Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes) (original) (raw)
. Author manuscript; available in PMC: 2014 Feb 1.
Published in final edited form as: Biochem Soc Trans. 2013 Feb 1;41(1):245–251. doi: 10.1042/BST20120265
Abstract
Body fluids of cancer patients contain TEXs (tumour-derived exosomes). Tumours release large quantities of TEXs, and the protein content of exosome or MV (microvesicle) fractions isolated from patients’ sera is high. TEXs down-regulate functions of immune cells, thus promoting tumour progression. We isolated TEXs from tumour cell supernatants and sera of patients with solid tumours or AML (acute myelogenous leukaemia). The molecular profile of TEXs was distinct from that of circulating exosomes derived from normal cells. TEXs were co-incubated with activated T-cells, conventional CD4 +CD25neg T-cells or CD56 +CD16 + NK (natural killer) cells respectively. TEXs down-regulated CD3_ζ_ and JAK3 (Janus kinase 3) expression in primary activated T-cells and mediated Fas/FasL (Fas ligand)-driven apoptosis of CD8 + T-cells. TEXs promoted CD4 +CD25neg T-cell proliferation and their conversion into CD4 +CD25hiFOXP3 + (FOXP3 is forkhead box P3) Treg cells (regulatory T-cells), which also expressed IL-10 (interleukin 10), TGF_β_1 (transforming growth factor _β_1), CTLA-4 (cytotoxic T-lymphocyte antigen 4), GrB (granzyme B)/perforin and effectively mediated suppression. Neutralizing antibodies specific for TGF_β_1 and/or IL-10 inhibited the ability of TEXs to expand Treg cells. TEXs obtained at diagnosis from AML patients’ sera were positive for blast-associated markers CD33, CD34, CD117 and TGF_β_1, and they decreased cytotoxic activity of NK cells isolated from NC (normal control) donors, induced Smad phosphorylation and down-regulated NKG2D receptor expression. Correlations between the TEX molecular profile or TEX protein levels and clinical data in cancer patients suggest that TEX-mediated effects on immune cells are prognostically important. In contrast with exosomes released by normal cells, TEXs have immunosuppressive properties and are involved in regulating peripheral tolerance in patients with cancer.
Keywords: immune suppression, natural killer cell (NK cell), T-cell, tumour-derived exosome (TEX), tumour escape
Background
In a recent Nature Medicine report, TEXs (tumour-derived exosomes) were featured as emerging mediators of tumorigenesis [1]. The report makes three important points: (i) the protein content of exosomes isolated from sera of subjects with stage 4 melanoma and short survival was significantly higher than that in patients who had longer survival; (ii) exosomes from plasma of subjects with melanoma had a melanoma-specific molecular signature that could be resolved in Western blots and distinguished patients with NED (no evident disease) after therapy from patients whose disease progressed; and (iii) in mice, melanoma-derived exosomes reprogrammed bone marrow progenitor cells towards a malignant phenotype, supporting tumour growth and metastasis [1,2]. This series of studies, emphasizing the critical role of TEXs in tumorigenesis, has caught the attention of the medical and scientific communities and more or less legitimized the rapidly expanding field of TEX biology [3].
The ability of tumours to escape from the host immune system has long been considered an obstacle to successful cancer immunotherapy [4]. Human tumours develop capabilities to down-regulate functions of immune cells and, especially, functions of anti-tumour effector cells, including CD8+ and CD4+ T-lymphocytes, NK (natural killer) cells and DCs (dendritic cells) [4,5]. Several years ago, we and others observed that sera of cancer patients, but not sera of NC (normal control) donors, can suppress functions of normal activated T-cells following a brief incubation period [6,7]. Subsequently, this suppressive effect was found to be mediated by a glycoprotein-containing fraction of small membranous vesicles with a diameter of 50–100 nm, which were identified as exosomes by TEM (transmission electron microscopy) and which had a molecular composition resembling that of cell-surface membranes in the mother tumour cells [6,7].
Most, if not all, viable cells secrete exosomes, and exosome biogenesis has been studied extensively [8]. Exosomes are not released by plasma membrane shedding; their biogenesis begins with endosomes, which fuse to form MVBs (multivesicular bodies). Through the inward budding of the MVB membrane, ILVs (intraluminal vesicles) are formed, which, in the process of invagination, enclose various endoplasmic components [8]. Upon MVB fusion with the cell membrane, exosomes are released through an ATP-dependent process into extracellular space as double-membraned vesicles often termed MVs (microvesicles) [9].
Secretion of exosomes is not a random process; it is highly regulated by cellular signals that direct proteins into the MVB pathway. ESCRT (endosomal sorting complex required for transport) plays a key role in exosome formation [8]. Some of the ESCRT-associated proteins such as Tsg101 (tumour susceptibility gene 101) or Alix [ALG-2 (apoptosis-linked gene 2)-interacting protein X] are characteristic components of exosomes [8]. Tumour cells secrete large quantities of TEXs, which are found in all body fluids, but those most extensively studied in humans come from the peripheral circulation. Sera of cancer patients are enriched in TEXs, but also contain exosomes originating from many other normal cells. As indicated above, TEXs are currently of great interest not only because they represent one of the mechanisms used by tumours for subversion of the host immune system, including anti-tumour activities of T-cells and NK cells, but also because of their potential as biomarkers of tumour progression.
Molecular composition of TEXs
Exosomes in sera of cancer patients can be separated by ultracentrifugation, quantified for protein content and evaluated further for their molecular composition. Interestingly, the molecular profile of TEXs isolated from patients’ sera is distinct from that of other exosomes [10]. As shown in Figure 1, TEXs are enriched in TAAs (tumour-associated antigens), MHC class I and II molecules, co-stimulatory molecules, various growth factor receptors, such as EGFR (epidermal growth factor receptor) or HER-2 (human epidermal growth factor receptor 2), as well as death receptor ligands such as FasL (Fas ligand), TRAIL (tumour-necrosis-factor-related apoptosis-inducing ligand) or PDL-1 (programmed cell death ligand 1) and inhibitory factors such as PGE2 (prostaglandin E2) [11]. This molecular profile suggests that TEXs have the capacity to both interact with DCs and induce T-cell responses as well as to inhibit these responses. This is one of the most intriguing aspects of their biology. Furthermore, since the molecular content of exosomes reflects a selective sorting process in the cell of their origin [12], it is likely that the parent cells define the target cell specificity of exosomes. In other words, TEXs secreted by tumour cells may be targeted to reach a predetermined target. Importantly, TEXs carry genetic information in the form of DNA, mRNA and miRNA (microRNA), which implies that TEXs have the potential to induce genetic changes in target cells.
Figure 1. A schematic diagram of TEXs showing some proteins and lipids generally found to be present in tumour-derived exosomes.

HSP, heat-shock protein; ICAM 1, intercellular adhesion molecule 1; LAMP-1, lysosome-associated membrane protein 1; miRNA, microRNA; PLAses, phospholipases; TAA, tumour-associated antigen; TRAIL, tumour-necrosis-factor-related apoptosis-inducing ligand. Modified with kind permission from Springer Science + Business Media, 2012, Emerging Concepts of Tumor Exosome-Mediated Cell–Cell Communication, pp. 149–168, Immune modulation of T cells and natural killer cells by tumor-derived exosomes, Whiteside, T.L., Figure 7.1, © 2012 Springer Science + Business Media, New York.
TEX isolation from tumour cell supernatants and patients’ sera
To obtain TEXs from sera or other body fluids of patients with cancer, we have used a two-step procedure consisting of size-exclusion chromatography on a Sepharose 2B column followed by ultracentrifugation of the exclusion fractions at 100000 g for 2 h to pellet exosomes [7,13]. The protein and lipid content of TEXs isolated from patients’ sera or tumour cell supernatants can be determined in Western blots, as illustrated in Figure 2, or by MS. Protein analyses of TEXs revealed the presence of components found in all exosomes, such as tetraspanins, and cell-type-specific proteins. TEXs from different tumour cells contain and concentrate a set of molecules that is unique or characteristic for each type of the parent cell (Figure 2). Consequently, the biochemical composition of TEXs resembles that of tumour cells from which they derive. TEXs carry membrane-associated enzymes such as an ATP hydrolase, CD39 and a 5′-ectonucleotidase, CD73 [14]. Importantly, TEXs derived from COX-2+ (cyclo-oxygenase 2) tumours carry PGE2 [11].
Figure 2. TEX isolation and characterization.
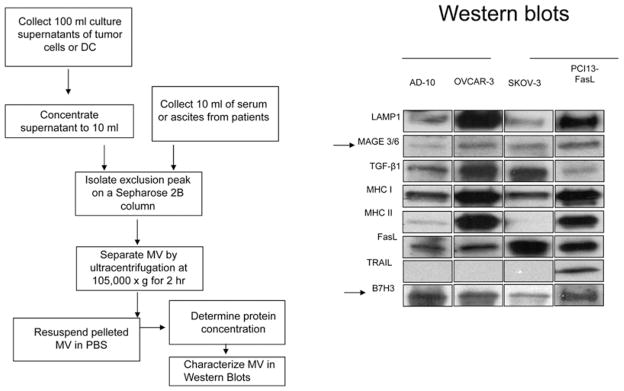
Method used for TEX isolation from tumour cell supernatants or sera of cancer patients (left). TEXs isolated from supernatants of various tumour cell lines were studied by Western blot [right; reproduced with permission from Szajnik, M., Czystowska, M., Szczepanski, M.J., Mandapathil, M. and Whiteside, T.L. (2010) Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 5, e11469]. Note the differences in the protein content of TEXs obtained from supernatants of various tumour cell lines.
TEX-mediated inhibition of anti-tumour immune responses
In vitro co-incubation experiments [15] in which TEXs purified from patients’ sera or tumour supernatants were added to freshly purified human CD4+ or CD8+ T-cells showed that TEXs failed to promote proliferation of CD8+ T-cells, although proliferation of CD4+ T-cells was not inhibited by TEXs [15]. In contrast, exosomes derived from activated T-cells or from _in vitro_-matured DCs used as controls readily induced T-cell proliferation [15]. More recent evidence indicated that transfer of exosomes from tumour-bearing mice to ovalbumin-immunized mice induced down-regulation of antigen-specific T-cell responses [16]. This in vivo evidence confirms that TEX-mediated immunosuppression observed in vitro can be reproduced in vivo.
On the basis of the literature and our data, several distinct mechanisms utilized by TEXs to inhibit immune responses have been recognized (Figure 3) as follows:
Figure 3. Some of the mechanisms used by TEXs to modulate functions of different immune cells.

See the text for details.
TEX-induced apoptosis of activated CD8 + T-cells
We showed that when OKT3 (anti-CD3 antibody)-activated T-cells were incubated with cancer patients’ sera, supernatants of cultured tumour cells, isolated TEX fractions or CH-11 (anti-CD9) antibody, which cross-links Fas, they underwent apoptosis as demonstrated by DNA fragmentation. Pre-treatment of T-cells with pan-caspase inhibitors [Z-VAD-FMK (benzyloxycarbonyl-Val-Ala-DL-Asp-fluoromethylketone)] partially, but significantly, blocked this effect [7]. Only TEXs, and not exosomes derived from normal cells, such as DCs or fibroblasts, induced apoptosis of activated CD8+ T-cells [10]. The ability of TEXs to induce CD8+ T-cell apoptosis was due to the presence of the membrane-associated form of FasL (42 kDa) and MHC class I molecules in TEXs [7,15,17–19]. TEXs with the highest content of these molecules were most active in inducing T-cell apoptosis, which could be partially blocked by anti-Fas or anti-(MHC class I) antibodies and completely blocked in the presence of both antibodies [7]. Although the role of FasL carried by TEXs in inducing apoptosis of activated Fas+ CD8+ T-cells is clear, that of MHC class I molecules remains speculative. Possibly, the direct engagement of MHC class I molecules with the CD8 receptor activates the endogenous Fas/FasL pathway, leading to T-cell apoptosis [20,21]. In the circulation of cancer patients, nearly all CD8+ lymphocytes express surface CD95 [22] and many express PD-1 (programmed cell death 1) [23]. Because TEXs present in these patients’ sera carry FasL [7,15] and/or PDL-1, the death pathways (Fas/FasL or PD-1/PDL-1 respectively) are likely to be responsible for ‘spontaneous apoptosis’ of CD8+ T-cells observed in vivo [24]. The ex vivo studies with human T-cells and purified TEXs clearly implicate TEXs in the CD8+ T-cell demise observed in cancer patients, and especially evident after immunotherapy, when CD8+ T-cells are activated and sensitive to apoptosis. TEX-mediated signals leading to apoptosis of CD8+ T-cells induce early membrane changes (annexin V binding), caspase 3 cleavage, cytochrome c release from mitochondria, loss of the mitochondrial membrane potential and, ultimately, DNA fragmentation [25,26]. The PI3K (phosphoinositide 3-kinase)/Akt pathway is a central TEX target in activated CD8+ T cells: their exposure to TEXs causes a dramatic time-dependent Akt dephosphorylation and its inactivation, which is accompanied by down-regulation of the expression of the anti-apoptotic Bcl-2 family members (i.e. Bcl-2, Bcl-xL and Mcl-1) and up-regulation of the pro-apoptotic protein Bax [26]. We have demonstrated that TEXs induce T-cell death by engaging both the extrinsic and intrinsic apoptotic pathways in these cells [26,27]. Our in vitro studies are consistent with reports of similar changes in Bcl-2, Bcl-xL or Bax in circulating T-cells of patients with different malignancies [22,28,29], potentially implicating TEXs in inducing these events in vivo.
TEXs deliver tolerogenic signals to immune cells
TEX-induced unresponsiveness of CD8+ T-cells is associated with: (i) inhibition of signalling via the TCR (T-cell receptor) and IL-2R [IL (interleukin)-2 receptor]; (ii) signals inhibiting cytokine production and T-cell proliferation; and (iii) signals inducing apoptosis of activated T-cells, as indicted above. In TCR-stimulated T cells, the CD3_ζ_ chain transfers activating signals to the nucleus. We have shown that TEXs and sera of cancer patients mediated dose- and time-dependent inhibition of CD3_ζ_ chain expression in T-cells [7,13,30]. TEXs also down-regulated mRNA for ζ in T-cells [7,13,30]. Co-incubation of T-cells with TEXs also resulted in reduced expression of JAK (Janus kinase) 3 [7]. As integrity of the JAK pathway is critical for function of receptors for cytokines expressing the common γ chain (IL-2, IL-7 and IL-15), its suppression results in a failure of T-cells to proliferate and produce cytokines. The addition of TEXs to activated CD8+ T-cells also reduced expression of phosphorylated STAT (signal transducer and activator of transcription) 5 in these cells. In contrast, TEXs increased phosphorylated STAT5 expression in activated CD4+ T-cells [15]. These data showed that TEXs selectively targeted activated CD8+ T-cells, interfering with TCR- and IL-2R-mediated signalling and inducing death of proliferating CD8+ T-cells [15].
TEXs promote differentiation, expansion and functions of Treg cells (regulatory T-cells)
Whereas TEXs added to pre-activated CD8+ T-cells inhibited proliferation, activated CD4+ T-cells increased in the presence of TEXs [15]. In the circulation of cancer patients, the frequency of CD4+ CD25highFOXP3+ (FOXP3 is forkhead box P3) Treg cells was elevated relative to that in NC donors (P < 0.0001), and these patients’ sera were highly enriched in exosomes [31]. Potentially, TEXs promoted Treg cell expansion, and they carried TGF_β_ (transforming growth factor β) and IL-10, the factors known to promote conversion of conventional T-cells into Treg cells. Only TEXs, but not exosomes or MVs, obtained from normal cells induced Treg cell expansion in culture. Interestingly, Treg cells were completely resistant to TEX-mediated apoptosis [31]. Instead, TEXs effectively mediated conversion of conventional CD4+ CD25neg T-cells into CD4+ CD25highFOXP3+ Treg cells [31]. Upon co-incubation with TEXs, Treg cells up-regulated expression of FasL, IL-10, TGF_β_1, CTLA-4 (cytotoxic T-lymphocyte antigen 4), GrB (granzyme B) and perforin (P < 0.05 for all) and mediated higher suppression of autologous responder cell proliferation (P < 0.01). TEXs were enriched in membrane-form TGF_β_1, and they increased expression of phosphorylated Smad2/3 and phosphorylated STAT3 in Treg cells. These TEX-mediated effects were dependent on TGF_β_1 and also on IL-10, as neutralizing antibodies specific for these cytokines blocked the ability of TEXs to expand Treg cells. In aggregate, these results indicated that TEXs had immunoregulatory properties [31].
TEXs interfere with DC maturation and favour MDSC (myeloid-derived suppression cell) differentiation
Effects of TEXs on human monocytes were studied by Rivoltini and co-workers [32]. These investigators showed that by blocking differentiation of human monocytes into DCs, TEXs interfered with CTL (cytotoxic T-lymphocyte) generation [32]. Human DCs generated in the presence of TEXs had low expression of co-stimulatory molecules, produced inhibitory cytokines (e.g. TGF_β_) as well as PGE2 and induced dose-dependent suppression of T-cell proliferation and anti-tumour cytotoxicity [32]. Furthermore, monocytes incubated in the presence of Treg cells differentiated into MDSCs, which are well known to play a key role in the suppression of anti-tumour immunity [33].
TEX-mediated interference with NK cell activity
It has been reported that NK cells of cancer patients mediate low levels of anti-tumour activity and have low expression levels of activating receptors, NKp30, NKp46, NKG2C and NKG2D [34,35]. TEXs were shown to inhibit cytolysis mediated by NK cells ex vivo, and our preliminary experiments suggest that the treatment of mice with TEXs decreased the percentage of NK cells in the spleen and lungs (T.L. Whiteside, unpublished work). We investigated the possibility that TGF_β_1 carried by TEXs impaired NK cell cytotoxicity and lowered NKG2D expression in patients’ NK cells [36,37]. TEXs isolated from sera of patients with AML (acute myelogenous leukaemia) at the time of diagnosis had similar effects on the NK cell phenotype and functions. AML patients’ sera were enriched in blast-derived exosomes carrying CD34, CD33 and CD117, as well as a membrane-form of TGF_β_1 [38]. These TEXs decreased cytolytic activity of normal NK cells. They down-regulated NKG2D receptor expression and induced Smad phosphorylation in NK cells [38]. Neutralization of TGF_β_1 carried by TEXs significantly, but not completely, abrogated these TEX-mediated effects. In addition, whereas AML patients’ sera contained elevated levels of soluble TGF_β_1, considerably more TGF_β_1 resided in TEXs and could be released and measured upon their disruption by detergents [38]. These findings suggest that TEX-associated TGF_β_1 is responsible for NK cell dysfunction in patients with AML and perhaps other malignancies.
TEXs and adenosine production
Adenosine is a well-known immunosuppressive factor [39]. Adenosine operates via its receptors (A1, A2A, A2B and A3) expressed on various cell types, including lymphocytes. Signalling via the A2A receptor, adenosine up-regulates cAMP levels in CD4+ effector T-cells, thereby reducing cellular functions [39]. The ability of murine Treg cells to produce adenosine is due to the presence of ectonucleotidases CD39 (ATP hydrolase) and CD73 (5′-nucleotidase) on the cell surface [40,41]. In humans, natural Treg cells express CD39, but rarely CD73, although inducible Treg cells found in the blood and tumour tissues of cancer patients co-express both these enzymes [42]. TEXs, which are ubiquitously present in body fluids of cancer patients, carry CD73 and have 5′-nuclotidase activity ([14], and P. Schuler and T.L. Whiteside, unpublished work). These TEXs can deliver membrane-tethered CD73 to CD39+ cells, enabling ATP hydrolysis to adenosine. As tumour cells are often enriched in CD73 [43], TEXs are especially well equipped to deliver CD73 to sites of ATP hydrolysis, enabling adenosine production and thus negatively modulating T-cells in the tumour microenvironment.
Implications of TEXs for anti-tumour immunity
TEXs present in body fluids of cancer patients may use various mechanisms to modulate anti-tumour functions of immune cells. Using annexin V, which detects a phosphatidylserine flip in the T-cell membrane, we observed that, in cancer patients, over 50% of circulating CD8+ T-cells bound annexin V, i.e. were in the early stages of apoptosis as measured by flow cytometry [24,25,29]. TEXs isolated from sera of patients with melanoma, breast cancer, ovarian cancer and HNSCC (head and neck squamous cell carcinoma) induced caspase activation in Jurkat cells (used as a surrogate for primary T-cells in cellular apoptosis assays), whereas exosomes from normal donors’ sera did not [44]. Whereas annexin V binding to CD8+ T-cells readily discriminated between normal donors and patients with HNSCC (P < 0.001), it did not distinguish patients with active disease from those with no evident disease after therapy [22]. Not surprisingly, the presence in sera of TEXs carrying high levels of FasL discriminated HNSCC patients with advanced (stage T3/T4) tumours (P < 0.009) from those with stage T1/T2 tumours whose TEXs had low levels of FasL [7]. TEXs from sera of patients with nodal metastases and poor prognosis were enriched in FasL and induced apoptosis in Jurkat cells discriminating this cohort of HNSCC patients (n = 32) from those with no nodal involvement and good prognosis (n = 28) at P < 0.02 [44]. TEXs isolated from AML patients’ sera at diagnosis varied widely in the levels of membrane-associated TGF_β_ [38]. Our preliminary results suggest that these levels correlate with the blast counts and thus could serve as prognostic biomarkers. On the basis of these results, it is possible to predict that TEXs will become increasingly important as biomarkers of disease progression, especially when technologies for separation of TEXs from other exosomes present in patients’ body fluids become available. Body fluids contain a mixture of exosomes, and molecular profiling of such a mixture, even if it is enriched in TEXs, might not be sufficiently informative. To be able to use TEXs as “a liquid biopsy”, as suggested recently by Taylor and Gercel-Taylor [45], will require the development of strategies for TEX separation from exosomes secreted by normal cells. One such strategy being developed in our laboratory employs immunoaffinity-based capture of TEXs from cancer patients’ body fluids using an antibody specific for a well-known tumour antigen, CSGP4 (chondroitin sulfate glycoprotein 4), or high-molecular-mass melanoma antigen, which is selectively expressed in a variety of tumour cells, but not in normal cells [46]. Capture of TEXs on beads coated with the antibody is followed by quantitative recovery and subsequent molecular analyses. Our immediate goal is to establish such selective capture procedures for TEXs in body fluids before committing to proteomics-based analyses of their molecular profiles and linking these profiles to clinical outcome.
Protection of immune cells from TEXs
Because TEXs appear to contribute to tumour escape by inducing dysfunction and death of immune effector cells in cancer patients, TEX elimination or blocking of their effects could be beneficial [47]. Several strategies for TEX removal have been considered. Ichim et al. [48] proposed a physical approach based on the extracorporeal removal of exosomes from plasma of cancer patients. Huber et al. [49] suggest interventions with TEX secretion by inhibiting upstream pathways, e.g. using drugs interfering with the microtubule stability or PPIs (proton pump inhibitors) to modify secretory metabolism. Yet another strategy aims to protect anti-tumour effector cells from functional impairments and death by using cytokines [50]. For example, survival cytokines IL-7 and IL-15, as well as the biologic IRX-2, which is produced by ex vivo_-stimulated human PBMCs (peripheral blood mononuclear cells) and contains natural cytokines and growth factors, can effectively protect T-cells from TEX-mediated apoptosis [26]. The pre-treatment of T-cells with these cytokines restored the balance between the pro- and anti-apoptotic Bcl-2 family members and normalized JAK3 and CD3_ζ expression in these cells, using the PI3K/Akt pathway as the key regulatory mechanism [26,27]. These results are in agreement with previously reported clinical and experimental data showing that the survival cytokines using the common receptor _γ_-chain are able to protect activated T-cells from tumour-induced death and enhance their anti-tumour activity [50].
Acknowledgments
Funding
Supported in part by the National Institutes of Health [grant number PO1-CA109688 (to T.L.W.)].
Abbreviations used
AML
acute myelogenous leukaemia
CTLA-4
cytotoxic T-lymphocyte antigen 4
DC
dendritic cell
ESCRT
endosomal sorting complex required for transport
FasL
Fas ligand
FOXP3
forkhead box P3
HNSCC
head and neck squamous cell carcinoma
IL
interleukin
IL-2R
IL-2 receptor
JAK
Janus kinase
MDSC
myeloid-derived suppression cell
MV
microvesicle
MVB
multivesicular body
NC
normal control
NK
natural killer
PD-1
programmed cell death 1
PDL-1
programmed cell death ligand 1
PGE2
prostaglandin E2
PI3K
phosphoinositide 3-kinase
STAT
signal transducer and activator of transcription
TCR
T-cell receptor
TEX
tumour-derived exosome
TGF_β_
transforming growth factor β
Treg cell
regulatory T-cell
References
- 1.Peinado H, Alecković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar C, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Somasundaram R, Herlyn M. Melanoma exosomes: messengers of metastasis. Nat Med. 2012;18:853–854. doi: 10.1038/nm.2775. [DOI] [PubMed] [Google Scholar]
- 3.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2011;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Whiteside TL, Mandapathil M, Szczepanski M, Szajnik M. Mechanisms of tumor escape from the immune system: adenosine-producing Treg, exosomes and tumor-associated TLRs. Bull Cancer. 2011;98:E25–E31. doi: 10.1684/bdc.2010.1294. [DOI] [PubMed] [Google Scholar]
- 5.Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol. 2006;16:3–15. doi: 10.1016/j.semcancer.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 6.Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, Zaccheddu A, Colone M, Arancia G, Gentile M. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology. 2005;128:1796–1804. doi: 10.1053/j.gastro.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 7.Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005;11:1010–1020. [PubMed] [Google Scholar]
- 8.Chaput N, Théry C. Exosomes: immune properties and potential clinical implementations. Semin Immunopathol. 2011;33:419–440. doi: 10.1007/s00281-010-0233-9. [DOI] [PubMed] [Google Scholar]
- 9.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 10.Wieckowski E, Whiteside TL. Human tumor-derived vs dendritic cell-derived exosomes have distinct biologic roles and molecular profiles. Immunol Res. 2006;36:247–254. doi: 10.1385/IR:36:1:247. [DOI] [PubMed] [Google Scholar]
- 11.Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem Pharmacol. 2011;81:1171–1182. doi: 10.1016/j.bcp.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 13.Taylor DD, Gercel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br J Cancer. 2005;92:305–311. doi: 10.1038/sj.bjc.6602316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. 2011;187:676–683. doi: 10.4049/jimmunol.1003884. [DOI] [PubMed] [Google Scholar]
- 15.Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8 + T lymphocytes. J Immunol. 2009;183:3720–3730. doi: 10.4049/jimmunol.0900970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang C, Ruffner MA, Kim SH, Robbins PD. Plasma-derived MHC class II + exosomes from tumor-bearing mice suppress tumor antigen-specific immune responses. Eur J Immunol. 2012;42:1778–1784. doi: 10.1002/eji.201141978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andreola G, Rivoltini L, Castelli C, Huber V, Perogo P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303–1316. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martínez-Lorenzo MJ, Anel A, Alava MA, Piñeiro A, Naval J, Lasierra P, Larrad L. The human melanoma cell line MelJuSo secretes bioactive FasL and APO2L/TRAIL on the surface of microvesicles: possible contribution to tumor counterattack. Exp Cell Res. 2004;295:315–329. doi: 10.1016/j.yexcr.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 19.Abrahams VM, Straszewski SL, Kamsteeg M, Hanczaruk B, Schwartz PE, Rutherford TJ, Mor G. Epithelial ovarian cancer cells secrete functional Fas ligand. Cancer Res. 2003;63:5573–5581. [PubMed] [Google Scholar]
- 20.Contini P, Ghio M, Poggi A, Filaci G, Indiveri F, Ferrone S, Puppo F. Soluble HLA-A,-B,-C and -G molecules induce apoptosis in T and NK CD8 + cells and inhibit cytotoxic T cell activity through CD8 ligation. Eur J Immunol. 2003;33:125–134. doi: 10.1002/immu.200390015. [DOI] [PubMed] [Google Scholar]
- 21.Contini P, Ghio M, Merlo A, Poggi A, Indiveri F, Puppo F. Apoptosis of antigen-specific T lymphocytes upon the engagement of CD8 by soluble HLA class I molecules is Fas ligand/Fas mediated: evidence for the involvement of p56lck, calcium calmodulin kinase II, and calcium-independent protein kinase C signaling pathways and for NF-κB and NF-AT nuclear translocation. J Immunol. 2005;175:7244–7254. doi: 10.4049/jimmunol.175.11.7244. [DOI] [PubMed] [Google Scholar]
- 22.Kim JW, Tsukishiro T, Johnson JT, Whiteside TL. Expression of pro- and antiapoptotic proteins in circulating CD8 + T cells of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004;10:5101–5110. doi: 10.1158/1078-0432.CCR-04-0309. [DOI] [PubMed] [Google Scholar]
- 23.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann TK, Dworacki G, Tsukihiro T, Meidenbauer N, Gooding W, Johnson JT, Whiteside TL. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res. 2002;8:2553–2562. [PubMed] [Google Scholar]
- 25.Saito T, Kuss I, Dworacki G, Gooding W, Johnson JT, Whiteside TL. Spontaneous ex vivo apoptosis of peripheral blood mononuclear cells in patients with head and neck cancer. Clin Cancer Res. 1999;5:1263–1273. [PubMed] [Google Scholar]
- 26.Czystowska M, Han J, Szczepanski MJ, Szajnik M, Quadrini K, Brandwein H, Hadden JW, Signorelli K, Whiteside TL. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ. 2009;16:708–718. doi: 10.1038/cdd.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czystowska M, Szczepanski MJ, Szajnik M, Quadrini K, Brandwein H, Hadden JW, Whiteside TL. Mechanisms of T-cell protection from death by IRX-2: a new immunotherapeutic. Cancer Immunol Immunother. 2011;60:495–506. doi: 10.1007/s00262-010-0951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groneberg C, Pickartz T, Binder A, Ringel F, Srock S, Sieber T, Schoeler D, Schriever F. Clinical relevance of CD95 (Fas/Apo-1) on T cells of patients with B-cell chronic lymphocytic leukemia. Exp Hematol. 2003;31:682–685. doi: 10.1016/s0301-472x(03)00109-7. [DOI] [PubMed] [Google Scholar]
- 29.Massaia M, Borrione P, Attisano C, Barral P, Beggiato E, Montacchini L, Bianchi A, Boccadoro M, Pileri A. Dysregulated Fas and Bcl-2 expression leading to enhanced apoptosis in T cells of multiple myeloma patients. Blood. 1995;85:3679–3687. [PubMed] [Google Scholar]
- 30.Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, Whiteside TL. T-cell apoptosis and suppression of T-cell receptor/CD3-ζ by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin Cancer Res. 2003;9:5113–5119. [PubMed] [Google Scholar]
- 31.Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg) PLoS ONE. 2010;5:e11469. doi: 10.1371/journal.pone.0011469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corebelli A, Fais S, Parmiani G, Rivoltini L. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-β-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66:9290–9298. doi: 10.1158/0008-5472.CAN-06-1819. [DOI] [PubMed] [Google Scholar]
- 33.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16:348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 34.Bauernhofer T, Kuss I, Henderson B, Baum AS, Whiteside TL. Preferential apoptosis of CD56dim natural killer cell subset in patients with cancer. Eur J Immunol. 2003;33:119–124. doi: 10.1002/immu.200390014. [DOI] [PubMed] [Google Scholar]
- 35.Szczepanski MJ, Szajnik M, Welsh A, Foon KA, Whiteside TL, Boyiadzis M. Interleukin-15 enhances natural killer cell cytotoxicity in patients with acute myeloid leukemia by upregulating the activating NK cell receptors. Cancer Immunol Immunother. 2010;59:73–79. doi: 10.1007/s00262-009-0724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol. 2008;180:7249–7258. doi: 10.4049/jimmunol.180.11.7249. [DOI] [PubMed] [Google Scholar]
- 37.Clayton A, Tabi Z. Exosomes and the MICA-NKG2D system in cancer. Blood Cells Mol Dis. 2005;34:206–213. doi: 10.1016/j.bcmd.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-β1. Haematologica. 2011;96:1302–1309. doi: 10.3324/haematol.2010.039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3′,5′-monophosphate and phosphatases. J Immunol. 2004;173:932–944. doi: 10.4049/jimmunol.173.2.932. [DOI] [PubMed] [Google Scholar]
- 40.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Höpner S, Centonze D, Bernardi G, Dell’Acqua ML, et al. Expression of ectonucleotidase CD39 by Foxp3 + Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 42.Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, Gorelik E, Lang S, Whiteside TL. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285:27571–27580. doi: 10.1074/jbc.M110.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B. CD73: a novel target for cancer immunotherapy. Cancer Res. 2010;70:6407–6411. doi: 10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bergmann C, Strauss L, Wieckowski E, Czystowska M, Albers A, Wang Y, Zeidler R, Lang S, Whiteside TL. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head Neck. 2009;31:371–380. doi: 10.1002/hed.20968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor DD, Gercel-Taylor C. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Semin Immunopathol. 2011;33:441–454. doi: 10.1007/s00281-010-0234-8. [DOI] [PubMed] [Google Scholar]
- 46.Rivera Z, Ferrone S, Wang X, Jube S, Yang H, Pass HI, Kanodia S, Gaudino G, Carbone M. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin Cancer Res. 2012;18:5352–5363. doi: 10.1158/1078-0432.CCR-12-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, Rivoltini L. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15:80–88. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 48.Ichim TE, Zhong Z, Kaushal S, Zheng X, Ren X, Hao X, Joyce JA, Hanley HH, Riordan NH, Koropatnick J, et al. Exosomes as a tumor immune escape mechanism: possible therapeutic implications. J Transl Med. 2008;6:37. doi: 10.1186/1479-5876-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huber V, Filipazzi P, Iero M, Fais S, Rivoltini L. More insights into the immunosuppressive potential of tumor exosomes. J Transl Med. 2008;6:63. doi: 10.1186/1479-5876-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev. 2004;202:275–293. doi: 10.1111/j.0105-2896.2004.00199.x. [DOI] [PubMed] [Google Scholar]