraction scale. Interestingly, DiIC18 undergoes similar molecular orientational order changes under cytoskeleton perturbations, except in the case of cytochalasin D, whose cell membrane exhibits unexpected patches, which we attribute to damage from interactions with the lipid probe (Fig. S9).
Figure 3.
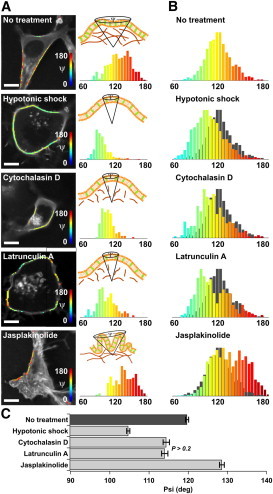
Effect of actin perturbation on molecular order in COS-7 cells labeled with di-8-ANEPPQ. (A) ψ maps (superimposed on the fluorescence intensity image) and corresponding histograms for a typical cell under conditions of (top to bottom) no treatment, and treatment with hypotonic shock, cytochalasin D, latrunculin A, and jasplakinolide. A schematic representation of the membrane local morphology is shown. (B) Histograms of the measured ψ values for 12 cells from each treatment group (gray represents the reference histogram with no treatment). (C) Mean ± SE values for each treatment. All cases show significant differences (P < 10−5) except for cytochalasin D and latrunculin A. Scale bars, 10 _μ_m.
Modification of lipid orientational order upon cholesterol depletion
Lipid orientational behavior is also governed by the membrane molecular content, which drives lipid packing (14). To explore the contribution of molecular orientational order to lipid organization when local polarity changes, we induced cholesterol depletion in the COS-7 cell membrane using cholesterol oxidase, which specifically converts cholesterol into cholestenone, and methyl-_β_-cyclodextrin (MBCD), which removes cholesterol from the membrane. Under low concentrations, both treatments induce a shift of the whole population toward a slightly higher molecular orientational order (Figs. 4 and S10), which indicates either a local morphology change or an increased rigidity of the local environment surrounding the lipid probes. At higher concentration, the behavior is reversed and orientational disorder is clearly appearing, even though the apparent macroscopic morphology of the cell does not change. This indicates that under high depletion conditions (which are known to induce a strong lipid disorder), lipid probes also tend to undergo more orientational freedom.
Figure 4.
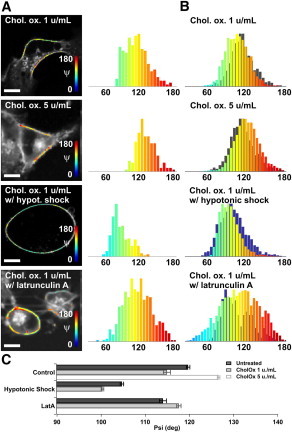
Effect of cholesterol depletion on molecular order in COS-7 cells labeled with di-8-ANEPPQ using cholesterol oxidase (Chol. ox.) and combined treatments. (A) ψ maps superimposed on the fluorescence intensity image and corresponding histograms for a typical cell for treatment conditions (top to bottom) Chol. ox. at 1 μ/mL and 5 μ/mL and at 1 μ/mL combined with hypotonic shock and latrunculin A. (B) Histograms of the measured ψ values of 12 cells for the respective treatment groups in A (gray, reference histogram with no cholesterol depletion; blue, reference histogram with hypotonic shock or latrunculin A). (C) Mean ± SE values represented for each treatment. All cases show significant differences (P < 10−5). Scale bars, 10 _μ_m.
Treatments combining cholesterol depletion with actin cytoskeleton perturbation were also performed to measure possible correlations between lipid packing and the nature of cytoskeleton filaments in close interaction with the membrane. When the treatments are applied in cells whose wrinkles are removed due to a preliminary hypotonic shock, a shift to even higher orientational order is visible, as if lipid packing orientational order could be probed with higher sensitivity thanks to membrane smoothing (Fig. 4). When cholesterol depletion is performed on a cell deprived of polymerized cytoskeleton (latrunculin A), the change of orientational order is slightly reversed and highly heterogeneous, indicating that an intact cytoskeleton filament is required to produce a reaction of lipid-packing ordering to cholesterol depletion. Note that these combined treatments lead to similar results for cholesterol oxidase and methyl-_β_-cyclodextrin.
Discussion
We implemented an excitation-polarization-resolved fluorescence technique that can quantify molecular orientational order along the whole contour of a cell membrane, offering the possibility of studying orientational behavior of lipids under conditions that can lead to heterogeneous populations of cells.
We show in particular that in unperturbed cells, the orientational order value, ψ, can be used as a direct signature of the local membrane morphology, able to report membrane accumulations (folds, attached internal membranes, or vesicles) of smaller scale than the diffraction limit. These accumulations are seen as regions richer in lipid probes and angularly more disoriented. ψ is, moreover, a sensitive signature to slight modifications of the local membrane morphology, given that a change of a few degrees (corresponding to the precision of the measurements; see Fig. S6) is the signature of changes of the membrane profile amplitude of ∼2 nm (see Materials and Methods). In particular, these wrinkles are globally removed by treatments affecting the actin cytoskeleton and locally enhanced when cells are treated with jasplakinolide (Fig. 3), which is known to induce confined regions with a high content of disordered polymeric actin (35). In this study, we demonstrate that these regions essentially occur at a subdiffraction scale, and we show a method of quantifying their spatial amplitude in a noninvasive way. This determination is an important piece of information that could, for instance, allow investigation into the role of topology effects in measurement methodologies potentially affected by them, such as FCS.
This mechanical interpretation supposes that there is no strong influence of a lower-scale lipid packing (driving lipid orientational freedom at the molecular scale) on the retrieved molecular orientational order. We implemented a measurement of lipid packing using GP, which exploits the spectral sensitivity of lipid probes to local polarity and is able to inform on the presence of lipid ordered domains in the membrane (36). GP measurements were performed on di-8-ANEPPQ in the same set-up, using the same methodology as in Owen et al. (36) (see Supporting Material). di-8-ANEPPQ exhibits a molecular structure similar to that of the well-known styryl-like lipid order probe di-4-ANEPPDHQ (36), and it is expected to reflect lipid ordered phases in its blue side emission (∼540 nm), whereas its presence in lipid disordered phases would enhance its red side emission (∼675 nm). In untreated cells, no significant difference in ψ values was observable between these two spectral channels, showing that if distinct lipid phases were present in the cell, the dye experienced similar orientational disorder in these domains. This is most probably due to the fact that the morphological complexity of the cell membrane prevails over all other possibly weaker molecular-scale disorder effects.
Forcing the membrane lipid packing to undergo stronger changes, however, revealed the sensitivity of orientational order to lipid packing. Measurements were performed under different conditions of cholesterol depletion, which is known to strongly modify lipid packing properties in cell membranes. Under relatively low depletion concentration, a slightly higher orientational order appears (Fig. 4), which corroborates previous observations of increased membrane stiffness (28), decreased lateral mobility of membrane proteins (37), and increased degree of lipid packing (30). For the cell membranes investigated in this work, a GP increase was detectable for both cholesterol oxidase and MBCD, clearly correlating with the increase in orientational order (Fig. S11). When cholesterol depletion was brought to a much higher level, a GP decrease was observed, correlating with a decrease in orientational order (Fig. S11). This decrease in lipid packing under a high dose of cholesterol depletion is also consistent with previous observations in cell membranes (29). Overall, these observations show the strong relationship between lipid packing and orientational order when cholesterol content is affected in cells, emphasizing the connection between local mechanical constraint and polarity.
It has been shown, however, that actin-filament attachment sites are strongly interrelated with the presence of lipid ordered domains (28,37), recognized as high packing sites with decreased fluidity (31). The mechanical property of the membrane therefore rather depends on both the organization of the submembrane cytoskeleton (and its attachment to the membrane) and on the physical properties of the lipid membrane itself. This statement is confirmed by our measurement of combined cytoskeleton disruption (using latrunculin A) and cholesterol depletion, which cancels the order effect and leads to a highly heterogeneous response. This indicates that the absence of polymerized actin in the proximity of the membrane hampers the action of lipid packing. This is in agreement with previous findings in similar conditions (38), reporting a faster lipid motion due to the absence of diffusion barriers (39) and an abrogation of the stiffening effect (28). Actin filaments attached to the membrane thus favor the existence of ordered lipid domains, which is directly visible in lipid packing and orientational order properties at the molecular scale. Finally, experiments performed on a disrupted cytoskeleton (hypotonic shock) in combination with cholesterol depletion show the opposite behavior with an increased order. Hypotonic shock probably preserves the stabilizing function of the cytoskeleton, which therefore maintains a strengthening effect on the membrane. Under such conditions, the membrane morphology is also smoothened, which probably makes the measured quantity more sensitive to lipid packing changes.
Overall, this technique appears to be capable of providing unique complementary information on subresolution-scale morphological features in cell membranes. However, to achieve a finer measurement of orientational freedom at the molecular scale in lipid domains would necessitate combining excitation-polarization-resolved fluorescence with lipid packing measurement in the membrane, and to account for the inevitable role of the cytoskeleton.
Conclusion
We have developed an excitation-polarization-resolved fluorescence microscopy technique based on an epiconfocal configuration that is able to circumvent the limitations of fluorescence anisotropy and linear dichroism schemes. This scheme allows the quantification of molecular orientational order in cell membranes of arbitrary shapes and shows its relation to local subdiffraction-scale morphology, local lipid environment, and membrane stiffness. These properties can be probed locally in a noninvasive way, which yields new (to our knowledge) perspectives on the architecture of biomolecular assembly. This technique can be exploited in a large variety of biological processes, addressing heterogeneous behaviors (such as proteins clustering in signalization sites, local perturbations induced by cell mechanics under stress, trafficking, or migration) and giving structural insight into biomolecular assemblies (such as insoluble fibril formation in cells or tissues).
Acknowledgments
The authors thank P. Réfrégier, J. Duboisset (Institut Fresnel, Marseille, France), as well as D. Marguet, H. T. He (Centre d’Immunologie de Marseille Luminy, Marseille, France), for helpful discussions and advice.
This work was supported by the Centre National de Recherche Scientifique, Agence Nationale de la Recherche grants ANR 2010 BLAN 150902 (ReceptORIENT), 121402 (NanoDIGICODE), 18818 (RADORDER), and ANR-10-INBS-04-01 (France-BioImaging Infrastructure network), Conseil Regional Provence Alpes Côte d’Azur, and the China Scholarship Council.
Supporting Material
Document S1. Eleven figures, one table, and supporting results
Document S2. Article plus Supporting Material
References
- 1.Cheresh D.A., Leng J., Klemke R.L. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. J. Cell Biol. 1999;146:1107–1116. doi: 10.1083/jcb.146.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anantharam A., Onoa B., Axelrod D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J. Cell Biol. 2010;188:415–428. doi: 10.1083/jcb.200908010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fooksman D.R., Grönvall G.K., Edidin M. Clustering class I MHC modulates sensitivity of T cell recognition. J. Immunol. 2006;176:6673–6680. doi: 10.4049/jimmunol.176.11.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber G. Rotational Brownian motion and polarization of the fluorescence of solutions. Adv. Protein Chem. 1953;8:415–459. doi: 10.1016/s0065-3233(08)60096-0. [DOI] [PubMed] [Google Scholar]
- 5.Dix J.A., Verkman A.S. Mapping of fluorescence anisotropy in living cells by ratio imaging. Application to cytoplasmic viscosity. Biophys. J. 1990;57:231–240. doi: 10.1016/S0006-3495(90)82526-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varma R., Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- 7.Vrabioiu A.M., Mitchison T.J. Structural insights into yeast septin organization from polarized fluorescence microscopy. Nature. 2006;443:466–469. doi: 10.1038/nature05109. [DOI] [PubMed] [Google Scholar]
- 8.Borejdo J., Burlacu S. Measuring orientation of actin filaments within a cell: orientation of actin in intestinal microvilli. Biophys. J. 1993;65:300–309. doi: 10.1016/S0006-3495(93)81060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gould T.J., Gunewardene M.S., Hess S.T. Nanoscale imaging of molecular positions and anisotropies. Nat. Methods. 2008;5:1027–1030. doi: 10.1038/nmeth.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberti M.J., Jovin T.M., Jares-Erijman E. Confocal fluorescence anisotropy and FRAP imaging of α-synuclein amyloid aggregates in living cells. PLoS ONE. 2011;6:e23338. doi: 10.1371/journal.pone.0023338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazar J., Bondar A., Firestein S.J. Two-photon polarization microscopy reveals protein structure and function. Nat. Methods. 2011;8:684–690. doi: 10.1038/nmeth.1643. [DOI] [PubMed] [Google Scholar]
- 12.Benninger R.K.P., Vanherberghen B., Önfelt B. Live cell linear dichroism imaging reveals extensive membrane ruffling within the docking structure of natural killer cell immune synapses. Biophys. J. 2009;96:L13–L15. doi: 10.1016/j.bpj.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Axelrod D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 1979;26:557–573. doi: 10.1016/S0006-3495(79)85271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Florine-Casteel K. Phospholipid order in gel- and fluid-phase cell-size liposomes measured by digitized video fluorescence polarization microscopy. Biophys. J. 1990;57:1199–1215. doi: 10.1016/S0006-3495(90)82639-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocheleau J.V., Edidin M., Piston D.W. Intrasequence GFP in class I MHC molecules, a rigid probe for fluorescence anisotropy measurements of the membrane environment. Biophys. J. 2003;84:4078–4086. doi: 10.1016/S0006-3495(03)75133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benninger R.K.P., Onfelt B., French P.M.W. Fluorescence imaging of two-photon linear dichroism: cholesterol depletion disrupts molecular orientation in cell membranes. Biophys. J. 2005;88:609–622. doi: 10.1529/biophysj.104.050096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kress A., Ferrand P., Brasselet S. Probing orientational behavior of MHC class I protein and lipid probes in cell membranes by fluorescence polarization-resolved imaging. Biophys. J. 2011;101:468–476. doi: 10.1016/j.bpj.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pentcheva T., Edidin M. Clustering of peptide-loaded MHC class I molecules for endoplasmic reticulum export imaged by fluorescence resonance energy transfer. J. Immunol. 2001;166:6625–6632. doi: 10.4049/jimmunol.166.11.6625. [DOI] [PubMed] [Google Scholar]
- 19.Chan F.T.S., Kaminski C.F., Kaminski Schierle G.S. HomoFRET fluorescence anisotropy imaging as a tool to study molecular self-assembly in live cells. ChemPhysChem. 2011;12:500–509. doi: 10.1002/cphc.201000833. [DOI] [PubMed] [Google Scholar]
- 20.Vishwasrao H.D., Trifilieff P., Kandel E.R. In vivo imaging of the actin polymerization state with two-photon fluorescence anisotropy. Biophys. J. 2012;102:1204–1214. doi: 10.1016/j.bpj.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gasecka A., Han T.J., Brasselet S. Quantitative imaging of molecular order in lipid membranes using two-photon fluorescence polarimetry. Biophys. J. 2009;97:2854–2862. doi: 10.1016/j.bpj.2009.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeMay B.S., Noda N., Oldenbourg R. Rapid and quantitative imaging of excitation polarized fluorescence reveals ordered septin dynamics in live yeast. Biophys. J. 2011;101:985–994. doi: 10.1016/j.bpj.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeMay B.S., Bai X., Gladfelter A.S. Septin filaments exhibit a dynamic, paired organization that is conserved from yeast to mammals. J. Cell Biol. 2011;193:1065–1081. doi: 10.1083/jcb.201012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesoine J.F., Lee J.Y., Hwang J. Quantitative scheme for full-field polarization rotating fluorescence microscopy using a liquid crystal variable retarder. Rev. Sci. Instrum. 2012;85:053705. doi: 10.1063/1.4717682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sund S.E., Swanson J.A., Axelrod D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys. J. 1999;77:2266–2283. doi: 10.1016/S0006-3495(99)77066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Rheenen J., Jalink K. Agonist-induced PIP2 hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol. Biol. Cell. 2002;13:3257–3267. doi: 10.1091/mbc.E02-04-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adler J., Shevchuk A.I., Parmryd I. Plasma membrane topography and interpretation of single-particle tracks. Nat. Methods. 2010;7:170–171. doi: 10.1038/nmeth0310-170. [DOI] [PubMed] [Google Scholar]
- 28.Byfield F.J., Aranda-Espinoza H., Levitan I. Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophys. J. 2004;87:3336–3343. doi: 10.1529/biophysj.104.040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaus K., Gratton E., Jessup W. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc. Natl. Acad. Sci. USA. 2003;100:15554–15559. doi: 10.1073/pnas.2534386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahammad S.S., Dinic J.J., Parmryd I. Limited cholesterol depletion causes aggregation of plasma membrane lipid rafts inducing T cell activation. Biochim. Biophys. Acta. 2010;1801:625–634. doi: 10.1016/j.bbalip.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Dinic J., Ashrafzadeh P., Parmryd I. Actin filaments attachment at the plasma membrane in live cells cause the formation of ordered lipid domains. Biochim. Biophys. Acta. 2013;1828:1102–1111. doi: 10.1016/j.bbamem.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Ferrand P., Pianta M., Marguet D. A versatile dual spot laser scanning confocal microscopy system for advanced fluorescence correlation spectroscopy analysis in living cell. Rev. Sci. Instrum. 2009;80:083702. doi: 10.1063/1.3205447. [DOI] [PubMed] [Google Scholar]
- 33.Collett E. CRC Press; Boca Raton, FL: 1993. Polarized Light: Fundamentals and Applications. [Google Scholar]
- 34.Wang X., Kress A., Ferrand P. High frame-rate confocal polarization resolved fluorescence microscopy. Rev. Sci. Instrum. 2013;84:053708. doi: 10.1063/1.4807318. [DOI] [PubMed] [Google Scholar]
- 35.Bubb M.R., Spector I., Fosen K.M. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 2000;275:5163–5170. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- 36.Owen D.M., Rentero C., Gaus K. Quantitative imaging of membrane lipid order in cells and organisms. Nat. Protoc. 2012;7:24–35. doi: 10.1038/nprot.2011.419. [DOI] [PubMed] [Google Scholar]
- 37.Kwik J., Boyle S., Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. USA. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun M., Northup N., Forgacs G. The effect of cellular cholesterol on membrane-cytoskeleton adhesion. J. Cell Sci. 2007;120:2223–2231. doi: 10.1242/jcs.001370. [DOI] [PubMed] [Google Scholar]
- 39.Lenne P.-F., Wawrezinieck L., Marguet D. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 2006;25:3245–3256. doi: 10.1038/sj.emboj.7601214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Eleven figures, one table, and supporting results
Document S2. Article plus Supporting Material