Ligand promiscuity through the eyes of the aminoglycoside N3 acetyltransferase IIa (original) (raw)
Abstract
Aminoglycoside-modifying enzymes (AGMEs) are expressed in many pathogenic bacteria and cause resistance to aminoglycoside (AG) antibiotics. Remarkably, the substrate promiscuity of AGMEs is quite variable. The molecular basis for such ligand promiscuity is largely unknown as there is not an obvious link between amino acid sequence or structure and the antibiotic profiles of AGMEs. To address this issue, this article presents the first kinetic and thermodynamic characterization of one of the least promiscuous AGMEs, the AG _N_3 acetyltransferase-IIa (AAC-IIa) and its comparison to two highly promiscuous AGMEs, the AG _N_3-acetyltransferase-IIIb (AAC-IIIb) and the AG phosphotransferase(3′)-IIIa (APH). Despite having similar antibiotic selectivities, AAC-IIIb and APH catalyze different reactions and share no homology to one another. AAC-IIa and AAC-IIIb catalyze the same reaction and are very similar in both amino acid sequence and structure. However, they demonstrate strong differences in their substrate profiles and kinetic and thermodynamic properties. AAC-IIa and APH are also polar opposites in terms of ligand promiscuity but share no sequence or apparent structural homology. However, they both are highly dynamic and may even contain disordered segments and both adopt well-defined conformations when AGs are bound. Contrary to this AAC-IIIb maintains a well-defined structure even in apo form. Data presented herein suggest that the antibiotic promiscuity of AGMEs may be determined neither by the flexibility of the protein nor the size of the active site cavity alone but strongly modulated or controlled by the effects of the cosubstrate on the dynamic and thermodynamic properties of the enzyme.
Keywords: aminoglycosides, acetyltransferase, protein dynamics, ligand promiscuity, isothermal titration calorimetry, nuclear magnetic resonance, aminoglycoside modification, antibiotic resistance, intrinsically disordered proteins
Introduction
Among the mechanisms by which bacterial strains sustain resistance to the actions of antibiotics, the most common is that of enzymatic modification. Bacteria often harbor one or more genes coding for proteins that catalyze a covalent modification to an antibiotic.1 One family of antibiotics, known as the aminoglycosides (AGs), are vulnerable to proteins that can phosphorylate, acetylate, and/or nucleotidylate them. These AG-modifying enzymes (AGMEs) are a large family of proteins where each member modifies a unique hydroxyl or amino functional group on the antibiotic. Such modifications prevent the antibiotic from associating with the bacteria's ribosome and interfering with protein translation; a normally bactericidal function.2,3 Many AGMEs are known to have a broad ligand selectivity as in the case of the AG phosphotransferase (3′)-IIIa (APH) which can modify >15 structurally diverse AGs.1,4
As AGs are most commonly used to treat severe infections like tuberculosis and meningitis,5,6 understanding the biological basis of antibiotic promiscuity by AGMEs and uncovering the molecular details by which they select their ligands holds importance in both medicinal and scientific communities. To this end, this article serves to introduce the basic kinetic and thermodynamic properties of a weakly promiscuous protein, the AG _N_3 acetyltransferase IIa (AAC-IIa), and to discuss its similarities and differences with two strongly promiscuous AGMEs, the AG _N_3 acetyltransferase IIIb (AAC-IIIb) and the APH that have been previously characterized.7–12 APH and AAC-IIa are highly dynamic enzymes and display properties of intrinsically disordered proteins in the absence of AGs. Upon binding of AGs, they adopt well-defined structures. However, APH has no sequence homology (<5%) or similarity (<10%) to AAC-IIa and has a much broader substrate profile. On the other hand, AAC-IIIb and AAC-IIa catalyze the same reaction, the acetyl coenzyme A (AcCoA)-dependent acetylation of the amine functional group at the C3 position of AGs, a structural feature shared by all members of this antibiotic family.13 AAC-IIIb and AAC-IIa are strikingly similar in both amino acid sequence and structure and yet demonstrate strong differences in their substrate profiles and kinetic and thermodynamic properties. This provides a unique situation to probe the molecular basis underlying the substrate promiscuity of the AGMEs that could be extrapolated to other protein–ligand systems and potentially aid antibacterial drug development.
Results
Steady State Kinetics
AAC-IIa is an _N_3 acetyltransferase and thus acetylates AG antibiotics at the amine functional group of carbon 3 on the 2-deoxystreptamine ring with the aid of AcCoA (Fig. 1). Kanamycin class AGs are the targeted substrates of this enzyme although extremely slow turnover of neomycins (200- to 5000-fold slower than kanamycins) was able to be detected only via product analysis by TLC and NMR after overnight incubation with a high concentration of enzyme (Supporting Information Fig. S1). This makes AAC-IIa one of the least promiscuous AGMEs with detailed characterization.
Figure 1.
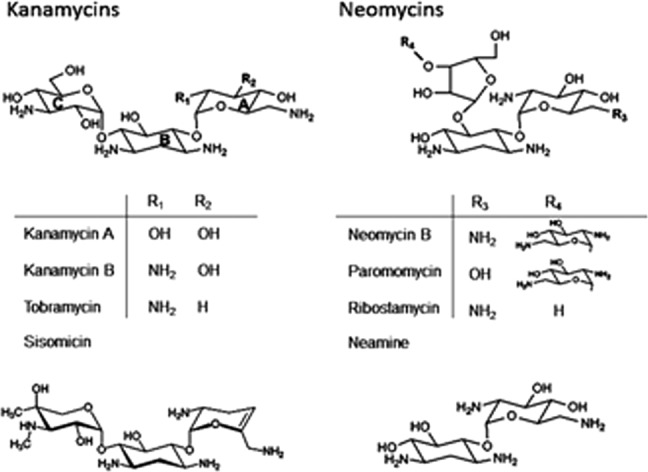
Aminoglycoside antibiotic structures. Aminoglycosides on the left are of the kanamycin class while those on the right belong to the neomycin class. A, B, and C notations inside the kanamycin on the left are the primed (′), 2-deoxystreptamine (unprimed), and double primed (′′) rings, respectively. Neomycins follow the same nomenclature. Sisomicin and neamine are shown separately at the bottom.
Within the kanamycins, turnover rates are quite varied revealing a strong degree of substrate selectivity by this enzyme (Table I). Tobramycin has the highest _k_cat value followed by kanamycin B, sisomicin, and kanamycin A. However, sisomicin has the lowest _K_m thus making it the best substrate (highest _k_cat/_K_m) of AAC-IIa. Tobramycin comes in second place at merely half the value of sisomicin. In addition, substrate inhibition was observed for AAC-IIa as is common among all AGMEs studied to date.
Table I.
Steady-State Kinetic Comparison Between AAC(3)-IIIb and AAC(3)-IIa
| _k_cat (/s) | K_m (μ_M) | k_cat/K_m (/M, /s) × 106 | K_i (μ_M) | |||||
|---|---|---|---|---|---|---|---|---|
| Antibiotic | IIIb | IIa | IIIb | IIa | IIIb | IIa | IIIb | IIa |
| Tobramycin | 48 | 30 | 1.3 | 3.6 | 36.9 | 8.3 | 17 | 73 |
| Kanamycin B | 55 | 10 | 2.6 | 6.5 | 21.2 | 1.5 | 26 | 300 |
| Kanamycin A | 51 | 1.1 | 4.8 | 6.1 | 10.6 | 0.2 | 380 | None |
| Sisomicin | 46 | 5.0 | 1.5 | 0.3 | 30.7 | 16.7 | >700 | ∼1000 |
| Neomycin | 10 | <0.006 | ||||||
| Paromomycin | 34 | <0.006 | ||||||
| Ribostamycin | 24 | <0.005 |
Thermodynamic Properties
Isothermal titration calorimetry was used to determine the thermodynamic parameters of association of antibiotics to AAC-IIa (Table II and Supporting Information Fig. S2). First, attempts to determine a dissociation constant for both neomycin and ribostamycin, with and without the cosubstrate present, yielded weak and unsaturable binding signals. Dissociation constants of the neomycin class antibiotics to AAC-IIa are roughly estimated to be >500 μ_M_. Thus, the extremely slow turnover rate observed for neomycins is likely a consequence of their low affinity to the protein.
Table II.
Thermodynamic Comparison of AAC-IIIb and AAC-IIa
| K_D (μ_M) | Δ_H_int (kcal/mol) | –T_Δ_S (kcal/mol) | Δ_G_ (kcal/mol) | Δ_n_ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | IIIb | IIa | IIIb | IIa | IIIb | IIa | IIIb | IIa | IIIb | IIa |
| Sisomicin (B)a | 10.3 | 1.5 | −5.5 | −49 | −1.3 | 41 | −6.8 | −8.0 | 0.34 | 2.8 |
| Sisomicin (T) | 1.7 | 0.1 | −14.9 | −9.7 | 7.2 | −0.30 | −7.7 | −10.0 | 0.66 | −0.34 |
| Tobramycin (B) | 33.0 | 34 | −8.4 | −16 | 2.3 | 9.9 | −6.2 | −6.1 | 0.60 | 1.1 |
| Tobramycin (T) | 2.9 | 1.7 | −22.2 | −7.6 | 14.6 | 0.04 | −7.6 | −7.6 | 1.0 | −0.10 |
| Kanamycin B (B) | 30.0 | 130 | −8.1 | −12 | 1.9 | 6.7 | −6.2 | −5.3 | 0.60 | 0.90 |
| Kanamycin B (T) | 3.9 | 7.8 | −26.0 | −8.6 | 18.6 | 1.6 | −7.4 | −7.0 | 1.5 | 0.30 |
| Kanamycin A (B) | 79.8 | 110 | −1.2 | −1.6 | −4.4 | −4.0 | −5.6 | −5.6 | −0.07 | −0.47 |
| Kanamycin A (T) | 16.0 | 2.2 | −12.8 | −9.9 | 6.2 | 2.1 | −6.6 | −7.8 | 1.3 | 0.02 |
| CoASHb | 1.9 | 9.7 | 0.4 | −23 | −8.2 | 17 | −7.8 | −6.0 | ND | 1.4 |
| CoASH (Tob) | 2.5 | 1.6 | −16.7 | −12 | 9.0 | 4.1 | −7.7 | −7.9 | 0.60 | 0.14 |
| CoASH (KanB) | 2.0 | 1.6 | −19.8 | −18 | 11.9 | 9.7 | −7.8 | −8.3 | 0.80 | 0.69 |
| CoASH (KanA) | 6.6 | 1.1 | −20.8 | −13 | 13.6 | 5.0 | −7.2 | −8.0 | 1.4 | 0.23 |
| CoASH (Siso) | 2.3 | 0.8 | −10.8 | −9 | 3.1 | 0.6 | −7.7 | −8.4 | 0.5 | −0.04 |
As expected for kanamycins, ITC signals were analyzable. Sisomicin, containing two methyl groups on ring C as well as a hydrogen atom at the C3′ position, is the tightest binding antibiotic and has the most negative (favorable) enthalpy of association (Fig. 1). Tobramycin, also with a C3′ hydrogen, but without the nonpolar methyl groups of sisomicin, is second. Kanamycins A and B, with C3′ hydroxyls, show the weakest binding affinity and the lowest enthalpies of binding among the kanamycins. Although their affinities are similar to one another, kanamycin B shows a sevenfold more favorable enthalpy suggesting its N2′ amine (p_K_a >8) is likely to be involved in an extra hydrogen bond and/or electrostatic interactions due to its positive charge at the experimental pH of 7.5 versus the C2′ hydroxyl of kanamycin A.
Taken together, these data suggest that hydrophobic interactions are the more dominating factors influencing both K_D and Δ_H. Indeed, AAC-IIa consists of 50% nonpolar residues with the antibiotic binding site postulated to contain several of these including tryptophan, tyrosine, and phenylalanine (Fig. 2).
Figure 2.
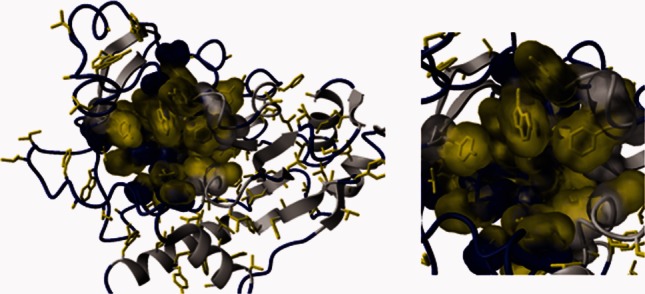
AAC-IIa contains many hydrophobic residues (yellow). Many of these are located on loops (blue) in or nearby to the antibiotic binding site. The molecular surface of the antibiotic binding pocket is shown in an expanded view in the right panel.
When antibiotic associates to the AAC-IIa-CoASH complex instead of apo-AAC-IIa, its affinity increases between 15- and 50-fold. However, Δ_H_ becomes less favorable for all except kanamycin A. The unfavorable contribution of entropy (T_Δ_S) to the formation of the AAC-IIa-antibiotic (binary) complexes becomes almost zero for the AAC-IIa-CoASH-antibiotic (ternary) complexes. This renders the overall Δ_G_ for the formation of ternary complexes to be more favorable than binary. The same is true for ternary complexes with AAC-IIIb; however, in this case, the entropic contribution becomes more disfavored compared to binary complexes, which is overcompensated by a more favorable increase in enthalpy. Ternary complexes forming with a more favorable Δ_G_ is therefore common for all AGMEs.12,14–17
Unlike AAC-IIIb, the binding of AGs to the AAC-IIa–CoASH complex is entirely driven by the enthalpic contribution with very small or no entropic compensation/contribution (Table II). As shown in Figure 3, enthalpy–entropy compensation plots for the binary and ternary complexes of AAC-IIIb and AAC-IIa show AG-dependent separation in enthalpy and entropy yielding slopes of 1.0–1.1. One exception is when an AG associates with the AAC-IIa-CoASH complex. Here, the values for the ternary complexes of AAC-IIa are clustered around T_Δ_S and show almost no dependence on AGs (Fig. 3). This property separates AAC-IIa not only from AAC-IIIb but also from all other AGMEs with available thermodynamic data where there is always a significant entropic contribution to the formation of the ternary complex regardless of the direction of change in the enthalpy.12,15–17 However, when the ternary complex is formed by the addition of CoASH to the AAC-IIa-AG complex, the enthalpy–entropy compensation plots become similar to AAC-IIIb yielding slopes of 1.0 and 0.99, respectively (Fig. 3).
Figure 3.
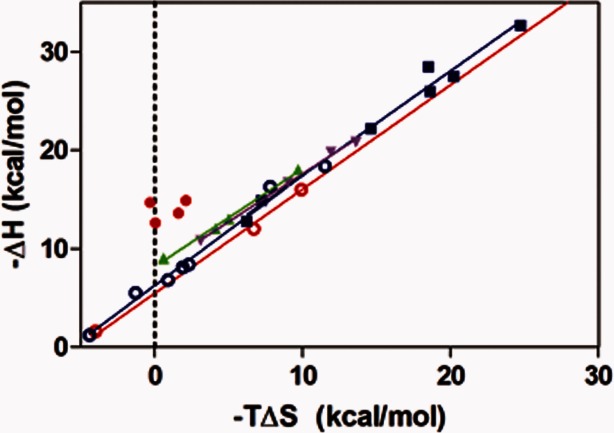
Enthalpy–entropy compensation plots for complexes of AAC-IIa-antibiotic (red, open); AAC-IIa-CoASH-antibiotic (red, filled, and points upshifted by 5 kcal/mol for clarity); AAC-IIIb-antibiotic (blue, open); AAC-IIIb-CoASH-antibiotic (blue, filled); AAC-IIa-antibiotic-CoASH (green); AAC-IIIb-antibiotic-CoASH (purple).
Another difference observed in thermodynamic parameters of AAC-IIIb and AAC-IIa is in the net protonation (Δ_n_) that accompanies the formation of binary and ternary complexes. First, the values observed for Δ_n_ are always larger with AAC-IIa than AAC-IIIb for the binding of AGs to the apo-enzyme. This means that AGs induce more changes in p_K_a values of titratable groups of AAC-IIa than AAC-IIIb. Next, binding of AGs to the AAC-IIIb–CoASH complex always occurs with larger Δ_n_ values than when they bind to the apo-enzyme. This indicates that CoASH causes an increase in the net protonation/deprotonation of the enzyme's titratable groups. In contrast, the binding of AGs to the AAC-IIa-CoASH complex shows smaller Δ_n_ values than those observed in binding to the apo-enzyme (kanamycin A excepted). These observations, although need not to, parallel the trend observed in changes in Δ_H_ between binary and ternary complexes of both enzymes [a more positive Δ_H_ (less favorable) in the ternary complex is accompanied by a more negative Δ_n_ and vice versa]. In fact, Δ_n_ becomes almost zero with AAC-IIa ternary complexes suggesting that there is no or very small contribution to Δ_H_ from the heat of ionization of the buffer used. We note that Δ_n_ ≍ 0 does not necessarily indicate the absence of protonation/deprotonation.10 These observations thus indicate that titratable groups make differential contributions depending on the nature of the complex and the enzyme.
Nuclear Magnetic Resonance and Structural Dynamics
The heteronuclear single quantum 1H-15N correlation spectrum (HSQC) of apo-AAC-IIa is extremely overlapped with few signals typical of intrinsically disordered or unfolded proteins. This correlates with the predicted structure of AAC-IIa which contains several large loops. In the absence of ligands, these may be fluctuating between numerous conformations causing the NMR peaks to broaden.
The presence of AGs or CoASH causes a ligand-dependent increase in the resolution of the HSQC spectrum of the apo-enzyme. The AAC-IIa-tobramycin complex yields the highest resolution of the three binary complexes, followed by CoASH and then sisomicin. These data are quite revealing about the nature of antibiotic selectivity by AAC-IIa. Tobramycin induces a significantly larger change in peak dispersion from the spectrum of apo-enzyme than sisomicin (Fig. 4, top). Indeed, the formation of the ternary AAC-IIa–tobramycin–CoASH complex does not cause any further increase in resolution but only shifts in several peak positions. Contrary to this, the addition of CoASH to the AAC-IIa–sisomicin complex is required to achieve similar peak dispersion to the ternary complex of tobramycin. As shown in the bottom panel of Figure 4, spectra for ternary complexes of both tobramycin and sisomicin are almost superimposable. This suggests that conformational changes play substantial roles in AAC-IIa's ability to recognize and interact with antibiotics.
Figure 4.
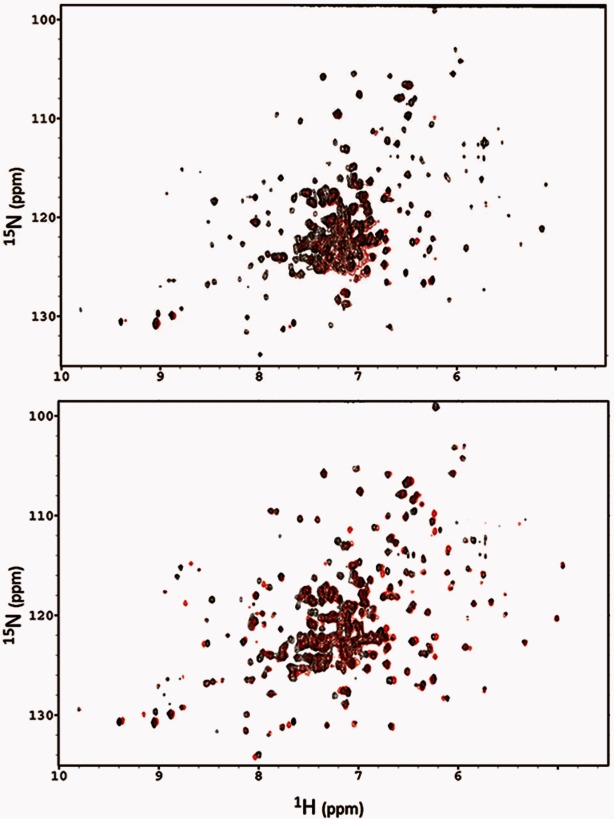
NMR spectra comparing the peak dispersion of binary (top) and ternary (bottom) complexes of sisomicin (red) and tobramycin (black) with AAC-IIa.
Discussion
The Acetylation Reaction and Its Kinetic Character
Both AAC-IIIb and AAC-IIa are _N_3 acetyltransferases and thus modify AG antibiotics at the amine functional group of carbon 3 on the 2-deoxystreptamine ring (Fig. 1) using AcCoA. Early studies involving cell-based assays showed that AAC-IIIb confers resistance to several AGs from both the kanamycin and neomycin classes while AAC-IIa was apparently limited to the kanamycin class only.1 These two classes of AGs differ by the pseudosugar substitution pattern on the 2-doexystreptamine ring where kanamycins are 4,6- and neomycins are 4,5-disubstituted. Also, ring C is a pentose-type ring in neomycins but a hexose derivative in kanamycins (Fig. 1).
Steady-state kinetic data supports the in vivo observations where the acetyltransferase activity of AAC-IIa with the neomycins is 200- to 5000-fold slower than the kanamycins (Table I and Supporting Information Fig. S3), which explains why this enzyme is unable to confer resistance to the neomycin class. Catalytic rates of acetylation for neomycins could not be determined by standard enzymatic assay but only by overnight incubation of neomycin group AGs with AAC-IIa and subsequent analysis by thin layer chromatography and NMR confirmed that neomycins are indeed substrates for this enzyme (Supporting Information Fig. S1). Ribostamycin, the smallest member of the neomycin class, is similar to the kanamycins in that it has only three rings in its structure, therefore, the kinetic differentiation between neomycin and kanamycin classes of AGs by AAC-IIa is not simply a matter of antibiotic size but must be related to the spatial orientation of the drug. AAC-IIIb also demonstrates a kinetic discrepancy between the two AG classes albeit only by 1.5–5.0-fold (Table I).16
Although both enzymes can catalyze modification of several members of the kanamycin group AGs, there are significant differences in kinetic parameters and their effects on the efficiency of the reaction. Turnover rates (_k_cat) and catalytic efficiencies (_k_cat/_K_m) for all AG substrates of AAC-IIa are consistently lower than those of AAC-IIIb (Table I). However, for AAC-IIIb, _k_cat values for the kanamycin group are all similar to each other while there is a 30-fold range of turnover rates with AAC-IIa. Despite these differences, both proteins demonstrate that AGs possessing an amine group at the C2′ position (i.e., kanamycin B) are favored substrates in terms of _k_cat/_K_m with a further preference for C3′-deoxy (i.e., tobramycin). Where the _k_cat/_K_m is dominated by _K_m for AAC-IIIb, _k_cat carries more weight for AAC-IIa; thus, the means by which these two proteins achieve their substrate preferences are different.
Thermodynamics of Antibiotic Association
The extremely slow turnover rate of the neomycins with AAC-IIa is likely a consequence of their low affinity to the protein. Attempts to determine a dissociation constant for both neomycin and ribostamycin, with and without the cosubstrate present, yielded weak and unsaturable binding signals. A rough estimate for dissociation constants of the neomycin class antibiotics to AAC-IIa is >500 μ_M_. This is in stark contrast not only to AAC-IIIb but also to all other AGMEs where neomycin B either has the highest affinity or is among those that show strongest binding affinity.12,14–17
When the cosubstrate analog, coenzyme A, is present in a preformed complex between either AAC-IIIb16 or AAC-IIa, subsequent antibiotic association occurs with a stronger affinity; as much as 50-fold in some cases. Most striking is that coenzyme A increases AG affinity between 2- and 10-fold more with AAC-IIa than with AAC-IIIb (Table II). Differences observed between these two enzymes also extend to the binding affinity of CoASH where the _K_D with AAC-IIa is about 5-fold higher than that of AAC-IIIb. The presence of AGs also has differential effects on the affinity of CoASH to each enzyme. They increase the affinity by 6- to 9-fold with AAC-IIa and have either weakening or no effect for AAC-IIIb (Table II).
Both enzymes also show significant differences in Δ_H_ and TΔ_S_. Enthalpies of antibiotic association to AAC-IIa are more favored (more negative) than to AAC-IIIb (Table II). Such an increase in favorable Δ_H_ between the two enzymes must be the result of interactions that do not influence antibiotic affinity as it is consistently weaker for AAC-IIa than AAC-IIIb. However, both enzymes strongly favor amine groups at the C2′ position which is common among many AGMEs studied to date.12,14–17 When the proteins are precomplexed with coenzyme A, enthalpies of antibiotic association show opposite trends. While Δ_H_ becomes more favored for AAC-IIIb, it becomes less favored (less negative) for AAC-IIa. Kanamycin A is the exception where its binding enthalpy becomes more favored for both proteins. We note that kanamycin A is the only AG that binds to both enzymes with favorable entropy. It is also the AG that binds to both enzymes with the least favorable enthalpy and low affinity. Reasons for this unique behavior are unclear at this time.
Δ_G_ values for the formation of the binary enzyme–AG and the ternary enzyme–AG–CoASH complexes vary by ≤ 3 kcal/mol. The determined values of Δ_G_ show that the formation of the ternary complexes are always more favorable than the binary complexes, which is also true for all other AGMEs.
AAC(3)-IIa and AAC(3)-IIIb Origins and Structure
To date, three genes coding for the AAC(3)-II type AGMEs have been found in more than 50% of all gram negative bacterial species.1 Among these, AAC-IIa is the most predominant as it is present in ∼85% of clinical isolates harboring an AAC(3)-II-type gene.18–21 The protein product is 30 kDa (286 amino acids) and shares ∼35% amino acid sequence identity and an additional ∼39% sequence similarity to the AAC(3)-IIIb protein (30 kDa and 274 amino acids) (Supporting Information Fig. S4). Using the AAC-IIa sequence in searches to find similar proteins of known structure, a strong match was found with the Yokd protein from Bacillus subtilis (PDB ID: 2NYG chain A) which, incidentally, is the same protein that was used as the template for building the homology model of AAC-IIIb.16 As such, models of AAC-IIa and AAC-IIIb are very similar and superimpose to 1.43 Å over the entire protein (Fig. 5). It is therefore likely that AAC-IIa, like AAC-IIIb, is a member of the GCN5-related acetyltransferase (GNAT) superfamily of acetyltransferases.22
Figure 5.
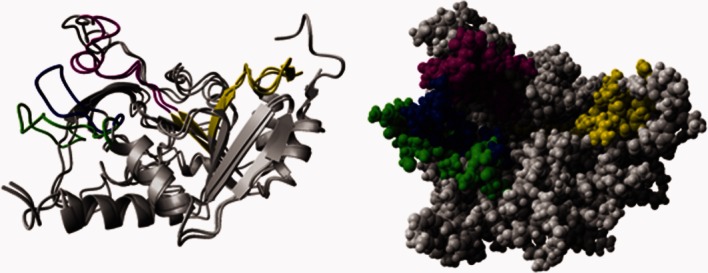
Homology models of AAC(3)-IIa and AAC(3)-IIIb. Shown in gray is the ribbon (left) and space filling (right) representations of the superimposed structures of AAC-IIa and AAC-IIIb. The GNAT conserved acetyl acceptor site (i.e., aminoglycoside), motif B, is shown in purple for both proteins. Motif A, the GNAT conserved region responsible for acetyl coenzyme A association, is in yellow. The blue and green loops are of AAC(3)-IIIb and AAC(3)-IIa, respectively. An interactive view is available in the electronic version of the article.
Between the two proteins, the region showing the most difference in amino acid sequence is the loop comprised of residues 202–222 in AAC-IIa and 209–226 in AAC(3)-IIIb (Fig. 5). AAC-IIa is also slightly longer with an additional 12 residues. As a result, the C-terminal 16 residues are not present in the model and so are of unknown structure and conformation.
Rationalizing the Substrate Selectivity Differences of AAC-IIIb and AAC-IIa
As mentioned above, AAC-IIa and AAC-IIIb are likely to be very similar in structure. Reasons then for the striking differences in ligand preference and thermodynamic/kinetic properties between these proteins are difficult to envision. As dynamics (protein motion) is emerging as a key component of ligand recognition among AGMEs as well as other protein systems,7,23–28 it is likely that this may have a strong hand in our observations. One candidate region is that of the aforementioned loop showing the most difference in amino acid sequence (Fig. 5, blue and green). This loop is positioned close to the antibiotic binding site and could easily undergo a conformational change. Either the positioning or dynamic behavior of this loop in AAC-IIa may largely be responsible for the observed substrate discrepancy with AAC-IIIb. It may be oriented in such a manner to hinder favorable interactions between neomycins and AAC-IIa. Also, based on the homology models, it appears that this loop in AAC-IIa is positioned further away from the active site than it is in AAC-IIIb, leaving its antibiotic binding site more open (Fig. 5, space filling). Such a situation could make it difficult for AAC-IIa to interact with antibiotics in a specific manner. Indeed, all AGs (except tobramycin and sisomicin) have much weaker affinities to AAC-IIa than to AAC-IIIb where the strongest dissociation constant for AAC-IIa is still a weak 33 μ_M_ (Table II). This is also consistent with highly variable _k_cat values observed with AAC-IIa with structurally very similar kanamycin group members, which may reflect differences in their alignments within the active site. However, differences between the overall dynamic properties of these two enzymes may also contribute to their differential substrate profiles.
Previous experimental and computational data with AAC-IIIb suggested that coenzyme A association induces a significant increase in dynamics of the antibiotic binding loop (motif B, Fig. 5, purple segment) to provide the most optimal AG interaction.8,27 Indeed, coenzyme A association to AAC-IIIb occurs with a Δ_H_ of ∼0.4 kcal/mol and an increase in entropy, which is consistent with the highly increased mobility of the AG binding loop.8 An increase in the flexibility of this loop may require breaking several intramolecular interactions between the loop residues and the rest of the enzyme, thus imposing an enthalpic penalty. For AAC-IIa, on the other hand, coenzyme A association occurs with a large change in enthalpy (−23 kcal/mol) which is suggestive of more bonds being formed. This is accompanied by a significant loss of entropy (−17 kcal/mol). Thus, the binding of CoASH to AAC-IIa must be inducing a number of new intramolecular and intermolecular interactions that contribute to the binding enthalpy favorably. It is logical then that the highly selective substrate profile of AAC-IIa is accomplished by coenzyme A causing the two loops (motif B and the green loop of Fig. 5) to interact either with one another or with other parts of the protein (more bonds and less disorder) in such a way as to form a restrictive AG binding site.
Regarding antibiotic association, affinity increases up to 50-fold when AAC-IIIb is precomplexed with coenzyme A. In addition, a more negative (favorable) Δ_H_ and a compensating more negative (unfavored) T_Δ_S is observed. This makes sense if coenzyme A is causing a conformational change to the protein that allows more bonds to form between AAC-IIIb and the antibiotic. However, antibiotic affinity to AAC-IIa also increases with coenzyme A but with a decrease in favorable enthalpy (less negative). This appears to be counterintuitive; however, it is the exact situation observed with another AGME, the AG phosphotransferase-(3′)-IIIb (APH), in which antibiotic association to APH occurred with a more favorable enthalpy than with the APH-nucleotide complex.12 In that case, H/D exchange using NMR spectroscopy demonstrated that the presence of the nucleotide causes a number of hydrogen bonds to be broken in several regions of the protein including between strands of a β-sheet in the nucleotide binding site and even in hydrophobic patches of the enzyme.7,23 Therefore, it is likely that similar effects are the cause of the determined decrease in the favorable binding enthalpy of AGs to AAC-IIa in the presence of CoASH.
It is also noteworthy that in the absence of any ligand, both APH and AAC-IIa show strong signal overlap in 1H-15N correlation NMR spectra indicative of a flexible and partially intrinsically disordered protein that gains a well-defined structure upon antibiotic association. Figure 6 shows 1H-15N HSQC spectra acquired with the apo- and AG-bound forms of both enzymes. There are also AG-dependent differences in spectra taken with both enzymes even for the structurally similar AGs. It is clear from these that dynamic changes resulting from the binding of AGs to these enzymes are not localized to the antibiotic binding site. Such data with AAC-IIIb suggests flexibility but not to the extent of AAC-IIa and APH.7,23 AAC-IIIb shows a well-dispersed NMR spectrum indicative a well-defined structure even in the absence of antibiotic (Fig. 6). However, ∼40–50 resonances are missing from the spectrum which means that these residues are in flexible regions, likely the antibiotic binding loop and the N/C termini, and are undergoing exchange between different conformations on a millisecond timescale and are therefore invisible (or exchange broadened) in the NMR spectrum. Amino acid-specific labeling of the protein yielded HSQC spectra that showed a number of missing resonances consistent with the number of the particular amino acids located in the flexible regions of the protein including the motif B.27
Figure 6.
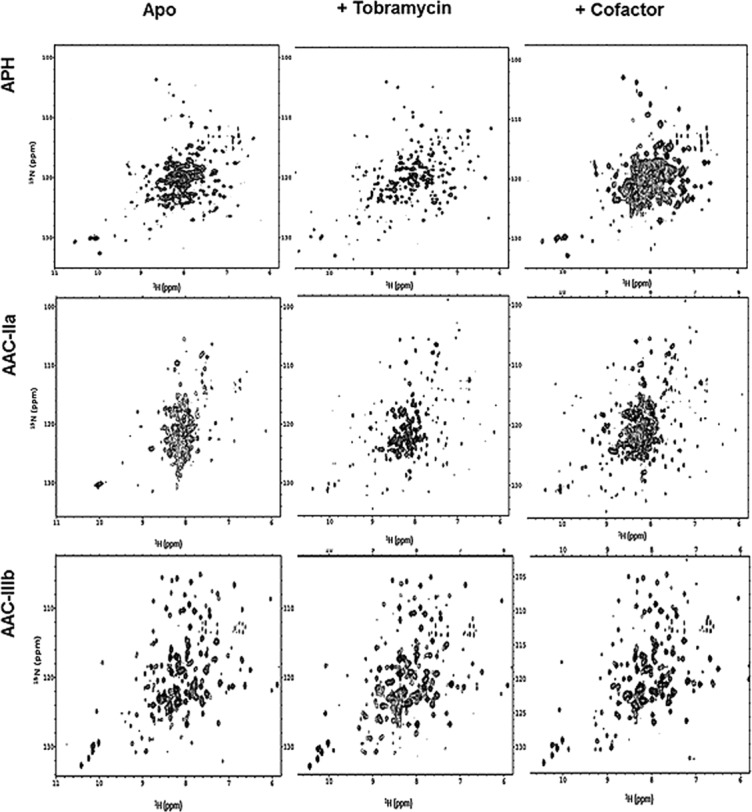
1H-15N HSQC NMR spectra of AGME complexes. APH in its apo, tobramycin, and MgAMPPCP bound forms are shown left to right in the top row. The middle row is the apo, tobramycin, and CoASH complexes with AAC-IIa and the bottom row is that of AAC-IIIb. All spectra were taken under similar experimental conditions and shown to matching contour levels.
In terms of substrate promiscuity, these observations are complicated to interpret. It would be most logical that proteins having parallel substrate profiles would have similar dynamic behavior and NMR spectra. This, however, is not the case. APH is highly promiscuous and thus more similar to AAC-IIIb than to AAC-IIa in terms of substrate profile. However, the NMR spectra of apo- and antibiotic-bound APH are more like those of the weakly promiscuous AAC-IIa rather than AAC-IIIb. The answer to this puzzle may lie with the cosubstrate. For APH, the NMR spectrum of the APH-nucleotide complex is highly overlapped just like that of apo-APH indicating that the cosubstrate MgATP causes minimal change to the overall APH structure (Fig. 6).7 It is most likely then that this protein is designed to conformationally and dynamically respond to antibiotics with little help from the cofactor. In contrast, the overlapped NMR spectrum of AAC-IIa becomes more resolved for both AAC-IIa–CoASH and AAC-IIa–antibiotic complexes. These suggest that while both APH and AAC-IIa are highly flexible in the absence of any ligand, their different levels of antibiotic selectivity is governed by how strong of a role the cofactor plays in altering the protein conformation. Where coenzyme A may shift the whole conformation of AAC-IIa to exclude certain antibiotics, APH may maintain a broad antibiotic selectivity by adjusting its structure mainly in response to antibiotic and not cosubstrate. Titrations with the smallest AG antibiotic, neamine (Fig. 1), also highlights differences between the adoptability of these two highly dynamic proteins to different ligands. Neamine binds APH with a very high affinity (K_D < 1 μ_M) while it cannot bind to AAC-IIa (Supporting Information Fig. S5). These data show that neither the flexibility of the protein or the size of the binding site cavity is sufficient for providing increased promiscuity. A coordinated dynamics may be more important instead, which may separate these two enzymes. Consistent with these data, a recent computational work also suggested that dynamics of each AGME and their interactions with the same AG may be different.29
AAC-IIIb is strongly promiscuous like APH but yet demonstrates an NMR spectrum indicative of a well-defined structure with or without ligands. However, the missing resonances attributed to the antibiotic binding loop still lend a degree of flexibility to this protein. It appears then that AAC-IIIb may be a descendent of AAC-IIa and has evolved the ability to control the effect of CoASH in order to increase the number of antibiotics that it can modify and enhance the resistance abilities of bacteria. In this case, the coenzyme may still produce a conformational change to AAC-IIIb but only enough to allow for an increase in optimal antibiotic interaction but not to the extent as to create the exclusive binding site of AAC-IIa.
Differential effects of the coenzyme on AAC-IIIb and AAC-IIa are also manifested in the binding stoichiometry of AGs to these enzymes. Binding of smaller (three ring) AGs to AAC-IIIb occurs with stoichiometry of >1 regardless of whether the AG belongs to the kanamycin or neomycin class. However, larger AGs such as neomycin and paromomycin bind with 1:1 stoichiometry. The presence of CoASH renders all binding stoichiometries to be 1:1.8,16 These observations clearly indicate that the large AG binding cavity is affected strongly by the presence of the cofactor in AAC-IIIb to optimize the binding site for all types and sizes of AGs. Contrary to this, kanamycins bind to AAC-IIa with a 1:1 stoichiometry regardless of the presence of CoASH (recall that this enzyme binds neomycins too weakly to determine thermodynamic parameters). Being more dynamic than AAC-IIIb, apo-AAC-IIa has a more difficult time binding to antibiotics. Like AAC-IIIb, CoASH increases affinity of AGs and thus optimizes the AG site. However, this optimization comes with creation of a structural preference for antibiotics as mentioned above to exclude neomycin class.
These effects are also visible in enthalpy–entropy compensation plots (Fig. 3) where the binding of AGs to the AAC-IIa–CoASH complex is almost completely enthalpic with very little or no contribution of entropy. The converse, however, is not true. In the binding of CoASH to the enzyme–AG complex, this plot yields a line with a slope of 1 indicating complete enthalpy–entropy compensation. This property separates AAC-IIa not only from AAC-IIIb but from the rest of AGMEs studied to date, which are all significantly more promiscuous than AAC-IIa.
The role of the cosubstrate in antibiotic interaction with AGMEs could be of strong significance for drug design. Indeed, attempts to synthesize bisubstrate analogs of the AG _N_-acetyltransferase (6′) have shown more inhibitory success with compounds that truncate the antibiotic moiety rather than that of coenzyme A.30,31 Future efforts of this type would benefit from knowledge of both the structural and dynamic consequences that the cosubstrate has on AGMEs.
Conclusions
AAC-IIIb demonstrates faster antibiotic turnover rates, higher antibiotic affinity, and less favored (less negative) enthalpy of antibiotic association than AAC-IIa. Also, AG binding becomes more enthalpically favored and entropically disfavored when CoASH is complexed to AAC-IIIb while the exact opposite occurs when CoASH is complexed with AAC-IIa. Both proteins, however, catalytically and enthalpically favor those AGs with amine groups at the 2′ carbon and show an increase in affinity of all AGs when CoASH is present.
Most importantly, our data suggest that the degree of promiscuity by an AGME is governed by the dynamics of the protein which is strongly influenced by the cosubstrate, either coenzyme A or Mg–nucleotide. Where APH adjusts its flexible structure around the antibiotic to promote broad substrate selectivity, AAC-IIa is adjusted by coenzyme A to favor only kanamycin class antibiotics. Two loops near the antibiotic binding site are likely the means by which this is achieved. We should note that the loop that interacts with the substrate to be acetylated is conserved in the GNAT superfamily of acetyltransferases.22 AAC-IIIb may be a descendent of AAC-IIa whose structure evolved just enough to maintain the more optimal antibiotic association caused by CoASH but decrease its ability to alter AAC-IIIb's conformation to restrict the antibiotic binding site.12,15–17
The above observations are unlikely to be predictable by many primary sequence based bioinformatics approaches. Static structures of these enzymes are also insufficient to explain the differential substrate profiles of these enzymes. For example, apo-APH and its ligand-bound forms have superimposable structures and yet dynamic properties of the apo-enzyme are dramatically different than the antibiotic-bound forms based on NMR spectra and H/D exchange data.7,23 Similarly, crystal structures of the AG _N_-acetyltransferase(2′)-Ic determined with three different AGs failed to explain the AG-dependent variations in _k_cat values.32 Incorporation of the type of data provided in this work may be necessary for such approaches to be more successful with flexible enzymes that show substrate promiscuity.
Materials and Methods
Materials
From Cambridge Isotope Laboratories (Andover, MA) were purchased 99.9% deuterium oxide and 99% 15N enriched salts. Dithiothreitol and isopropyl β-d-1-thiogalactopyranoside (IPTG) was acquired from Inalco Spa (Milano, Italy). Ion exchange matrix (Macro Q) was purchased from Bio-Rad Laboratories (Hercules, CA), while high-performance Ni-Sepharose resin was purchased from Amersham Biosciences (Piscataway, NJ). Purified thrombin was graciously provided by E. Fernandez (The University of Tennessee). Neamine was a kind gift from Julian Davies of the University of British Columbia and used after further purification by HPLC. All other AGs, coenzymes, and reagents were purchased at the highest possible purity from Sigma.
Protein Overexpression and Purification
An Escherichia coli strain containing the gene coding for the AG N3 acetyltransferase-IIa was graciously provided by George Miller (Achaogen, San Francisco, CA). Using standard PCR techniques, the AAC gene was cloned with an N-terminal, six-His tag and thrombin cleavage site and inserted into a pET15b vector for overexpression under the control of lactose operon. The pET15b-AAC plasmid was transformed into E. coli TOP10 cells for long-term storage as well as BL21 (DE3) cells for overexpression. The AAC-IIa protein was overexpressed with IPTG using standard protocols in either Luria Broth or M9 minimal media for isotope labeling (see NMR methods). The overexpressed protein is stable in harvested cells stored at −80°C for only 1 month, and as such purification was performed promptly after cell harvest.
Isolation of the AAC-IIa protein was achieved using standard nickel affinity chromatography. Briefly, cells were suspended in 15 mL of lysis buffer (100 m_M_ NaCl, 20 m_M_ imidazole, 200 μ_M_ PMSF, and 50 m_M_ Tris-HCl (pH 7.6) at 4°C), lysed with a French press, and centrifuged for 1 h at 34,000_g_. The supernatant was then passed through 1 mL of Ni-Sepharose resin where the AAC-IIa protein was ultimately eluted to >98% purity with a 100–350 m_M_ gradient of imidazole after extensive resin washing (100 m_M_ NaCl, 50 m_M_ Tris-HCl pH 7.6 at 4°C). The 6x-Histidine tag was removed by incubation with thrombin for 3 h at 25°C. Free 6x-Histidine tag was removed by passing through a Ni-Sepharose resin. Subsequent removal of thrombin by ion exchange chromatography required that the pH be increased from 7.6 to 8.0, and all NaCl was removed from the protein solution through extensive dialysis against 50 m_M_ Tris-HCl, pH 8.0 at 4°C. MacroPrep Q strong anion exchange media was used for the chromatography. AAC-IIa was eluted with a 0–750 m_M_ NaCl gradient. Finally, the AAC solution was dialyzed extensively against the appropriate buffer at 50 m_M_ (pH 7.6) at 4°C and 100 m_M_ NaCl. Final protein yields were consistently between 12 and 18 mg/L of induced culture. The concentration of AAC-IIa was determined spectrophotometrically using an _ɛ_0.1% (280 nm) of 1.16.
Steady-State Kinetics
Kinetic parameters for AAC-IIa activity were determined by a continuous assay utilizing a coupled reaction that produces increasing concentrations of pyridine 4-thiolate monitored at 324 nm using a Cary-Win UV–vis spectrophotometer (Varian, Palo Alto, CA) as previously described.16,33,34 Samples consisted of 100 m_M_ NaCl and 50 m_M_ 3-(N_-morpholino)propanesulfonic acid (MOPS) (pH 7.6) at 25°C. AAC (10 n_M) was used in each assay, and the reaction was initiated by addition of AG at various concentrations while the AcCoA concentration was held at 100 μ_M_. Reactions followed Michaelis–Menten type kinetics with all substrates tested where substrate inhibition, common among several AGMEs, allowed data to fit to the equation, v = (_V_max[_S_])/(_K_m + [_S_] + [_S_]2/_K_i). Here, v is the initial velocity, _V_max is the maximal velocity, _K_m is the Michaelis constant, and _K_i is the substrate inhibition constant. Turnover rates (_k_cat) were calculated from the relationship _V_max = _k_cat[_E_]T.
Isothermal Titration Calorimetry
ITC experiments were performed on a VP-ITC microcalorimeter from Microcal, Inc. (Northampton, MA), at 25°C. AAC-IIa concentrations were between 20 and 30 μ_M_ for the maintenance of c values ([binding sites] association constant) within the range of 1–100 for greater accuracy of association constant (K_A) measurements. Titrant solutions were prepared with desulfated AGs35 or coenzyme A (CoASH) diluted into the final dialysis buffer used in enzyme purification, allowing both cell and syringe solutions to contain 100 m_M NaCl and the appropriate buffer at 50 m_M_ (pH 7.6 or pH 6.6) at 25°C. In ternary experiments, either CoASH or antibiotic was included in the cell solution with AAC-IIa at ∼98% saturation levels for subsequent titration of the opposite ligand. Samples were degassed for 10 min before being loaded. Titrations of ligands into buffer were performed as a control, and the resulting heats of ligand dilution were subtracted from the experimental data before curve fitting. The pH of each cell and syringe solution was checked before and after each experiment, and no change was observed. The integrity of AAC-IIa was ensured by measurement of the enzymatic activity and concentration before and after each experiment. In all cases, the enzymatic activity remained greater than 85% of the starting activity at the end of titrations.
Thermograms were integrated using Origin software provided by the instrument manufacturer, and the best fits were obtained with one-site binding. Although sisomicin binary data fit best to the one-site binding equation, fits were not optimal. We therefore used Sedphat36,37 software to eliminate the possibility of an idiosyncrasy of fitting software. Data fitted in this way was consistent with those obtained with Origin software for all titrations. Therefore, all data reported herein are from Origin fits.
As binding of AGs to the enzyme causes shifts in the p_K_a values of several functional groups, experiments at pH 7.5 were performed individually in Tris-HCl, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and piperazine-1,4-bis(2-ethanesulfonic acid) buffers with heats of ionization (Δ_H_ion) of 11.4, 5.02, and 2.74 kcal/mol, respectively, to determine the intrinsic enthalpy (Δ_H_int) as described previously.8,10,38,39 Δ_G_ values were calculated from association constants (K_A) derived from fitted titration curves using the equation Δ_G = −RT ln(K_A). T_Δ_S values were then determined from the relationship Δ_G = Δ_H_int − T_Δ_S.
Nuclear Magnetic Resonance
AAC(3)-IIa was uniformly 15N-labeled using M9 minimal media containing 15N salts. The protein concentration was 150 μ_M_ in 50 m_M_ MOPS and 100 m_M_ NaCl, pH 7.6. Where applicable, ligands were present in high enough concentrations to ensure >95% binding site saturation as calculated from the dissociation constants. Nuclear magnetic resonance experiments were performed using a 600-MHz, Varian Inova spectrometer equipped with a 1H, 13C, 15N triple-resonance, salt-tolerant cryogenic probe at the University of Tennessee. Sensitivity enhanced 1H-15N HSQC (heteronuclear single-quantum coherence) correlation spectra40 were recorded with 64 scans of 64 increments in the 15N dimension with the TROSY41 option and a delay of 1.5 s between scans; 2048 data points were collected with an acquisition time of 128 ms. Data were processed with NMRpipe software.42 FID was multiplied with a sin2 window function in the acquisition dimension before Fourier transformation. No baseline correction or other cosmetic procedures were applied. Spectra were exported to Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco) for analysis and display. Enzymatic activities were determined before and after experiments where >85 % of the original activity was maintained. Data pertaining to APH and AAC-IIIb were collected and processed similarly as described elsewhere.7,8,11
Homology Modeling
The AAC-IIa amino acid sequence was submitted to the protein data bank (PDB) search tool of Molecular Operating Environment (MOE) at the University of Tennessee. From this, the top hit was the Bacillus subtilis protein Yokd (PDB ID: 2NYG chain A). MOE was then utilized to make 10 energy-minimized models of AAC-IIa using Yokd as the template structure. All models yielded similar results where only the C-terminal 16 residues were missing. The model with the least φ and ψ angle outliers and atom clashes was then transformed into PDB format for qualitative comparisons with AAC-IIIb.
Acknowledgments
The authors thank Yufei Yue for the cloning of the AAC-IIa gene and initial transformation of the expression plasmid which was later transformed in the current cell line by A.L.N.
Glossary
APH (3′)-IIIa or APH
aminoglycoside phosphotransferase-(3′)-IIIa
ANT (2′′)-Ia or ANT
aminoglycoside nucleotidyltransferase-(2′′)-Ia
AAC-IIIb
aminoglycoside acetyltransferase (3)-IIIb
AAC-IIa
aminoglycoside acetyltransferase (3)-IIa
DTT
dithiothreitol
GNAT
GCN5-related acetyltransferase
IPTG
isopropyl β-d-1-thiogalactopyranoside
PMSF
phenylmethanesulfonylfluoride
Tris-HCl
2-amino-2-hydroxymethyl-propane-1,3-diol hydrochloride
PIPES
piperazine-1,4-bis(2-ethanesulfonic acid)
HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
2-DOS
2-deoxystreptamine
AG
aminoglycoside
AGME
aminoglycoside-modifying enzyme
AcCoA
acetyl coenzyme A
CoASH
coenzyme A
ITC
isothermal titration calorimetry
BLAST
basic local alignment search tool
HSQC
heteronuclear single quantum coherence
MOE
molecular operating environment
NMR
nuclear magnetic resonance.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Shaw KJ, Rather PN, Hare RS, Miller GH. Molecular-genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev. 1993;57:138–163. doi: 10.1128/mr.57.1.138-163.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spotts CR, Stanier RY. Mechanism of Streptomycin action on bacteria—unitary hypothesis. Nature. 1961;192:633–637. doi: 10.1038/192633a0. [DOI] [PubMed] [Google Scholar]
- 3.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16s ribosomal-RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 4.McKay GA, Thompson PR, Wright GD. Broad spectrum aminoglycoside phosphotransferase type III from Enterococcus: overexpression, purification and substrate specificity. Biochemistry. 1994;33:6936–6944. doi: 10.1021/bi00188a024. [DOI] [PubMed] [Google Scholar]
- 5.Mitja O, Pigrau C, Ruiz I, Vidal X, Almirante B, Planes AM, Molina I, Rodriguez D, Pahissa A. Predictors of mortality and impact of aminoglycosides on outcome in listeriosis in a retrospective cohort study. J Antimicrob Chemo. 2009;64:416–423. doi: 10.1093/jac/dkp180. [DOI] [PubMed] [Google Scholar]
- 6.Schraufnagel DE. Tuberculosis treatment for the beginning of the next century. Intl J Tuberculosis Lung Dis. 1999;3:651–662. [PubMed] [Google Scholar]
- 7.Norris AL, Serpersu EH. NMR detected hydrogen-deuterium exchange reveals differential dynamics of antibiotic and nucleotide-bound aminoglycoside phosphotransferase 3'-IIIa. J Am Chem Soc. 2009;131:8587–8594. doi: 10.1021/ja901685h. [DOI] [PubMed] [Google Scholar]
- 8.Norris AL, Serpersu EH. Interactions of coenzyme A with the aminoglycoside acetyltransferase (3)-IIIb and thermodynamics of a ternary system. Biochemistry. 2010;49:4036–4042. doi: 10.1021/bi1001568. [DOI] [PubMed] [Google Scholar]
- 9.Norris AL, Serpersu EH. Antibiotic selection by the promiscuous aminoglycoside acetyltransferase-(3)-IIIb is thermodynamically achieved through the control of solvent rearrangement. Biochemistry. 2011;50:9309–9317. doi: 10.1021/bi2011916. [DOI] [PubMed] [Google Scholar]
- 10.Ozen C, Malek JM, Serpersu EH. Dissection of aminoglycoside-enzyme interactions: a calorimetric and NMR study of neomycin B binding to the aminoglycoside phosphotransferase(3′)-IIIa. J Am Chem Soc. 2006;128:15248–15254. doi: 10.1021/ja0643220. [DOI] [PubMed] [Google Scholar]
- 11.Ozen C, Norris AL, Land ML, Tjioe E, Serpersu EH. Detection of specific solvent rearrangement regions of an enzyme: nMR and ITC studies with aminoglycoside phosphotransferase(3′)-IIIa. Biochemistry. 2008;47:40–49. doi: 10.1021/bi701711j. [DOI] [PubMed] [Google Scholar]
- 12.Ozen C, Serpersu EH. Thermodynamics of aminoglycoside binding to aminoglycoside-3'-phosphotransferase IIIa studied by isothermal titration calorimetry. Biochemistry. 2004;43:14667–14675. doi: 10.1021/bi0487286. [DOI] [PubMed] [Google Scholar]
- 13.Davies JE. Aminoglycoside-aminocyclitol antibiotics and their modifying enzymes. In: Lorian V, editor. Antibiotics in Laboratory Medicine. Baltimore, MD: Williams and Wilkins; 1991. pp. 691–713. [Google Scholar]
- 14.Hegde SS, Dam TK, Brewer CF, Blanchard JS. Thermodynamics of aminoglycoside and acyl-coenzyme A binding to the Salmonella enterica AAC(6′)-Iy aminoglycoside N-acetyltransferase. Biochemistry. 2002;41:7519–7527. doi: 10.1021/bi020190l. [DOI] [PubMed] [Google Scholar]
- 15.Jing XM, Wright E, Bible AN, Peterson CB, Alexandre G, Bruce BD, Serpersu EH. Thermodynamic characterization of a thermostable antibiotic resistance enzyme, the aminoglycoside nucleotidyitransferase (4′) Biochemistry. 2012;51:9147–9155. doi: 10.1021/bi301126g. [DOI] [PubMed] [Google Scholar]
- 16.Norris AL, Ozen C, Serpersu EH. Thermodynamics and kinetics of association of antibiotics with the aminoglycoside acetyltransferase (3)-IIIb, a resistance-causing enzyme. Biochemistry. 2010;49:4027–4035. doi: 10.1021/bi100155j. [DOI] [PubMed] [Google Scholar]
- 17.Wright E, Serpersu EH. Molecular determinants of affinity for aminoglycoside binding to the aminoglycoside nucleotidyltransferase(2″)-Ia. Biochemistry. 2006;45:10243–10250. doi: 10.1021/bi060935d. [DOI] [PubMed] [Google Scholar]
- 18.Allmansberger R, Brau B, Piepersberg W. Genes for gentamicin-(3)-N-acetyl-transferase-Iii and transferase-Iv. 2. Nucleotide-sequences of 3 Aac(3)-Iii genes and evolutionary aspects. Mol Gen Genet. 1985;198:514–520. doi: 10.1007/BF00332949. [DOI] [PubMed] [Google Scholar]
- 19.Barg NL. Construction of a probe for the aminoglycoside 3-V-acetyltransferase gene and detection of the gene among endemic clinical isolates. Antimicrob Agents Chemo. 1988;32:1834–1838. doi: 10.1128/aac.32.12.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teran FJ, Alvarez M, Suarez JE, Mendoza MC. Characterization of 2 aminoglycoside-(3)-N-acetyltransferase genes and assay as epidemiologic probes. J Antimicrob Chemo. 1991;28:333–346. doi: 10.1093/jac/28.3.333. [DOI] [PubMed] [Google Scholar]
- 21.Vliegenthart JS, Ketelaarvangaalen PAG, Vandeklundert JAM. Nucleotide-sequence of the Aacc2 gene, a gentamicin resistance determinant involved in a hospital epidemic of multiply resistant members of the family enterobacteriaceae. Antimicrob Agents Chemo. 1989;33:1153–1159. doi: 10.1128/aac.33.8.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Ann Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serpersu EH, Norris AL. Effect of protein dynamics and solvent in ligand recognition by promiscuous aminoglycoside-modifying enzymes. In: Derek H, editor. Advances in carbohydrate chemistry and biochemistry. Vol. 67. Waltham, MA: Academic Press; 2012. pp. 221–248. [DOI] [PubMed] [Google Scholar]
- 24.Wieninger SA, Serpersu EH, Ullmann GM. ATP binding enables broad antibiotic selectivity of aminoglycoside phosphotransferase(3′)-IIIa: an elastic network analysis. J Mol Biol. 2011;409:450–465. doi: 10.1016/j.jmb.2011.03.061. [DOI] [PubMed] [Google Scholar]
- 25.Bahar I, Lezon TR, Yang LW, Eyal E. Global dynamics of proteins: bridging between structure and function. Ann Rev Biophys. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tzeng SR, Kalodimos CG. Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol. 2011;21:62–67. doi: 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Hu XH, Norris AL, Baudry J, Serpersu EH. Coenzyme A binding to the aminoglycoside acetyltransferase (3)-IIIb increases conformational sampling of antibiotic binding site. Biochemistry. 2011;50:10559–10565. doi: 10.1021/bi201008f. [DOI] [PubMed] [Google Scholar]
- 28.Freire E, Vanosdol WW, Mayorga OL, Sanchezruiz JM. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Ann Rev Biophys Biophys Chem. 1990;19:159–188. doi: 10.1146/annurev.bb.19.060190.001111. [DOI] [PubMed] [Google Scholar]
- 29.Romanowska J, Reuter N, Trylska J. Comparing aminoglycoside binding sites in bacterial ribosomal RNA and aminoglycoside modifying enzymes. Proteins. 2013;81:63–80. doi: 10.1002/prot.24163. [DOI] [PubMed] [Google Scholar]
- 30.Gao F, Yan XX, Baettig OM, Berghuis AM, Auclair K. Regio- and chemoselective 6'-N-derivatization of aminoglycosides: bisubstrate inhibitors as probes to study aminoglycoside 6'-N-acetyltransferases. Angew Chem. 2005;44:6859–6862. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]
- 31.Gao F, Yan XX, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K. Synthesis and structure-activity relationships of truncated bisubstrate inhibitors of aminoglycoside 6'-N-acetyltransferases. J Med Chem. 2006;49:5273–5281. doi: 10.1021/jm060732n. [DOI] [PubMed] [Google Scholar]
- 32.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. Aminoglycoside 2'-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nature Struct Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 33.Williams JW, Northrop DB. Synthesis of a tight-binding, multisubstrate analog inhibitor of gentamicin acetyltransferase-I. J Antibiot. 1979;32:1147–1154. doi: 10.7164/antibiotics.32.1147. [DOI] [PubMed] [Google Scholar]
- 34.Owston MA, Serpersu EH. Cloning, overexpression, and purification of aminoglycoside antibiotic 3-acetyltransferase-IIIb: conformational studies with bound substrates. Biochemistry. 2002;41:10764–10770. doi: 10.1021/bi0261241. [DOI] [PubMed] [Google Scholar]
- 35.Wu LZ, Serpersu EH. Deciphering interactions of the aminoglycoside phosphotransferase(3′)-IIIa with its ligands. Biopolymers. 2009;91:801–809. doi: 10.1002/bip.21251. [DOI] [PubMed] [Google Scholar]
- 36.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Analyt Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 37.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atha DH, Ackers GK. Calorimetric determination of the heat of oxygenation of human hemoglobin as a function of pH and the extent of reaction. Biochemistry. 1974;13:2376–2383. doi: 10.1021/bi00708a022. [DOI] [PubMed] [Google Scholar]
- 39.Baker BM, Murphy KP. Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys J. 1996;71:2049–2055. doi: 10.1016/S0006-3495(96)79403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 41.Weigelt J. Single scan, sensitivity- and gradient-enhanced TROSY for multidimensional NMR experiments. J Am Chem Soc. 1998;120:10778–10779. [Google Scholar]
- 42.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe—a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.