Substrate Binding Stabilizes a Pre-translocation Intermediate in the ATP-binding Cassette Transport Protein MsbA (original) (raw)
Background: During substrate transport, ATP-binding cassette exporters switch between an inward-facing and an outward-facing state in a nucleotide-dependent fashion.
Results: Substrate binding to bacterial MsbA initiates dimerization of nucleotide-binding domains without opening the membrane domains at their external side.
Conclusion: Substrate binding to MsbA stabilizes an “inward-facing closed” pre-translocation state that binds ATP.
Significance: Our observations suggest a fundamental mechanism by which substrates stimulate ATP hydrolysis.
Keywords: ABC Transporter, Lipid Transport, Membrane Transport, Multidrug Transporters, Protein Conformation, Substrate-stimulated ATPase
Abstract
ATP-binding cassette (ABC) transporters belong to one of the largest protein superfamilies that expands from prokaryotes to man. Recent x-ray crystal structures of bacterial and mammalian ABC exporters suggest a common alternating access mechanism of substrate transport, which has also been biochemically substantiated. However, the current model does not yet explain the coupling between substrate binding and ATP hydrolysis that underlies ATP-dependent substrate transport. In our studies on the homodimeric multidrug/lipid A ABC exporter MsbA from Escherichia coli, we performed cysteine cross-linking, fluorescence energy transfer, and cysteine accessibility studies on two reporter positions, near the nucleotide-binding domains and in the membrane domains, for transporter embedded in a biological membrane. Our results suggest for the first time that substrate binding by MsbA stimulates the maximum rate of ATP hydrolysis by facilitating the dimerization of nucleotide-binding domains in a state, which is markedly distinct from the previously described nucleotide-free, inward-facing and nucleotide-bound, outward-facing conformations of ABC exporters and which binds ATP.
Introduction
ATP-binding cassette (ABC)3 exporters are found in all living organisms and are responsible for the active efflux from the cell of a wide range of molecules such as ions, drugs, lipids, peptides, and proteins. Their dysfunction has been linked to many disease conditions, including multidrug resistance, cystic fibrosis, Stargardt disease, age-related macular degeneration, adrenoleukodystrophy, Tangier disease, Dubin-Johnson syndrome, and progressive familial intrahepatic cholestasis. All ABC exporters share a conserved dimeric architecture in which each monomer is composed of a nucleotide-binding domain (NBD) and a cognate membrane domain (MD) (1). For multidrug ABC transporters, the structural conservation between the mammalian full-transporter ABCB1 and bacterial half-transporter homologs LmrA, Sav1866, and MsbA translates into functional similarities, including overlapping substrate specificities (2–5).
MsbA functions as a homodimer and has a recognized role in mediating lipid A export in Escherichia coli (6). Two MsbA orthologs were recently crystallized in distinct but complementary conformations as follows: nucleotide-free inward-facing (E. coli MsbA) and AMP-PNP (a nonhydrolyzable ATP-analog)-bound outward-facing (Salmonella typhimurium MsbA); inward and outward refer to cytoplasmic and periplasmic side of the plasma membrane, respectively (7). ADP·Vi (hydrolysis intermediate)-trapped MsbA was also observed in the outward-facing conformation, similar to the AMP-PNP-bound structure (7). Biochemical verifications of these conformations include electron paramagnetic resonance (EPR) studies on MsbA (8–10) and LmrA (11), hydrogen/deuterium exchange-mass spectrometry on BmrA (12), inter-molecular cysteine cross-linking on MsbA in membrane vesicles (13, 14), and most recently, by luminescence resonance energy transfer on MsbA (15).
These studies provide an overall view of the dynamic movements associated with an ATP hydrolysis (ATPase) cycle that is in agreement with an alternating access model of transport (1, 7, 16). Briefly, this model suggests that an ABC exporter alternates between an inward-facing and outward-facing conformation to present the substrate-binding pocket in the MDs to the inside and outside of the cell, respectively. These movements are guided by the binding and hydrolysis of ATP that cause alternating dimerization and dissociation, respectively, of the two NBDs (1). Although compelling, the current model does not address a fundamental property of ABC exporters, namely the substrate-dependent stimulation of the ATPase activity (17–20). Our understanding of this phenomenon requires insight into the conformation adopted by an ABC exporter upon substrate binding, and its relation to the cycle of ATP binding and hydrolysis at the NBDs.
At present, detailed structural information regarding substrate binding by ABC transporters is limited. Mouse ABCB1a was recently crystallized with cyclic peptide inhibitors (21). Although the peptide-bound ABCB1a structures identify a substrate-binding surface (1, 21, 22), these structures appear identical to the drug/nucleotide-free inward-facing ABCB1a and MsbA conformations. The suggestion that the protein structure is unaltered when progressing from a substrate/nucleotide-free apo-state to a substrate-bound state is inconsistent with previously published biochemical data, nearly all of which point to a measurable conformational change (19, 23–27). Furthermore, it is unclear how an inward-facing conformation with disengaged NBDs could accelerate ATP hydrolysis.
We recently found that the disruption of molecular contacts within a structurally conserved element, termed the tetrahelix bundle, inhibits the formation of a stable, ATP-bound, outward-facing state of E. coli MsbA (14). In this previous study, we used two cysteine reporters in MsbA-cl (cysteine-less) to measure nucleotide-dependent conformational changes, E208C (reports NBD dimerization) and A281C (reports the separation of two groups of TMs (TM1 and -2, TM3′, -4′, -5′, and -6′ separate from TM1′ and -2′ and TM3, -4, -5, and -6)) during “wing” formation in the MsbA dimer.
Here, we have conducted cysteine cross-linking, Förster/fluorescence resonance energy transfer (FRET), and cysteine accessibility studies on these reporters to biochemically study substrate binding-induced conformational changes in MsbA. We have further performed substrate-binding assays and substrate-stimulated ATP binding and ATPase measurements to assign a role for substrate binding in the current alternating access model for substrate transport.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
Lactococcus lactis strain NZ9000 Δ_lmrA_ Δ_lmrCD_, which is devoid of the major endogenous multidrug transporters LmrA and LmrCD, was used as a host for pNZ8048-derived plasmids; E. coli strain XL1 Blue was a host for the pGEM-5Zf(+) (Promega) cloning vector. L. lactis and _E. coli c_ells were grown in M17 (Oxoid) and LB (Formedium) medium, respectively, as described previously (28).
Site-directed Mutagenesis
Construction of N-terminal His6-tagged cysteine-less MsbA (MsbA-cl), E208C MsbA-cl, and A281C MsbA-cl has been reported (13). pGEM-A281C MsbA-cl was used as template to generate the E208C/A281C MsbA-cl double mutant. The gene was subcloned as an NcoI-SacI fragment into pNZ8048 downstream of the nisin A-inducible promoter, yielding pNH-E208C/A281C MsbA-cl. The mutated msbA gene was sequenced to confirm that only the intended mutations were introduced.
Inside-out Membrane Vesicles
The expression of MsbA proteins in lactococcal cells, and the preparation of inside-out membrane vesicles (ISOVs) in 100 mm K-HEPES buffer, pH 7.0, were performed as reported previously (28).
Cysteine Cross-linking
Cysteine cross-linking on the E208C, A281C, and E208C/A281C mutants was performed in ISOVs as reported previously (13). For each cross-linking reaction, ISOVs were diluted to 5 μg of protein/μl in 100 mm K-HEPES buffer, pH 7.0, containing 5 mm MgSO4 in a reaction volume of 90 μl in microcentrifuge tubes. To reduce background signals due to cross-links formed before nucleotide addition, 0.5 mm dithiothreitol (DTT) was added, and the samples were incubated at 20 °C for 2 min. Nucleotides (2 mm ADP, 2 mm AMP-PNP, or 2 mm ATP plus 2 mm sodium orthovanadate, referred to as ADP·Vi) or substrates (Hoechst 33342, verapamil, lipid A) or an equal volume of solvent was added following incubation at 20 °C for 5 min. The cross-linking reactions were initiated by the addition of 0.5 mm copper phenanthroline solution, which was added from a 50 mm stock made by mixing CuSO4 and 1,10-phenanthroline in a ratio 1:4 (w/w) in ultrapure H2O. The tube lids were pierced, and the reaction was allowed to occur for 5 min in a 30 °C shaker incubator (model 3032, GFL Gesellschaft fur Labortechnik mbH, Germany) at 200 rpm. The reactions were stopped by the addition of excess (10 mm) of the thiol alkylator _N_-ethylmaleimide (NEM) (added from a freshly prepared stock of 100 mm in ultrapure H2O) and incubation at 20 °C for 1–2 min. Next, 10–15 μg of ISOVs from the cross-linking reactions were mixed with 5× SDS sample loading buffer devoid of DTT and separated on an SDS-PAGE without any incubation. Band intensities on Western blots were compared by densitometry analyses using ImageJ software version 1.43 (National Institutes of Health).
FRET
The purification of His-tagged MsbA, as described previously (28), was modified as follows. For each experiment, after the overnight binding step the Ni2+ affinity resin was washed once with 3 ml of buffer B and suspended in 3 ml of buffer B. Then 3.3 μm Atto 590 + 3.3 μm Atto 665 (Atto-tec, Germany; prepared as 20 mm stocks in DMSO) were added, and after incubation on ice for 5 min, 1 μl of copper phenanthroline (prepared as above) was added, and the sample was incubated on a rotating wheel for 1 h at 4 °C. Subsequent wash and elution steps were identical to the above-mentioned. 7–10 μg of the labeled protein per measurement and ligands, 2 mm ATP plus 2 mm sodium orthovanadate or 2 mm ADP or 40 μg/ml lipid A or equal volume of DMSO, were used in a 2-ml reaction set up in SOG cuvettes. The LS 55B luminescence spectrometer (PerkinElmer Life Sciences) settings were as follows: excitation at 594 nm, slit width 5 nm, emission 600–800 nm, slit width 10 nm, scan speed 500 nm/min, stirrer “low.” All measurements reported here have been made after 10 min of stirring.
Cysteine Cross-linking and Accessibility
For cysteine cross-linking combined with Atto590 labeling, ISOVs were diluted to 7 μg of total membrane protein/μl in 100 mm K-HEPES buffer, pH 7.0, containing 5 mm MgSO4 in a reaction volume of 90 μl in microcentrifuge tubes. Nucleotides (2 mm AMP-PNP) or substrates (Hoechst 33342, verapamil) or lipid A or an equal volume of DMSO was added following incubation at 20 °C for 5 min. 20 μm Atto590 was then added (from a 2 mm stock in DMSO), and after incubation at room temperature for 5 min, cross-linking reactions were initiated by the addition of 0.5 mm copper phenanthroline solution (prepared as above). After piercing their lids, the tubes were kept in a 30 °C shaker (200 rpm) incubator for 15 min. For E208C, prior to the addition of nucleotides, the ISOVs were incubated for 3 min with 0.5 mm DTT. After incubation with nucleotides for 5 min, 0.5 mm copper phenanthroline was added; the tube lids were pierced, and samples were placed in a 30 °C shaker (200 rpm) incubator for 5 min. Next, 20 μm Atto590 was added, and the reaction was further continued in a 30 °C shaker (200 rpm) incubator for 10 min. All reactions were stopped by the addition of 10 mm NEM, followed by incubation at 20 °C for 1–2 min. Subsequently, 25 μg of ISOVs from the cross-linking reactions were mixed with 3–5× SDS sample loading buffer devoid of DTT and separated on an SDS-PAGE without any incubation. Gels were first viewed under UV light to detect the Atto590-labeled protein bands and subsequently Coomassie Brilliant Blue-stained to visualize all protein bands. Monomer intensities from UV images and dimer intensities from Coomassie Brilliant Blue-stained gel scans were subjected to densitometry analyses using the ImageJ software, version 1.43 (National Institutes of Health).
Ethidium Transport
Ethidium transport in intact cells was measured by fluorimetry as described (28).
Hoechst 33342 Binding
Binding of Hoechst 33342 to purified MsbA in solution was detected fluorimetrically (28). MsbA cysteine single/double mutants or MsbA-cl was purified as described before (28) with the following modifications. After the overnight binding step, the resin was washed once with buffer B and resuspended in 3 ml of buffer B. 16 μm CuPhen was added, and the suspension was incubated on the rotating wheel for 1 h at 4 °C, before proceeding with the washes and elution as described above. 10–12 μg of purified pre-cross-linked protein was diluted in 2 ml of assay buffer (100 mm KPi, pH 7.0, 5 mm MgSO4), and an equal volume of the elution buffer diluted in the assay buffer was used as a negative control. Hoechst 33342 was added as follows: 2× 10 μl of 5 μm Hoechst 33342, 2× 10 μl of 25 μm Hoechst 33342, followed by 10-μl additions of 50 μm Hoechst 33342 until a plateau in the fluorescence intensity was obtained. All steps of Hoechst 33342 were separated by 30 s. Fluorescence was monitored on an LS 55B luminescence spectrometer (PerkinElmer Life Sciences) using excitation and emission wavelengths of 355 and 457 nm and slit widths of 10 and 5 nm, respectively. Fluorescence intensities of the elution buffer control were subtracted.
TNP-ATP Binding
WT MsbA was purified as described (28), and 40 μg of protein was diluted in a 2-ml reaction buffer (50 mm KPi, pH 7.0, 10% glycerol, 0.01% _n_-dodecyl-β-d-maltoside) with or without 50, 100, or 200 μm verapamil. TNP-ATP (Molecular Probes, Invitrogen) was added as follows: 2× 5 μl of 100 μm TNP-ATP, followed by 5-μl additions of 500 μm TNP-ATP until a plateau in fluorescence emission was obtained. All increments were separated by 30 s. Equal volume of elution buffer was used as negative control. Fluorescence was monitored in an LS 55B luminescence spectrometer (PerkinElmer Life Sciences) using excitation and emission wavelengths of 407 and 535 nm, and slit widths of 10 and 15 nm, respectively.
Substrate-stimulated ATPase Activity
ATPase activity of purified MsbA was measured using the malachite green colorimetric method (13). Briefly, for each data point, 1–5 μg of protein was added to 100 mm K-HEPES buffer, pH 7.0, with 5 mm MgSO4, containing either 100 μg/ml lipid A (Sigma, diphosphoryl from E. coli F583 (Rough strain lipopolysaccharide (Rd) mutant); prepared as a 1 mg/ml stock in anhydrous DMSO) or equal volume DMSO (anhydrous, Sigma) for 5 min on ice. For incubations with verapamil, 10 μm of the compound was added instead of lipid A. Then the mixture was added to a range of ATP concentrations, 0–8 mm (high grade ATP from Sigma), in a total volume of 60 μl at 4 °C. The assay was then incubated for 5 min at 30 °C, after which _A_600 was determined following the addition of malachite green-ammonium molybdate, freshly activated with 0.1% Triton X-100.
Curve-fitting and Statistical Analysis
Graphs were plotted using Origin Pro 8 (OriginLab). Equilibrium Hoechst 33342 binding, TNP-ATP binding, and ATPase kinetics data were fit using the built-in hyperbola function (y = P1·x/P2 + x). P1 and P2 obtained for the resulting fits were designated _B_max (maximum binding)/_V_max (maximum rate of hydrolysis) and Kd (dissociation constant)/Km (ATP hydrolysis affinity constant), respectively. All nonlinear fits were subjected to the built-in fit statistics, including reduced χ2 and adjusted _R_2 to determine the fit status. Data from FRET, Hoechst 33342 binding, and ATPase kinetics were statistically analyzed using the Student's t tests, in which p < 0.05 was taken as significant (*), and p < 0.04 as highly significant (**).
RESULTS
Substrate Binding to MsbA Causes NBD Dimerization
We previously described the use of inter-molecular cysteine cross-linking between E208C and E208C′ in the cysteine-less MsbA (MsbA-cl) homodimer in ISOVs to dissect the conformational changes associated with the ATPase cycle in the absence of added transport substrates (13, 14, 16). Here, we used this assay to test the effect of typical MsbA substrates, including Hoechst 33342 and verapamil and the physiological substrate lipid A (2, 3, 5, 6, 26, 28), in the absence of added nucleotides. Upon the exposure of the E208C mutant to these substrates at room temperature prior to performing oxidative cross-linking, significant concentration-dependent increases in the proportions of cross-linked dimers were observed (Fig. 1, a–c). As the proportion of E208C cross-links reports the conformational movements associated with the dimerization of the NBDs (13), our results suggest that substrate binding to MsbA causes a closure of the NBDs.
FIGURE 1.

Substrate binding by the MsbA dimer causes NBD dimerization. a and b, cysteine-less MsbA containing a single cysteine residue near the NBD (E208C MsbA-cl) shows a substrate-dependent enhancement in the intensity of E208C-E208C′ cross-linked MsbA dimers (D) in ISOVs. To study the conformational changes due to substrate binding, ISOVs containing E208C MsbA-cl were subjected to cysteine cross-linking in the presence of the substrates verapamil or Hoechst 33342 (both in water) or lipid A (controlled against an equal volume of the solvent DMSO). Briefly, ISOVs at 5–7 mg/ml total membrane protein, in 100 mm K-HEPES buffer, pH 7.0, containing 5 mm MgSO4 were first mixed with 0.5 mm DTT for 2 min at 20 °C to reduce preformed cross-links. Substrate was then added at the indicated concentrations. Following 5 min of incubation at 20 °C, cross-linking was initiated by the addition of 0.5 mm CuPhen. After 5 min of incubation in a 30 °C shaker incubator, reactions were stopped by the addition of 10 mm NEM. 10–15 μg of protein from each sample was mixed with 6× SDS sample loading buffer devoid of reducing agents, separated on a 10% SDS-polyacrylamide gel, and transferred to Western blot probed with anti-His tag antibody. This assay was performed previously in the presence of nucleotides instead of substrates (13, 14). c, densitometry on dimer bands is presented as -fold change in intensity relative to the no substrate control for verapamil or Hoechst 33342 and relative to the DMSO control for lipid A (**, p < 0.03; *, p < 0.05 Student t test; n = 3, error bars represent mean ± S.E.).
To test the effect of substrate on conformational changes in the MsbA dimer by an alternative technique, we applied a FRET-based method to purified protein. In this method, the Ni2+ affinity resin-bound, His6-tagged E208C/E208C′ MsbA-cl homodimer was labeled through an oxidative cross-linking reaction with a FRET pair of thiol-reactive dyes (donor (D), Atto590, and acceptor (A), Atto665; termed E208C-DA). The predicted Förster distance (_R_o, the distance at which FRET is 50% efficient) of ∼70 Å for this pair of dyes was deemed suitable for our purpose, because the nucleotide-free, inward-facing crystal structure of E. coli MsbA predicts a distance of ∼55 Å between Glu-208 and Glu-208′ (7). The washed and eluted and probe-labeled protein samples were analyzed by SDS-PAGE. As per the manufacturer, and as confirmed in our experiments, D (but not A) absorbs strongly in the UV region (260–280 nm) and is fluorescent at 610–630 nm (Fig. 2, a and b). The observation that the UV fluorescence of D, when reacted with the E208C mutant in the presence of equimolar amounts of A, was reduced to half that in the absence of A, led us to conclude that D and A were equally reactive with an incorporation at an approximate ratio of 1:1 when applied together at equal concentrations (Fig. 2, b and c). Incubation of E208C labeled with D or A or D and A (termed E208C-D, E208C-A, and E208C-DA, respectively) with the oxidizing agent CuPhen did not yield any increase in cysteine cross-linking or D or A labeling efficiency (Fig. 2, a and b). This indicated that the oxidation reaction was complete, i.e. no free cysteine residues and unreacted probe molecules remained in the eluted protein samples. Upon exciting E208C-D, E208C-A, or E208C-DA at the excitation maximum for D (594 nm) and following the emitted fluorescence over 600–800 nm, we found a strong signal at ∼620 nm for E208C-D but not for E208C-A (Fig. 2, d and e). The fluorescence intensity for E208C-DA at ∼620 nm was reduced to about 0.5–0.75 times that of E208C-D, although an additional signal at ∼680 nm was observed that corresponds to the emission region of A (Fig. 2, d and e). These results point to the transfer of fluorescence energy from D to A in E208C-DA. Our control experiments revealed that the FRET signal at 680 nm could only be obtained with E208C-DA, as opposed to background signals for the elution buffer, the dyes D and A free in the elution buffer solution, or MsbA-cl incubated with D and A (Fig. 2, d and e). Taken together, the findings suggest that, although labeling of the E208C/E208C′ MsbA dimer with D and A will yield dimers populations that contain AA (25%), DD (25%) or DA or AD (50%), FRET signal is only obtained for the latter populations containing both A and D.
FIGURE 2.

Analyses of FRET between Atto-dye-labeled E208C and E208C′ in the MsbA-cl dimer. a and b, Ni2+ affinity resin-bound E208C MsbA-cl was labeled with Atto590 (donor, D) or Atto665 (acceptor, A) or both (DA). To test whether the labeling reaction was complete (and that no free labels/unreacted thiol groups were present in eluted samples), 1 μg of the purified labeled protein fractions, with or without excess (5 mm) CuPhen, was analyzed by SDS-PAGE in the absence of DTT. Coomassie Brilliant Blue staining (A) shows total protein in the sample (Mo, monomers; Di, dimers) and followed analysis of the gel by in-gel UV fluorescence (b) to detect _D_-labeled monomers. a and b represent the same gel and are cropped to enhance readability. c, band intensities in b were measured using densitometry and are presented as percentage of monomers labeled with D. Labeling of E208C-D was set at 100%. d, at the excitation maximum for D (594 nm), buffer (trace 1) or DA in the absence of protein (trace 2) did not produce any noticeable fluorescence at the emission maximum for A (680 nm). e, such a signal was also not obtained for E208C-D (trace 1) and E208C-A (trace 2), whereas clear evidence for transfer of fluorescence energy from D to A under these conditions was observed for E208C-DA (trace 4). In another control experiment, fluorescence emission by A was also observed for E208C-A at the excitation maximum of A (663 nm) (trace 3). f, MsbA-cl mixed with DA did not yield any FRET signal at 680 nm (trace 1). The addition of lipid A (40 μg/ml) did not affect fluorescence at 680 nm (trace 2). Similarly, the addition to E208C-A of the lipid A or ATP plus sodium orthovanadate (2 mm each, to trap MsbA with ADP·Vi) or 2 mm ADP did not cause any appreciable alterations in the fluorescence emission at 680 nm (traces 3–6).
To validate the use of FRET for detection of conformational changes as a complement to cysteine cross-linking at position Glu-208 in MsbA, FRET signals for E208C-DA were tested in the presence of ADP·Vi and ADP (Fig. 3). These were found to be in excellent agreement with the cysteine cross-linking results reported for E208C (13). Namely, ADP·Vi-trapped E208C-DA yielded a significantly higher FRET intensity compared with the no-nucleotide control, in significant contrast to ADP binding (p < 0.04, Fig. 3), suggesting that purified MsbA, similar to MsbA in ISOVs, is in the outward-facing conformation when trapped with ADP·Vi, although it is in the inward-facing state with ADP alone.
FIGURE 3.

FRET between E208C-DA demonstrates NBD closure upon substrate binding. a, addition of lipid A (40 μg/ml) or 2 mm ADP·Vi to E208C-DA led to significant elevations in the FRET intensity at 680 nm, compared with equal volume of the assay buffer or DMSO (solvent) or 2 mm ADP. b, each ligand condition was replicated three times with protein, purified, and labeled from independently prepared batches of ISOVs. Relative intensities at 680 nm compared with the buffer condition are shown (**, p < 0.04; n = 3, mean ± S.E.).
When 40 μg/ml lipid A was added to E208C-DA instead of the substrates, and in the absence of added nucleotides, we also observed a significant increase in the FRET signal compared with the DMSO (solvent) control or the ADP-bound condition (p < 0.04 for both comparisons, Fig. 3). This observation confirmed our conclusions from substrate-responsive E208C cross-linking (Fig. 1, a–c), which point to substrate binding-dependent NBD dimerization. Furthermore, we also conducted control experiments where MsbA-cl mixed with DA was incubated with lipid A or where E208C-A was incubated with ADP·Vi, ADP, or lipid A, but all these spectra were near-background (Fig. 2f), suggesting that the inclusion of nucleotides or lipid A did not produce any major background interferences in the region where FRET was observed for E208C-DA, i.e. 680 nm. The only fluorescent signals contributed by lipid A were found to be near ∼600 nm (Fig. 2f). Taken together, our cross-linking and FRET data on E208C suggest that substrate binding to the MsbA dimer brings the NBDs into close proximity.
Substrate Binding to MsbA Does Not Cause MD Separation
NBD closure has previously been reported for the steps of ATP binding and ATP hydrolysis in the catalytic cycle of MsbA (7, 8, 10–16). However, ATP binding and ATP hydrolysis were also reported to cause MD separation in MsbA and homologs (8, 9, 11, 12, 29). To investigate the effects of substrate binding on the conformation of MsbA at the MDs, we used our previously described cysteine reporter at the extracellular end of the MDs, A281C in MsbA-cl.
We have successfully used A281C at the extracellular end of TM6 in MsbA-cl to report nucleotide binding-dependent conformational changes in the MDs by measuring its accessibility to the thiol-reactive probe Atto590 in conjunction with cysteine cross-linking (14). In this assay, an increase in cysteine cross-linked dimers suggests MD closure/inward-facing MsbA, whereas an increase in Atto590-labeled monomers, detected through the UV fluorescence of Atto590 (explained under “Substrate Binding to MsbA Causes NBD Dimerization”), reports MD separation/outward-facing MsbA.
When we performed this cysteine cross-linking Atto590-labeling assay using A281C with substrates in the absence of added nucleotides, we observed no major changes in cysteine cross-linked dimers or Atto590-labeled monomers (Fig. 4, a–c). We used the incubation with AMP-PNP as a control in this experiment, where the inward- to outward-facing conformational shift was successfully detected (14). Our results suggest that as opposed to the dimerization near the NBDs caused by substrate binding (Figs. 1 and 3), the MDs do not undergo a detectable conformational change at the same substrate concentrations (Fig. 4). This observation distinguishes the substrate binding-led conformation from the nucleotide-dependent conformations reported previously, e.g. AMP-PNP/ADP·Vi binding gives NBD dimerization and concomitant MD separation (8–14). We further confirmed that the disparity in substrate-responsive cross-linking observed between E208C and A281C was not due to the use of Atto590, by reproducing some of the substrate-dependent data by crossing-over methods, i.e. using cysteine cross-linking accessibility for E208C (Fig. 5, a and b) and using cysteine cross-linking alone for A281C (Fig. 5, c and d).
FIGURE 4.

Substrate binding by MsbA does not cause MD separation. a and b, cysteine cross-linking-Atto590 labeling with A281C, which is located at the extracellular side of the MDs. We previously used this reaction to detect nucleotide-dependent conformational changes in the MsbA dimer in ISOVs (14). Here, we did not observe any marked changes in the proportion of dimers (D, dimers visualized on Coomassie Blue-stained SDS-polyacrylamide gels) or Atto590-labeled monomers (M, monomers visualized through in-gel UV fluorescence from Atto590) when increasing concentrations of the substrates verapamil, Hoechst 33342, or lipid A, or the solvent controls were included in the reaction instead of nucleotides. In contrast, incubations with AMP-PNP served as a robust positive control for the detection of MD separation in the outward-facing state, and gave clear alterations in dimer and monomer band intensities. c, densitometry on dimer (left) and monomer (right) signals is presented as -fold change in intensity relative to the no substrate/nucleotide control, which was taken as 1 in each experiment (**, p < 0.04 AMP-PNP versus verapamil/Hoechst 33342/lipid A; n = 3, mean ± S.E.).
FIGURE 5.

Confirmation of the disparity between substrate-dependent cross-linking between E208C-E208C′ and A281C-A281C′ in MsbA-cl. a and b, the lowest verapamil concentration (5 μm) already enhanced cysteine cross-linked E208C–E208C′ dimers (D) and decreased in Atto590-labeled monomers (M) (n = 3, mean ± S.E.). This assay was performed similar to that detailed under “Materials and Methods,” with the exception that 0.5 mm DTT was first added to reduce pre-formed E208C cross-links, and 0.5 mm CuPhen was added (5 min, 30 °C shaker incubator) prior to the addition of Atto590. c and d, level of cross-linked A281C-A281C′ dimers did not change in the presence of verapamil up to concentrations of 15 μm. No changes in monomer intensities or cross-linked dimer intensities were observed when cysteine cross-linking was performed in the presence of Atto590 (see Fig. 4).
Taken together, our data from the studies on the two reporters, E208C and A281C, suggest that the substrate-bound MsbA dimer exists in an inward-facing closed conformation, in which both NBDs and MDs are in close proximity.
Hoechst 33342 Binding to MsbA Is Preferred in Inward-facing Closed Conformation
To further study the inward-facing closed state, we introduced both the cysteine mutations in MsbA-cl to create the double mutant E208C/A281C, which was expressed at a similar level as MsbA-cl (Fig. 6a) and which, in the absence of cysteine cross-linking, was equally active in ethidium efflux activity as MsbA-cl (Fig. 6b).
FIGURE 6.

Expression and activity of the double-cysteine mutant E208C/A281C MsbA-cl. a, total membrane proteins in ISOVs harboring the E208C/A281C mutant were separated on an SDS-polyacrylamide gel and analyzed by Western blot. The double mutant protein expressed as well as MsbA-cl and underwent spontaneous Cys-Cys cross-linking in the absence of DTT, similar to the single mutant proteins E208C and A281C (13, 14). The high molecular weight signals arising due to this spontaneous cross-linking could be abolished by the use of DTT in the sample loading buffer. b, both MsbA-cl and the noncross-linked E208C/A281C mutant catalyzed metabolic energy-dependent ethidium export from L. lactis cells that were pre-loaded with 2 μm ethidium bromide. Fluorescence traces are typical for data obtained in three independent experiments using different batches of cells.
A281C-A281C′ cross-links in MsbA-cl are highly intense in the inward-facing conformation of MsbA under nucleotide-free or substrate-bound conditions (Fig. 5), compared with the nucleotide-bound conditions. In contrast, we previously found that E208C-E208C′ cross-links are less intense in the nucleotide-free inward-facing conformation (13, 14). Thus, we predicted that in the absence of added nucleotides, the double mutant E208C/A281C could possibly exist as two kinds of inward-facing species, either as A281C-A281C′ single cross-linked (inward-facing “open”) or E208C-E208C′ + A281C-A281C′ double cross-linked (inward-facing “closed”). We performed equilibrium Hoechst 33342 binding on purified and pre-cross-linked single cysteine mutants, E208C and A281C, and double cysteine mutant E208C/A281C, alongside MsbA-cl. The results revealed that the dissociation constants (Kd) for substrate binding to E208C and E208C/A281C MsbA-cl were ∼1.7- and ∼2.0-fold lower, respectively, than for A281C (p < 0.01 for both comparisons), with MsbA-cl itself lying in between (Table 1). Because both pre-cross-linked protein samples E208C and E208C/A281C have comparatively higher proportions of the inward-facing closed species compared with pre-cross-linked A281C or noncross-linked MsbA-cl and E208A MsbA-cl proteins (14), these data suggest that the inward-facing closed conformation of MsbA is the preferred high affinity substrate-binding state.
TABLE 1.
Hoechst 33342 binding to cysteine cross-linked purified MsbA proteins
| MsbA protein | Kda | _B_maxa |
|---|---|---|
| μ_m_ | a.u. | |
| MsbA-cl | 0.65 ± 0.04 | 33.5 ± 4.0 |
| MsbA-cl E208C | 0.47 ± 0.06b | 34.7 ± 2.7 |
| MsbA-cl A281C | 0.79 ± 0.03_b,c_ | 31.9 ± 7.8 |
| MsbA-cl E208C/A281C | 0.39 ± 0.04c | 34.3 ± 2.5 |
These data also show that the double E208C/A281C mutant was functionally and conformationally different from the single A281C mutant, thus distinguishing the inward-facing closed conformation from the inward-facing open conformation. It is important to note here that the overall Cys-Cys cross-linking efficiencies in purified proteins were found to be much lower than for proteins embedded in ISOVs (e.g. purified A281C cross-linking efficiency was ∼31% versus >75% in ISOVs; purified E208C cross-linking efficiency was ∼22% versus >40% in ISOVs, based on densitometry of monomer and dimer signals on Western blot). This could be one of the reasons for the modest changes in Kd that were observed in these experiments. Another reason might be interferences from the detergent itself, which may also bind as a substrate (30, 31), reducing the Hoechst 33342 binding-related signals. Our result from the substrate-binding experiments, i.e. inward-facing closed MsbA is the preferred substrate binding conformation, follows and complements our cysteine cross-linking and FRET data demonstrating that substrate binding on MsbA stabilizes an inward-facing closed state.
Substrate Binding to MsbA Does Not Affect the Nucleotide Binding Affinity but Enhances the Maximum Rate of ATP Hydrolysis
To investigate the mechanism by which substrate binding stimulates ATP hydrolysis, we studied the effect of substrate binding on the binding of ATP to MsbA in a fluorescent TNP-ATP-based assay (14). Significant hydrolysis of TNP-ATP in this assay was prevented by the exclusion of Mg2+ from the assay buffer. Despite adding increasing amounts of verapamil in the TNP-ATP binding assay, we were unable to find any detectable change in the TNP-ATP binding properties of WT MsbA (Fig. 7a). The dissociation constant (Kd) for TNP-ATP binding by WT MsbA was 0.36 ± 0.02 μm TNP-ATP in the absence of verapamil as follows: +50 μm verapamil, 0.33 ± 0.03 μm TNP-ATP; +100 μm verapamil, 0.35 ± 0.01 μm TNP-ATP; +200 μm verapamil, 0.31 ± 0.01 μm TNP-ATP), and the maximum TNP-ATP binding (_B_max) was 119.3 ± 6.1 a.u. in the absence of verapamil; +50 μm verapamil, 116.2 ± 5.6 a.u.; +100 μm verapamil, 128.0 ± 8.0 a.u.; +200 μm verapamil, 125.7 ± 3.4 a.u. (Fig. 7a). The effects of lipid A on TNP-ATP binding could not be tested due to the direct quenching of TNP fluorescence by lipid A.
FIGURE 7.

Effect of substrate binding on the kinetics of ATP binding and hydrolysis. a, binding curves (with _R_2 values >0.99) and binding parameters (Kd and _B_max, see text for details) of purified WT MsbA for the fluorescent analog of ATP, TNP-ATP, were unaffected by the inclusion of increasing amounts of verapamil up to 200 μm (n = 3, mean ± S.E.). b, lipid A (100 μg/ml)-stimulated ATPase activity of MsbA-cl was measured over a range of ATP concentrations, after which the data were fitted to a hyperbola (_R_2 values for both curves were >0.99). An equal volume of DMSO was used as the solvent control in the absence of lipid A. The stimulation of ATPase activity was due to a significant (p < 0.03) ∼1.24-fold increase in the maximum rate of hydrolysis (_V_max), with no observable changes in the apparent affinity for ATP (Km) (see text for parameter details; mean ± S.E., n = 3; error bars, where not visible, are hidden within the symbols).
We further assessed the effect of substrate binding on the kinetics of ATP hydrolysis by MsbA. To study this, we added a fixed concentration of lipid A or an equal volume of DMSO (solvent control) to purified MsbA-cl in Mg2+-containing buffer, with increasing concentrations of ATP. Hyperbolic fits could be generated from the resultant data for ATPase activity against ATP concentration (Fig. 7b). The plots revealed that lipid A-stimulated ATPase activity of MsbA was the result of a significant increase (∼1.24-fold, p < 0.03) in the maximum rate of ATP hydrolysis (_V_max = 229.1 ± 1.1 nmol/min/mg in presence of lipid A versus 191.1 ± 9.4 nmol/min/mg in presence of DMSO (solvent control)), whereas the apparent affinity for ATP in the hydrolysis reaction was unaltered (Km = 1.24 ± 0.05 mm ATP in presence of lipid A versus 1.31 ± 0.17 mm ATP in presence of DMSO) (Fig. 7b). Analogous to the observations for lipid A, verapamil (10 μm) stimulated the ATP activity, but the maximum stimulation of 1.13-fold was too low to allow a detailed kinetic analysis.
The TNP-ATP binding data on MsbA with verapamil and the ATPase results on MsbA with lipid A collectively led us to conclude that substrate binding to MsbA did not alter the transporter's affinity to bind ATP but enhanced the maximum rate of ATP hydrolysis. These observations are in agreement with those reported previously for ABCB1 (32–34).
DISCUSSION
The current alternating access model for substrate transport does not yet explain a key observation made for many ABC exporters that transported substrates stimulate the rate of ATP hydrolysis. To investigate this further, we assessed the conformational changes at the NBDs and MDs following substrate binding by the prototypical multidrug/lipid A ABC exporter MsbA. In addition, we studied the effects of substrate binding to MsbA on the kinetics of ATP binding and hydrolysis.
We show that substrate binding by MsbA stimulates the maximum rate of ATP hydrolysis by facilitating the dimerization of NBDs in an overall conformation of the MsbA dimer that is markedly distinct from the previously described nucleotide-free, inward-facing open and nucleotide-bound, outward-facing conformations of ABC exporters. This new state of MsbA might share features with the inward-facing closed conformation depicted by an x-ray crystal structure of Vibrio cholerae MsbA, in which both the MDs and NBDs are in closer proximity (Fig. 8) (7). The physiological relevance of this inward-facing closed conformation was not clarified, but it was proposed to act as an intermediate between the inward-facing open state and outward-facing state (7). This suggestion is supported by our finding that substrate binding to MsbA enhances the maximum rate of ATP hydrolysis without changing the binding affinity for the nucleotide. Inward-facing closed, pre-translocation conformations that are stabilized by substrate binding were also recently observed for the ABC importer MalFEGK2 (35) and the sodium-coupled hydantoin transporter Mhp1 (36).
FIGURE 8.
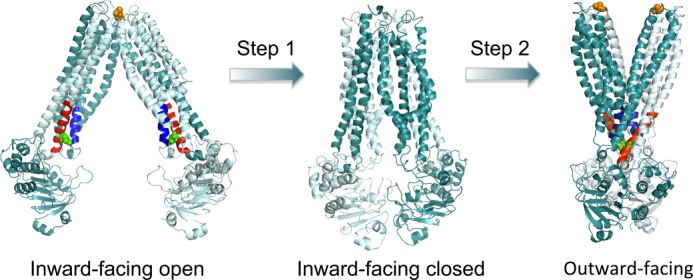
Proposed conformational changes of the MsbA dimer in the transition from inward-facing to outward-facing. Current mechanistic models for substrate transport by the MsbA dimer suggest substrate binding to the inward-facing conformation of dimeric MsbA (Protein Data Bank code 3B5W, full model) and its transition to the ATP-bound, outward-facing structure (Protein Data Bank code 3B60) (7, 21). The two A281C residues (in orange) are in close proximity in the inward-facing open state, whereas the two E208C residues (in green) are close in the outward-facing state. Our analyses suggest that substrate binding to MsbA in step 1 stabilizes an intermediate state that precedes the outward-facing conformation. In this intermediate state, both pairs of A281C/A281C′ and E208C/E208C′ residues are in close proximity. ATP binding to this intermediate state in step 2 switches MsbA into the outward-facing conformation by allowing the formation of stabilizing tetrahelix bundle interactions (helices in blue and red) (14). ATP hydrolysis is then required to resolve the outward-facing conformation back to an inward-facing conformation. The substrate-bound intermediate with dimerized NBDs as in the outward-facing state, but without separation of the MDs at the external side, might share similarities with the crystal structure of inward-facing closed MsbA (Protein Data Bank code 3B5X) (7), although the resolution of this structure is too low to allow accurate predictions of Cys-Cys cross-linking.
The increased proximity of the reporter residues E208C and E208C′ in the MsbA-cl dimer that is associated with the substrate binding-induced NBD closure (this work) was also observed in response to ATP binding (14). This earlier study on E. coli MsbA focused on the role of the tetrahelix bundle that is formed by the cytoplasmic extensions of TMs 3 and 4 and that contains Glu-208 and Glu-208′. Disruption of critical inter-monomer interactions by alanine substitution mutations in the tetrahelix bundle did not significantly affect the binding affinity for nucleotides or Hoechst 33342 (14). However, this disruption strongly inhibited the maximum rate of substrate transport and ATP hydrolysis due to impaired formation of the outward-facing state (14). Taken together, these published findings and our current observations suggest that substrate binding to MsbA stabilizes an intermediate state (an inward-facing closed conformation) that precedes the outward-facing conformation. ATP binding to this intermediate state switches MsbA into the outward-facing conformation through the formation of stabilizing tetrahelix bundle interactions (Fig. 8). ATPase activity is then required to resolve the outward-facing conformation back to inward-facing.
In conclusion, substrate binding to MsbA stimulates progression of the catalytic cycle by promoting the formation of an inward-facing closed, pre-translocation state that binds ATP.
*
This work was supported in part by the Biotechnology and Biological Sciences Research Council.
3
The abbreviations used are:
ABC
ATP-binding cassette
MD
membrane domain
NBD
nucleotide-binding domain
TM
transmembrane helix
CuPhen
copper phenanthroline
TNP-ATP
2′,3′-_O_-(2,4,6-trinitrophenyl)adenosine-5′-triphosphate tetra(triethylammonium)
AMP-PNP
adenosine 5′-(β,γ-imido)triphosphate)
Vi
sodium orthovanadate
NEM
_N_-ethylmaleimide
ISOV
inside-out membrane vesicle
a.u.
arbitrary unit.
REFERENCES
- 1.Gutmann D. A., Ward A., Urbatsch I. L., Chang G., van Veen H. W. (2010) Understanding polyspecificity of multidrug ABC transporters: closing in on the gaps in ABCB1. Trends Biochem. Sci. 35, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velamakanni S., Yao Y., Gutmann D. A., van Veen H. W. (2008) Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry 47, 9300–9308 [DOI] [PubMed] [Google Scholar]
- 3.Woebking B., Reuter G., Shilling R. A., Velamakanni S., Shahi S., Venter H., Balakrishnan L., van Veen H. W. (2005) Drug-lipid A interactions on the Escherichia coli ABC transporter MsbA. J. Bacteriol. 187, 6363–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Veen H. W., Venema K., Bolhuis H., Oussenko I., Kok J., Poolman B., Driessen A. J., Konings W. N. (1996) Multidrug resistance mediated by a bacterial homolog of the human multidrug transporter MDR1. Proc. Natl. Acad. Sci. U.S.A. 93, 10668–10672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reuter G., Janvilisri T., Venter H., Shahi S., Balakrishnan L., van Veen H. W. (2003) The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem. 278, 35193–35198 [DOI] [PubMed] [Google Scholar]
- 6.Doerrler W. T., Reedy M. C., Raetz C. R. (2001) An Escherichia coli mutant defective in lipid export. J. Biol. Chem. 276, 11461–11464 [DOI] [PubMed] [Google Scholar]
- 7.Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borbat P. P., Surendhran K., Bortolus M., Zou P., Freed J. H., Mchaourab H. S. (2007) Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 5, e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou P., McHaourab H. S. (2009) Alternating access of the putative substrate-binding chamber in the ABC transporter MsbA. J. Mol. Biol. 393, 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou P., Bortolus M., McHaourab H. S. (2009) Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J. Mol. Biol. 393, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellmich U. A., Lyubenova S., Kaltenborn E., Doshi R., van Veen H. W., Prisner T. F., Glaubitz C. (2012) Probing the ATP hydrolysis cycle of the ABC multidrug transporter LmrA by pulsed EPR spectroscopy. J. Am. Chem. Soc. 134, 5857–5862 [DOI] [PubMed] [Google Scholar]
- 12.Mehmood S., Domene C., Forest E., Jault J.-M. (2012) Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc. Natl. Acad. Sci. U.S.A. 109, 10832–10836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doshi R., Woebking B., van Veen H. W. (2010) Dissection of the conformational cycle of the multidrug/lipidA ABC exporter MsbA. Proteins 78, 2867–2872 [DOI] [PubMed] [Google Scholar]
- 14.Doshi R., Ali A., Shi W., Freeman E. V., Fagg L. A., van Veen H. W. (2013) Molecular disruption of the power stroke in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 288, 6801–6813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper R. S., Altenberg G. A. (2013) Association/dissociation of the nucleotide-binding domains of the ATP-binding cassette protein MsbA measured during continuous hydrolysis. J. Biol. Chem. 288, 20785–20796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doshi R., Gutmann D. A., Khoo Y. S., Fagg L. A., van Veen H. W. (2011) The choreography of multidrug export. Biochem. Soc. Trans. 39, 807–811 [DOI] [PubMed] [Google Scholar]
- 17.Senior A. E., al-Shawi M. K., Urbatsch I. L. (1995) The catalytic cycle of P-glycoprotein. FEBS Lett. 377, 285–289 [DOI] [PubMed] [Google Scholar]
- 18.Higgins C. F., Linton K. J. (2004) The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926 [DOI] [PubMed] [Google Scholar]
- 19.Szabó K., Welker E., Bakos, Müller M., Roninson I., Váradi A., Sarkadi B. (1998) Drug-stimulated nucleotide trapping in the human multidrug transporter MDR1. J. Biol. Chem. 273, 10132–10138 [DOI] [PubMed] [Google Scholar]
- 20.Eckford P. D., Sharom F. J. (2008) Functional characterization of Escherichia coli MsbA: Interaction with nucleotides and substrates. J. Biol. Chem. 283, 12840–12850 [DOI] [PubMed] [Google Scholar]
- 21.Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnaud O., Koubeissi A., Ettouati L., Terreux R., Alamé G., Grenot C., Dumontet C., Di Pietro A., Paris J., Falson P. (2010) Potent and fully noncompetitive peptidomimetic inhibitor of multidrug resistance P-glycoprotein. J. Med. Chem. 53, 6720–6729 [DOI] [PubMed] [Google Scholar]
- 23.Liu R., Sharom F. J. (1996) Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry 35, 11865–11873 [DOI] [PubMed] [Google Scholar]
- 24.Loo T. W., Bartlett M. C., Clarke D. M. (2003) Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. J. Biol. Chem. 278, 13603–13606 [DOI] [PubMed] [Google Scholar]
- 25.Loo T. W., Bartlett M. C., Clarke D. M. (2003) Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 278, 1575–1578 [DOI] [PubMed] [Google Scholar]
- 26.Siarheyeva A., Sharom F. J. (2009) The ABC transporter MsbA interacts with lipid A and amphipathic drugs at different sites. Biochem. J. 419, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G., Pincheira R., Zhang J.-T. (1998) Dissection of drug-binding-induced conformational changes in P-glycoprotein. Eur. J. Biochem. 255, 383–390 [DOI] [PubMed] [Google Scholar]
- 28.Woebking B., Velamakanni S., Federici L., Seeger M. A., Murakami S., van Veen H. W. (2008) Functional role of transmembrane helix 6 in drug binding and transport by the ABC transporter MsbA. Biochemistry 47, 10904–10914 [DOI] [PubMed] [Google Scholar]
- 29.Booth I. R., Edwards M. D., Black S., Schumann U., Miller S. (2007) Mechanosensitive channels in bacteria: signs of closure? Nat. Rev. Microbiol. 5, 431–440 [DOI] [PubMed] [Google Scholar]
- 30.Li-Blatter X., Nervi P., Seelig A. (2009) Detergents as intrinsic P-glycoprotein substrates and inhibitors. Biochim. Biophys. Acta 1788, 2335–2344 [DOI] [PubMed] [Google Scholar]
- 31.Infed N., Hanekop N., Driessen A. J., Smits S. H., Schmitt L. (2011) Influence of detergents on the activity of the ABC transporter LmrA. Biochim. Biophys. Acta 1808, 2313–2321 [DOI] [PubMed] [Google Scholar]
- 32.Scarborough G. (1995) Drug-stimulated ATPase activity of the human P-glycoprotein. J. Bioenerg. Biomembr. 27, 37–41 [DOI] [PubMed] [Google Scholar]
- 33.Ambudkar S. V., Lelong I. H., Zhang J., Cardarelli C. O., Gottesman M. M., Pastan I. (1992) Partial purification and reconstitution of the human multidrug-resistance pump: characterization of the drug-stimulatable ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 89, 8472–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.al-Shawi M. K., Senior A. E. (1993) Characterization of the adenosine triphosphatase activity of Chinese hamster P-glycoprotein. J. Biol. Chem. 268, 4197–4206 [PubMed] [Google Scholar]
- 35.Oldham M. L., Chen J. (2011) Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332, 1202–1205 [DOI] [PubMed] [Google Scholar]
- 36.Shimamura T., Weyand S., Beckstein O., Rutherford N. G., Hadden J. M., Sharples D., Sansom M. S., Iwata S., Henderson P. J., Cameron A. D. (2010) Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]