Effects of rosuvastatin on expression of angiotensin-converting enzyme 2 after vascular balloon injury in rats (original) (raw)
Abstract
Objective
To investigate the effects and mechanisms of rosuvastatin on angiotensin -converting enzyme 2 (ACE2) in the process of neointimal formation after vascular balloon injury in rats, and to explore the effects of ACE2 and rosuvastatin in restenosis.
Methods
Thirty-six Wistar rats were randomly allocated into three groups: control group (n = 12), surgery group (n = 12), and statin group (n = 12). Aortic endothelial denudation of rats was performed using 2F balloon catheters. At days 14 and 28 after injury, aortic arteries were harvested to examine the following. Intimal thickening was examined by hematoxylin and eosin staining. We measured angiotensin II (Ang II) and angiotensin 1-7 (Ang-[1–7]) levels by a radioimmunological method or enzyme-linked immunosorbent assay. Protein and mRNA expression of ACE2 and Ang II type 1 receptor (AT1) were investigated by immunohistochemistry, Western blots, and Reverse transcriptase-polymerase chain reaction (RT-PCR). We measured changes in proliferating cell nuclear antigen (PCNA) by immunohistochemistry. The level of phosphorylated extracellular signal regulated kinase 1/2 (P-ERK1/2) was evaluated by Western blotting.
Results
Proliferation of vascular smooth muscle cells (VSMC) and intimal thickening were higher at day 14 after vascular balloon injury in the surgery group compared with the control group. Proliferation of VSMC was decreased by day 28 after injury, while intimal thickening continued. With rosuvastatin treatment, the extent of VSMC proliferation and intimal thickening was reduced at day 14 and 28 after injury. Ang II and P-ERK levels were significantly increased, Ang-(1–7) levels were significantly decreased, mRNA and protein expressions of ACE2 were significantly decreased, and AT1 expression was significantly increased at days 14 and 28 after vascular balloon injury in the surgery group compared with the control group. PCNA expression was higher in the surgery group than in the control group, and it was significantly decreased after being given rosuvastatin. Expression of ACE2 mRNA and protein, and Ang-(1–7) levels were significantly increased, while AT1 expression and levels of Ang II and P-ERK were significantly decreased in the statin group compared with the surgery group.
Conclusions
Expression of ACE2 mRNA and protein is decreased in the process of intimal thickening after balloon injury. The inhibitory effect of rosuvastatin on intimal thickening is related to upregulation of ACE2, an increase in Ang-(1–7), downregulation of AT1, and activation of the P-ERK pathway.
Keywords: Balloon injury, Angiotensin converting enzyme 2, Extracellular signal regulated kinase, Rosuvastatin
1. Introduction
Percutaneous transluminal coronary angioplasty (PTCA) and stent implantation are the most commonly used coronary interventions worldwide. However, restenosis, one common complication of PTCA, seriously compromises the efficacy of treatment. Restenosis can occur in 30% to 50% of patients within 6 months after PTCA, and its occurrence is still up to 17% to 32% after stent implantation. Currently, there is no effective method to prevent restenosis. Drug-eluting stents can reduce the restenosis rate to 0% to 10% in some relatively simple lesions. However, the efficacy and safety of drug-eluting stents in complex cases still need to be verified by further investigations. The main mechanism of restenosis is the migration and proliferation of medial vascular smooth muscle cells (VSMC).[1] Angiotensin II (Ang II) takes part in this process, causing neointimal hyperplasia mainly through its type 1 receptor (AT1). Angiotensin-converting enzyme 2 (ACE2), a homologue of ACE compounds found in recent years, can efficiently catalyze Ang II into angiotensin 1-7 (Ang-[1–7]), which can antagonize Ang II through its MAS receptor, interfering in a series of reactions mediated by Ang II.[2] Extracellular signal-regulated kinase (ERK), an important member of the mitogen activated protein kinase (MAPK) family, is a common pathway or intersection of intracellular information transmission for cell proliferation and differentiation, responding to a variety of extracellular signal stimuli. ERK plays an important role in conducting signals from surface receptors to the nucleus, and also participates in functions, such as cell proliferation and differentiation.
Statins have other beneficial functions besides lipid regulation. Research has shown that rosuvastatin inhibits neointimal formation after vascular endothelial injury[3] and atorvastatin inhibits Ang II-induced vascular remodeling.[4] However, little is known about changes in ACE2 expression and its mechanisms in the process of neointimal formation.
Therefore, in this study, we investigated the effects and mechanisms of rosuvastatin on ACE2 in the process of neointimal formation after vascular balloon injury in rats. We also explored the effects of ACE2 and rosuvastatin in restenosis.
2. Methods
2.1. Animal model
Thirty-six male Wistar rats (purchased from Qingdao Animal Center), weighing 300 to 350 g, were randomly allocated into three groups: control group (n = 12), surgery group (n = 12), and statin group (n = 12). Aortic tissues were harvested on days 14 and 28 after surgery. There were six rats in each group at each time point. The surgery group had aortic endothelial denudation performed using self-made 2F balloon catheters. The statin group also had aortic endothelial denudation. Rosuvastatin (AstraZeneca Pharmaceutical Co., Ltd., UK) was administered by gastric gavage at a dosage of 5 mg/kg per day from 1 day before injury to day 14 or day 28 after injury. The control group received the same procedures, except for inflation of the balloon. Five milliliters of 0.9% physiological saline was administered to the control group and the surgery group once a day by gastric gavage.
Evan's blue 0.5% (2 mL/kg) was injected into the left common carotid vein immediately after the operation and the rats were sacrificed in 1 h. We observed endothelial denudation, and documented that this procedure effectively and completely denuded the aortic endothelium.
2.2. Histological examinations for VSMC migration, proliferation, and intimal hyperplasia
Approximately 4–5 cm of an aortic segment was harvested under aseptic conditions. A length of 5 mm of the abdominal aorta near the aortic arch end was obtained and fixed in 10% formalin, and embedded in paraffin for hematoxylin and eosin (HE) staining and immunohistochemical examination. The remaining vessel segment was cryopreserved at −80°C. HE slides were observed under a light microscope for VSMC migration, proliferation, and intimal hyperplasia. The thickness of the media and intima was measured by an image analyzer.
2.3. Radioimmunoassay and enzyme-linked immunosorbent assay for measurement of Ang II and Ang-(1–7) levels
A total of 35 mg of cryopreserved vessel segment was ground and homogenated. The supernatant was stored at -80°C. Ang II levels in vascular tissue were detected by the radioimmunoassay (RIA) method. The RIA was performed in the Technology Development Center of PLA General Hospital. Ang-(1–7) levels were measured by enzyme-linked immunosorbent assay (ELISA) (Shanghai Xi Tang Biotechnology Co., Ltd., China). The procedures were conducted rigorously in accordance with the instructions in the kits.
2.4. Immunohistochemistry for protein expression of ACE2, AT1, and proliferating cell nuclear antigen (PCNA)
The streptavidin-peroxidase method was used for the detection of protein expression of ACE2, AT1, and PCNA in the aorta. Antibodies against ACE2, AT1, and PCNA were purchased from Santa Cruz Company (USA). Aortic paraffin sections were processed by the following procedures: xylene dewaxing, ethanol hydration, blocking and inactivation of endogenous peroxidase, antigen retrieval, primary antibody diluent, a PBS rinse, secondary antibody incubation, diaminobenzidine (DAB) coloration after rinsing, hematoxylin, conventional dehydration, transparency, and drying and sealing of the slides. The number and distribution of brown particles were observed under a microscope after mounting the slides. The primary antibodies of ACE2, AT1, and PCNA were diluted to 1: 200, 1: 50, and 1: 100, respectively, and the second antibodies were diluted to 1: 1250.
Each slice was observed in five high power fields by an automatic image analysis system (Jieda Technology Company, Jiangsu Province, China) and the percentage of PCNA-positive cells in the total cells was counted.
2.5. Western blots for ACE2, AT1, and P-ERK
Protein was extracted from the aorta by a protein extraction reagent. The sample was loaded after being boiled for 5 min. After sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoresis blotting, diluted antibodies of ACE2 (1:200), AT1 (1: 100), P-ERK (1: 100), and actin (1: 500) (Santa Cruz Company, USA) were added for primary antibody reaction. Blots were incubated on a shaker at room temperature for 2 h, and membranes were washed for three times before they were used for secondary antibody reaction (1:1250) and DAB staining. The expression of protein was photographed and analyzed by a digital gel imaging system (Alpha Innotech Company, USA).
2.6. Detection of ACE2 and AT1 mRNA expression
2.6.1. Total RNA extraction
Single-cell deposition of VSMC was prepared from a segment of cryopreserved aorta. One milliliter of Trizol reagent (Invitrogen Company, USA) was added and total RNA was extracted using chloroform and isopropyl alcohol. RNA A260/A280 was tested by an UV spectrophotometer to detect its purity. The A260/A280 of all the samples was greater than 1.7.
2.6.2. Reverse transcriptase-polymerase chain reaction (RT-PCR)
A reverse transcription reaction was rigorously carried out according to the instructions in the RT kits (Promega Corporation, USA). Reaction conditions in a water bath were 42°C for 1 h, 4°C for 5 min, and 99°C for 5 min. The sequences of primers for ACE2 and the internal reference β-actin for amplification of cDNA in PCR were as follows: ACE2 upstream primer, 5′-CAAAGTTCACTTGCTTCTT GG-3′ and downstream primer, 5′-TACTGTAAATGGTGC TCATGG -3′, with a length of amplified fragments of 263 bp; AT1 upstream primer, 5′-CACCTATGTAAGATCGC TTC-3′, and downstream primer, 5′-GCACAATCGCCAT AATTATCC-3′, with a length of amplified fragments of 444 bp; and β-actin upstream primer, 5′-GAGGGAAATC GTGCGTGAC-3′, and downstream primer, 5′-GGAGCCA GGGCAGTAATC-3′, with a length of amplified fragments of 350 bp. The reaction conditions for PCR were 94°C for 1 min, 58°C for 1 min, and 72°C for 1 min, with a total of 35 cycles and a final extension of 7 min at 72°C.
2.6.3. Analysis
After 1.5% agarose gel electrophoresis, the results were analyzed by a gel imaging analyzer. ACE2, AT1, and β-actin fragments from RT-PCR amplification were in line with the expected length. The mean optical density ratio of amplification bands between the target gene and the internal reference β-actin was regarded as the relative value of target gene mRNA.
2.7. Statistical analysis
Measurement data of normal distribution are expressed as mean±SE. Two-way ANOVA was used to determine the significance of differences between groups with respect to data of repeated measurements and the Student-Newman-Keuls test was used as a post-hoc test. A P value of less than or equal to 0.05 was considered statistically significant. SPSS 16.0 software was used for all the analyses.
3. Results
3.1. VSMC proliferation and intimal hyperplasia
Fourteen days after balloon injury, we observed that endothelial cells covered most of the surface of the aorta, intimal VSMC had proliferated, the intima and media were hyperplasic and the internal elastic fibers were bent, while intimal hyperplasia predominated. Twenty-eight days after the injury, endothelial cells covered almost all of the damaged area, VSMC proliferation was significantly reduced, but extracellular matrix components were increased and the intima continued to grow. After administration of rosuvastatin, migration of VSMC to the intima was decreased, VSMC proliferation and intimal hyperplasia were reduced, and the extracellular matrix was reduced. At the same time, intimal thickness in the statin group was significantly lower compared with that in the surgery group (P < 0.01, Figure 1 and Table 1).
Figure 1. Effects of rosuvastatin on histological changes at day 14 after balloon injury.
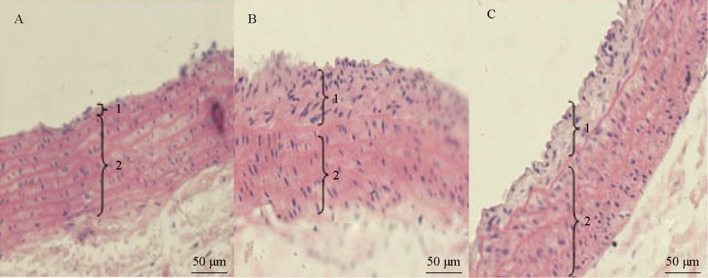
1: intima. 2: media. Pathological sections of arterial lesions by HE staining showed that the intima and vessel walls were thin in the control group (A), but the intima was thick and there were more proliferated VSMCs than in the surgery group (B). In the rosuvastatin group (C), the intima was thinner and there were less proliferated VSMCs than in the surgery group (× 200). VSMCs: vascular smooth muscle cells.
Table 1. Change in thickness of the intima and media after injury (µm).
| Group, n = 12 | Position | Day 14 after injury | Day 28 after injury |
|---|---|---|---|
| Control group | Intima | 29.32 ± 10.15 | 28.67 ± 9.67 |
| Media | 205.36 ± 12.93 | 211.35 ± 17.63 | |
| Surgery group | Intima | 91.66 ± 8.45** | 106.72 ± 12.59** |
| Media | 291.25 ± 14.32** | 251.61 ± 15.01** | |
| Statin group | Intima | 66.53 ± 7.29**,# | 76.67 ± 9.65**,# |
| Media | 247.62 ± 13.27**,# | 226.38 ± 12.55**,# |
3.2. Ang II and Ang-(1–7) levels
Ang II levels in the surgery group were significantly higher (P < 0.01, P < 0.05), and Ang-(1-7) levels were significantly lower (P < 0.01 for both time points) on days 14 and 28 after aortic endothelial balloon injury compared with the control group. Ang II levels were significantly lower and Ang-(1–7) levels were significantly higher in the rosuvastatin group compared with the surgery group (P < 0.01 for both time points, Figure 2).
Figure 2. Ang II and Ang-(1–7) levels in the aorta.
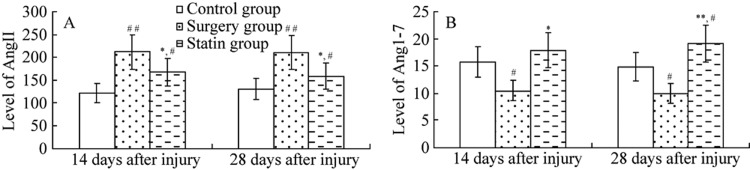
At days 14 (A) and 28 (B), surgery group rats showed higher plasma Ang II levels and lower Ang-(1–7) levels compared with the control group. Pretreatment with rosuvastatin decreased plasma Ang II and increased Ang-(1–7) levels. Values are expressed as mean ± SE (n = 6). *P < 0.05, **P < 0.01 vs. the control group; #P<0.05, ##P<0.01 vs. the surgery group. Ang II: angiotensin II; Ang-(1–7): angiotensin 1–7.
3.3. ACE2 and AT1 mRNA expression
On days 14 and day 28 after injury, ACE2 mRNA expression was significantly lower (P <0.01 for both time points) in the surgery group compared with the control group, and it was significantly higher after the use of rosuvastatin (P <0.05, P <0.01, Figure 3). AT1 mRNA expression was significantly higher (P < 0.01 for both time points) in the surgery group compared with the control group, and it was significantly lower after the use of rosuvastatin (P < 0.05 for both time points).
Figure 3. ACE2 and AT1 mRNA levels.

Expression of ACE2 (A) and AT1 (B) mRNA are shown at day 14 after injury; (C) ACE2 and AT1 mRNA at days 14 and 28 after injury. ACE2 mRNA expression was decreased and AT1 mRNA was increased after injury, but ACE2 mRNA was increased and AT1 mRNA was decreased after rosuvastatin treatment. Values are expressed as mean ± SE (n = 6). *P < 0.05, **P < 0.01 vs. the control group; #P < 0.05, ##P < 0.01 vs. the surgery group. 1: control group; 2: surgery group; 3: statin group; 4: marker. ACE2: angiotensin -converting enzyme 2; AT1: Ang II type 1 receptor.
3.4. Protein expression of ACE2 and AT1
The results of Western blotting showed that on days 14 and 28 after injury, protein expression of ACE2 was significantly lower (P < 0.01 for both time points), and AT1 was significantly higher in the surgery group than in the control group (P <0.01 for both time points). After rosuvastatin treatment (statin group), ACE2 expression was significantly higher (P < 0.05, P < 0.01) on days 14 and 28 after injury and AT1 protein expression was significantly lower (P < 0.05 for both time points) than in the surgery group. Immunohistochemistry showed that ACE2 and AT1 protein were expressed in the membrane and cytoplasm (brown-yellow particles) in vascular endothelial cells and smooth muscle cells (Figures 4 and 5).
Figure 4. ACE2 and AT1 protein levels by Western blot.

(A): ACE2 and AT1 levels shown by Western blot at day 14 after injury; (B): ACE2 and AT1 protein levels shown by Western blot at days 14 and 28 after injury. ACE2 levels were decreased and AT1 levels were increased after injury, but ACE2 levels were increased and AT1 levels were decreased after rosuvastatin treatment. Values are expressed as mean ± SE (n = 6).*P < 0.05, **P < 0.01 compared with the control group; #P < 0.05, ##P < 0.01 compared with the surgery group. ACE2: angiotensin -converting enzyme 2; AT1: Ang II type 1 receptor.
Figure 5. ACE2 (A) and AT1 (B) expression in the control group, surgery group and statin group.

ACE2 and AT1 protein are expressed in the membrane and cytoplasm (brown-yellow particles) in vascular endothelial cells and smooth muscle cells. ACE2 protein expression was lower (brown granules were significantly lower) and AT1 protein expression was higher (brown granules were significantly higher) in the surgery group compared with the control group. After rosuvastatin treatment, ACE2 expression was significantly higher and AT1 expression was significantly lower than in the surgery group. ACE2: angiotensin -converting enzyme 2; AT1: Ang II type 1 receptor.
3.5. Expression of PCNA
The nucleus of PCNA-positive cells was a buffy color. On day 14, PCNA expression was 9.60% ± 1.61% in the control group, 69.64% ± 4.56% in the surgery group, and 47.53% ± 3.22% in the statin group. On day 28, PCNA expression was 8.96% ± 1.52% in the control group, 51.35% ± 4.33% in the surgery group, and 35.23% ± 3.19% in the statin group. PCNA expression was significantly higher in the surgery group than in the control group as shown by immunohistochemistry (P < 0.01 both time points), and it was significantly decreased after being given rosuvastatin (P < 0.01 for both time points, Figure 6). PCNA expression was lower on day 28 than on day 14 in the surgery and statin groups (both P < 0.05).
Figure 6. PCNA expression in VSMCs 14 days after aortic injury (× 400).

(A): Control group; B: surgery group; C: statin group. Black lines show the neointima and red arrows indicate a brown positive nucleus. PCNA expression was significantly higher in the surgery group than in the control group, but it was lower in the statin group. PCNA: proliferating cell nuclear antigen; VSMCs: vascular smooth muscle cells.
3.6. P-ERK levels
P-ERK1/2 protein expression was significantly higher in the surgery group on days 14 and 28 after injury compared with the control group (P < 0.01, P < 0.05), and it was significantly inhibited in the statin group compared with the control group (P < 0.05, P < 0.05, Figure 7).
Figure 7. Expression of P-ERK1/2 by Western blot at day 14 after injury.

(A) Western blot showing P-ERK expression; (B) P-ERK protein levels. P-ERK1/2 protein expression was higher in the surgery group than in the control group, and it was significantly lower than in the statin group. Values are expressed as mean ± SE (n = 6).*P<0.05, **P<0.01 compared with the control group; #P < 0.05, ##P < 0.01 compared with the control group. P-ERK: phosphorylated extracellular signal regulated kinase.
4. Discussion
Restenosis is essentially a localized excessive healing response to vascular balloon injury. The mechanisms of restenosis include vascular elastic recoiling, negative vascular remodeling, thrombosis, and organization, VSMC hyperplasia, and extracellular matrix accumulation. Stents can prevent vessels from recoiling and negative vascular remodeling, but they fail to restrict VSMC proliferation, resulting in the occurrence of in-stent restenosis.[1]
Ang II, which is the main active peptide in the renin-angiotensin system, leads to VSMC migration and proliferation through the AT1 receptor when the vascular endothe-lium is injured, thereby resulting in intimal hyperplasia and accelerating occurrence of restenosis.[5] Our study showed that 14 days after aortic endothelial injury, Ang II protein levels and AT1 mRNA and protein expression were increased. The combination of an increase in Ang II and AT1 could accelerate VSMC proliferation and intimal hyperplasia.
ACE2 was the first discovered homologue of ACE, and is scattered in the heart, kidney, testis, lung, prostate, gastrointestinal tissue, vascular endothelial cells, VSMC, and some adventitia. ACE2 catalyzes the conversion of Ang II into Ang-(1–7). ACE2 and its catalytic products Ang-(1–7) are important bio-active substances of RAS. Ang-(1–7) exerts its anti-Ang II function by acting as ligands of the G protein-coupled receptor Mas.[6] Ang-(1–7) are cardiovascular protective peptides with vasodilatory, anti-growth, and anti-proliferative effects. ACE2 can increase the degradation of Ang II, reducing the adverse effects of Ang II, and it can also efficiently catalyze Ang II into Ang-(1–7), antagonizing Ang II and interfering with Ang II-mediated cell proliferation and vascular remodeling.[6],[7] Lovren reported that Ang-(1–7) can inhibit Ang II-induced VSMC proliferation and migration.[8] Our study showed that ACE2 expression was decreased after aortic endothelial injury. As a result, less Ang II was converted into Ang-(1–7) and the antagonism of Ang II was decreased, leading to an enhanced effect on VSMC migration, proliferation, and intimal hyperplasia.
Abnormal proliferation of VSMC is an important pathological feature of restenosis after percutaneous coronary intervention. The MAPK-mediated signal transduction pathway is a common pathway for transferring information of proliferation for a variety of cells. ERK1/2, an important extracellular signal-regulated kinase of the MAPK family, plays an important role in promoting VSMC proliferation and migration by acting as an important signal molecule in mediating cell differentiation, division, and proliferation. An increasing amount of evidence has shown that this signal transduction pathway is involved in restenosis after angioplasty.[9] Our study showed that the level of P-ERK was increased after aortic endothelial injury, which indicated activation of the ERK pathway after injury. This pathway can transmit information of cell proliferation and differentiation into cells, resulting in migration and proliferation of VSMC and neointimal hyperplasia. Lercanidipine (a calcium antagonist) can inhibit VSMC proliferation and neointimal formation through inactivating the Ras-ERK1/2 signal transduction pathway and reducing intracellular active oxygen.[10] Cilostazol inhibits the proliferation of VSMC via ERK1/2 pathways.[11]
Statins have some other beneficial functions besides lipid regulation. Clinical and experimental research have shown that statins inhibit RAS activation and improve vascular remodeling.[4],[12] Rosuvastatin decreases neointimal formation after vascular endothelial injury[3] and plays an important role in preventing restenosis.
Our study showed that rosuvastatin reduced intimal hyperplasia after aortic endothelial injury, while it increased ACE2 mRNA expression and Ang-(1–7) protein levels, suggesting that increased expression of ACE2 can catalyze more Ang II into Ang-(1–7) and antagonize Ang II-induced VSMC migration and proliferation. In addition, rosuvastatin downregulated the expression of AT1 and the levels of Ang II and P-ERK, which indicated that rosuvastatin reduced intimal proliferation after aortic endothelial injury through enhancing the ACE2-Ang-(1–7) pathway, as well as through inhibiting the Ang II-AT1 pathway. Therefore, vascular remodeling was improved and the incidence of restenosis was reduced. ERK pathway activation is also involved in this function. Further studies are required for exploring the extent of the effect of ACE2 in restenosis and its direct relation with ERK signaling pathways.
5. Conclusions
ACE2 mRNA and protein expressions are decreased in the process of intimal thickening after balloon injury. The inhibitory effect of rosuvastatin on intimal thickening is related to the upregulation of ACE2 expression, increased levels of Ang1-7, downregulation of AT1, and activation of the ERK pathway.
Acknowledgments
This study was supported by the National Nature Science Foundation of China (No: 81071246). We thank the following persons who have made the completion of this research and paper possible: Professor Lei Wang from the Department of Pharmacology, for her direction and assistance in the laboratory, and Dr. Hai-Yan Feng for her assistance with English translation.
References
- 1.Curcio A TD, Indolfi C. Mechanisms of smooth muscle cell p oliferation and endothelial regeneration after vascular injury and stenting. Circ J. 2011;75:1287–1296. doi: 10.1253/circj.cj-11-0366. [DOI] [PubMed] [Google Scholar]
- 2.Rabelo LA, Alenina N, Bader M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res. 2011;34:154–160. doi: 10.1038/hr.2010.235. [DOI] [PubMed] [Google Scholar]
- 3.Preusch MR, Vanakaris A, Bea F, et al. Rosuvastatin reduces neointima formation in a rat model of balloon injury. Eur J Med Res. 2010;15:461–467. doi: 10.1186/2047-783X-15-11-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briones AM, Rodríguez-Criado N, Hernanz R, et al. Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress. Hypertension. 2009;54:142–149. doi: 10.1161/HYPERTENSIONAHA.109.133710. [DOI] [PubMed] [Google Scholar]
- 5.Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4:104–111. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santos RA, Ferreira AJ, Verano-Braga T, et al. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216:R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 7.Shi L, Mao C, Xu Z, et al. Angiotensin-converting enzymes and drug discovery in cardiovascular diseases. Drug Discov Today. 2010;15:332–341. doi: 10.1016/j.drudis.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, Hu Y, Xu Q, et al. Different effects of angiotensin II and angiotensin-(1-7) on vascular smooth muscle cell proliferation and migration. PLoS One. 2010;5:e12323. doi: 10.1371/journal.pone.0012323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W, Guo T, Zhang Y, et al. The inhibitory effect of dexamethasone on platelet-derived growth factor-induced vascular smooth muscle cell migration through up-regulating PGC-1α expression. Exp Cell Res. 2011;317:1083–1892. doi: 10.1016/j.yexcr.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Wu JR, Liou SF, Lin SW, et al. Lercanidipine inhibits vascular smooth muscle cell proliferation and neointimal formation via reducing intracellular reactive oxygen species and inactivating Ras-ERK1/2 signaling. Pharmacol Res. 2009;59:48–56. doi: 10.1016/j.phrs.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Yoo AR, Koh SH, Cho GW, et al. Inhibitory effects of cilostazol on proliferation of vascular smooth muscle cells (VSMCs) through suppression of the ERK1/2 pathway. J Atheroscler Thromb. 2010;17:1009–1018. doi: 10.5551/jat.4309. [DOI] [PubMed] [Google Scholar]
- 12.Qiang B, Toma J, Fujii H, et al. Statin therapy prevents expansive remodeling in venous bypass grafts. Atherosclerosis. 2012;223:106–113. doi: 10.1016/j.atherosclerosis.2012.03.013. [DOI] [PubMed] [Google Scholar]