Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis (original) (raw)
. Author manuscript; available in PMC: 2013 Aug 5.
Published in final edited form as: Oncogene. 2007 Aug 27;27(9):1231–1242. doi: 10.1038/sj.onc.1210738
Abstract
Cyclin D1 levels are maintained at steady state by phosphorylation-dependent nuclear export and polyubiquitination by the SCFFBX4-αB crystallin E3 ligase. Inhibition of cyclin D1 proteolysis has been implicated as a causative factor leading to its overexpression in carcinomas of the breast and esophagus; however evaluation of the contribution of stable cyclin D1 to the genesis of such carcinomas has not been performed. We therefore generated transgenic mice wherein expression of either wild-type or a stable cyclin D1 allele (D1T286A) is regulated by MMTV-LTR. MMTV-D1T286A mice developed mammary adenocarcinomas at an increased rate relative to MMTV-D1 mice. Similar to human cancers that overexpress cyclin D1, D1T286A tumors were estrogen receptor positive and exhibited estrogen-dependent growth. MMTV-D1T286A tumors specifically overexpressed genes involved in DNA replication and DNA damage checkpoints suggesting that stabilization and nuclear accumulation of cyclin D1-dependent kinase contributes to genomic instability through perturbations in DNA replication. Collectively, these results suggest that temporal control of cyclin D1 subcellular localization and proteolysis is critical for the maintenance of homeostasis within the mammary epithelium.
Keywords: αB crystallin, CDK4, cyclin D1, FBX4, mammary gland
INTRODUCTION
Uncontrolled cellular proliferation is a hallmark of cancer. Cyclin D1, together with its catalytic partner CDK4/6, promotes G1-S phase transition via site-specific phosphorylation of the retinoblastoma family members and through stoichiometric titration to cell cycle inhibitors p27Kip1 and p21Cip1. These functions of cyclin D1 ensure efficient initiation of DNA synthesis (S phase) (Cheng et al., 1998; Harbour et al., 1999; Sherr & Roberts, 1999). During late G1 and S phases, cyclin D1 is phosphorylated at thr-286 (p-286) by GSK3β, which triggers two events (Diehl et al., 1998). First, p-286 cyclin D1 is targeted by the nuclear exportin, CRM1, thus resulting in cyclin D1 nuclear export (Alt et al., 2000). Second, once in the cytoplasm, the E3 ubiquitin ligase, SCFFBX4-αB crystallin, binds to p-286 cyclin D1 and targets it for ubiquitin-mediated proteasomal degradation (Lin et al., 2006). Subversion of either event results in uncontrolled cellular proliferation and altered cellular homeostasis (Alt et al., 2000; Benzeno & Diehl, 2004; Benzeno et al., 2006; Lin et al., 2006).
Cyclin D1 plays an essential role in mammary gland development and carcinogenesis thereby making it an attractive target for human breast cancer. The mammary epithelial compartment of cyclin D1 null mice fails to undergo the proliferative burst associated with pregnancy, and as a result, cyclin D1 null female mice are unable to nurse their pups (Sicinski et al., 1995). Additionally, cyclin D1 null mice are resistant to mammary tumorigenesis induced by erbB2 and Ras revealing a requirement for cyclin D1 downstream of these oncogenes (Yu et al., 2001). The necessity of cyclin D1 in erbB2 and Ras-dependent mammary carcinogenesis, together with the recent demonstration that cyclin D1-dependent tumorigenesis depends upon its activation of CDK4, suggests that pharmacological inhibitors of the cyclin D1-CDK4 kinase could be effective therapeutic agents for human breast cancer (Landis et al., 2006; Yu et al., 2006). Since cyclin D1 is overexpressed in approximately 50% of human breast carcinomas (Butt et al., 2005), development of CDK4 inhibitors could prove significant in efforts to treat breast cancer patients. In general, human breast cancers that overexpress cyclin D1 are estrogen receptor positive indicating that targeting both cyclin D1-associated kinases and the estrogen receptor may be an effective therapeutic option for breast cancer (Buckley et al., 1993; Butt et al., 2005; Utsumi et al., 2000).
Wild-type cyclin D1 is weakly oncogenic; this likely reflects the capacity of cells to efficiently maintain threshold levels of active, nuclear cyclin D1/CDK4 through phosphorylation-dependent nuclear export and degradation. Indeed, mice expressing wild-type cyclin D1 in the mammary epithelium under control of the MMTV-LTR promoter develop mammary adenocarcinomas with a protracted latency while expression in lymphocytes via the Eμ-enhancer does not elicit a tumorigenic phenotype demonstrating that cyclin D1 is a weak oncogene in vivo (Bodrug et al., 1994; Wang et al., 1994). In contrast, overexpression of the constitutive nuclear, non-degradable cyclin D1T286A mutant results in cellular transformation of NIH-3T3 fibroblasts in vitro and of lymphocytes in vivo (Alt et al., 2000; Gladden et al., 2006). These studies suggest that nuclear export and cytoplasmic proteolysis reduce the oncogenicity of cyclin D1.
To evaluate the role of constitutively nuclear and non-degradable cyclin D1 molecules in mammary cancer development, we generated transgenic mice expressing either wild-type cyclin D1 or a phosphorylation-deficient cyclin D1 mutant (D1T286A) under the control of the MMTV-LTR promoter. In this study, we provide evidence demonstrating that cyclin D1 phosphorylation, localization and ubiquitination are regulated events in mammary epithelium in vivo. We also demonstrate that disruption of normal cyclin D1 phosphorylation, nuclear export and cytoplasmic proteolysis accelerates mammary carcinogenesis, thus indicating that a nuclear, non-degradable cyclin D1 allele is a more potent oncogene than wild-type cyclin D1 in mammary epithelium in vivo.
RESULTS
Regulation of cyclin D1 thr-286 phosphorylation, subcellular localization and ubiquitination in mammary epithelium in vivo
We assessed the proliferative status of the mammary epithelium relative to cyclin D1 phosphorylation, subcellular localization and total cyclin D1 protein levels to gain a better understanding of cyclin D1 regulation in mammary epithelium. Immunohistochemical staining revealed that total cyclin D1 was intensely nuclear in epithelial cells during pregnancy, a period of mammary epithelial cell proliferation (Fig. 1A, panel i, solid arrows; Fig. 1B, top panel). Tertiary ducts (Fig. 1A, panel i, hollow arrows) contained less nuclear cyclin D1 staining compared to proliferative alveolar cells (Fig. 1A, panel i, solid arrows). In contrast, during lactation, some differentiated alveolar cells retained weak nuclear staining for total cyclin D1 (Fig. 1A, panel ii, solid arrow). However, overall cyclin D1 levels were reduced and largely cytoplasmic during lactation (Fig. 1A, panel ii, hollow arrow; Fig. 1B, bottom panel), a state of non-proliferation and terminal differentiation, suggesting a high rate of phosphorylation-dependent nuclear export and proteolysis during this stage. Indeed IHC staining with a p-286 specific antibody revealed weak cytoplasmic phospho-cyclin D1 staining during pregnancy (Fig. 1A, panel iii), and increased cytoplasmic staining during lactation (Fig. 1A, panel iv). This is consistent with in vitro analysis suggesting p-286 promotes cytoplasmic localization of cyclin D1 (Alt et al., 2000).
Figure 1. Cyclin D1 thr-286 phosphorylation, subcellular localization and protein levels are developmentally regulated in mammary epithelium in vivo.
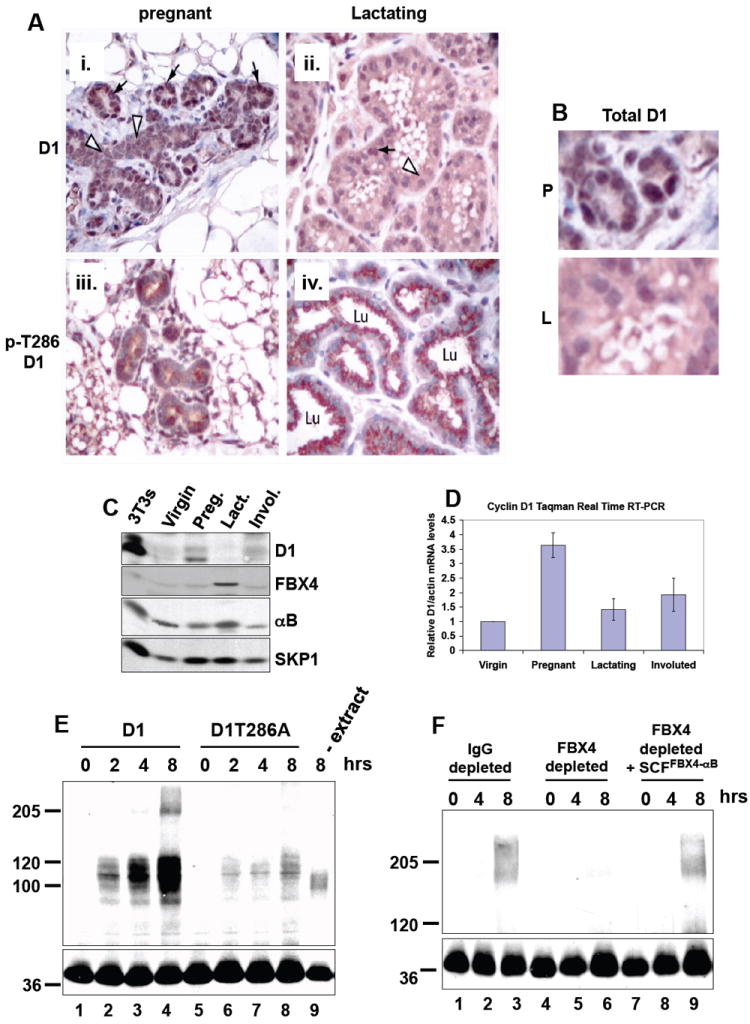
(A) IHC staining for total cyclin D1 at day 7 of pregnancy (i) and at lactation (ii) or staining with a phospho-specific antibody for cyclin D1 phosphorylated at Thr-286 at day 7 of pregnancy (iii) or during lactation (iv). (B) High magnification of total D1 staining during pregnancy (P) and lactation (L). (C) Total cyclin D1, FBX4 and αB crystallin levels are regulated during mammary gland development. Lysates were prepared from age-matched mammary glands of virgin, pregnant (day 10), lactating (day 10) and involuted (day 15) female, wild-type mice and blotted with the indicated antibodies. (D) Cyclin D1 mRNA levels during mammary gland development. (E and F) Cyclin D1 in vitro ubiquitination assays with lactating (day 10) mammary gland extracts.
We confirmed increased accumulation of cyclin D1 during pregnancy and loss of cyclin D1 during lactation by western analysis of whole mammary gland extracts (Fig. 1C). Cyclin D1 p-286 phosphorylation and cytoplasmic localization inversely correlated with total cyclin D1 levels (Fig. 1A, 1B and 1C) indicating that phosphorylation of cyclin D1 induces its nuclear export and proteolysis in mammary epithelial cells in vivo. Because the SCFFBX4-αB crystallin ubiquitin ligase regulates phosphorylation-dependent proteolysis of cyclin D1 (Lin et al., 2006), we assessed the role of FBX4 and αB crystallin in the regulation of cyclin D1 in mammary epithelium. Cyclin D1, FBX4 and αB crystallin protein levels were assessed in whole tissue extracts prepared from age-matched virgin, pregnant, lactating and involuted mammary glands (Fig. 1C). Cyclin D1 levels were low in the virgin glands, increased during pregnancy, and subsequently decreased during lactation and involution (Fig. 1C). Cyclin D1 mRNA levels were also high during pregnancy revealing that cyclin D1 transcription contributes to its accumulation during mammary gland development (Fig. 1D). Strikingly, FBX4 and αB crystallin protein levels were specifically high in the lactating mammary gland as compared to virgin, pregnant and involuted mammary glands (Fig. 1C). Collectively, these results indicate that cyclin D1 is regulated by thr-286 phosphorylation, nuclear export and ubiquitination by the SCFFBX4-αB crystallin complex in the mammary epithelial cells in vivo.
To directly address the role of p-286 for cyclin D1 ubiquitination in the mammary gland, we prepared ubiquitination competent extracts from lactating mammary glands. Robust in vitro ubiquitination cyclin D1 was observed in a time and p-286-dependent manner (Fig. 1E, lanes 1-4 vs. 5-8). Ubiquitinating activity was dependent on mammary gland extracts since omission of extracts inhibited cyclin D1 ubiquitination (Fig. 1E, lane 9). Cyclin D1 ubiquitination was also inhibited by performing the reaction at 4 degrees, chelating magnesium with EDTA, or replacing ATP with an ATP inhibitor (AMP-PNP) (data not shown).
To determine whether the SCFFBX4-αB crystallin ubiquitin ligase is required for p-286-dependent ubiquitination of cyclin D1, we immunodepleted FBX4 from mammary gland extracts and assessed cyclin D1 ubiquitinating activity. Eighty percent depletion of FBX4 from lactating mammary gland extracts (data not shown) significantly abrogated cyclin D1 ubiquitination (Fig. 1F, lanes 1-3 vs. lanes 4-6). Importantly, reconstitution of FBX4-depleted extracts with purified SCFFBX4-αB crystallin complexes prepared from Sf9 insect cells restored cyclin D1 ubiquitination demonstrating the specificity of the ubiquitinating activity (Fig. 1F, lanes 7-9). Collectively, these data demonstrate that thr-286 phosphorylation, nuclear export and ubiquitination by the SCFFBX4-αB crystallin ubiquitin ligase are important regulatory mechanisms of cyclin D1 in mammary epithelium in vivo.
Generation of MMTV-D1 and MMTV-D1T286A mice
To determine whether disruption of cyclin D1 proteolysis contributes to the neoplastic potential of cyclin D1 in the mammary gland, we generated transgenic mice harboring either wild type cyclin D1 or the phosphorylation-deficient cyclin D1 mutant, D1T286A, under the control of the MMTV-LTR promoter. Transgenic lines were initiated by breeding founders with non-transgenic FVB mice. Founders exhibiting germline transmission and expression of the transgene were identified. Vector design was identical for both MMTV-D1 and MMTV-D1T286A lines (Fig. 2A). Expression of wild-type cyclin D1 and mutant D1T286A transgenes was confirmed by western analysis of tissue harvested from pregnant and lactating female mice (Fig. 2B, 2C, 2D). Both transgenes were expressed specifically in the mammary gland with slightly greater levels of D1T286A than D1 as expected due to its increased stability. Additionally, both transgenic proteins assembled with CDK4, p27Kip1, p21Cip1 and retained similar kinase activity against the retinoblastoma protein (Fig. 2E and data not shown).
Figure 2. Generation of MMTV-D1 and MMTV-D1T286A transgenic mice.
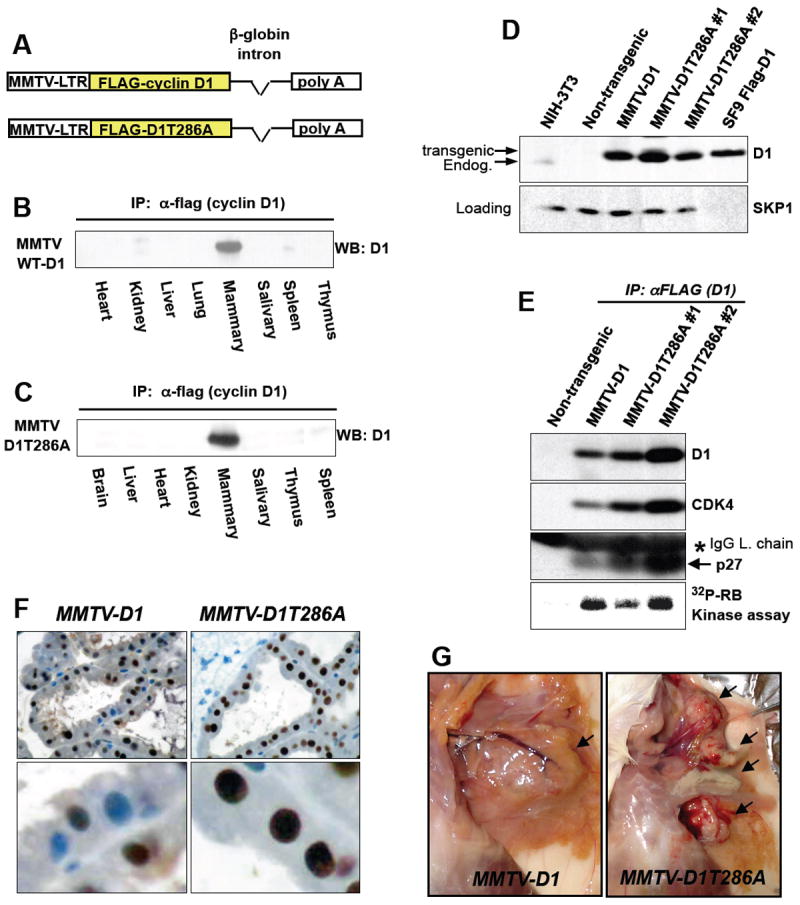
(A) Flag-tagged wild-type cyclin D1 or D1T286A were placed under the regulation of the MMTV-LTR. (B), (C) Lysates were prepared from the indicated tissue of pregnant female mice, precipitated with the anti-flag (M2) antibody and blotted with a cyclin D1 specific antibody (B, MMTV-D1 line; C, MMTV-D1T286A line). (D) Mammary gland lysates were prepared from age and parity matched lactating female mice and probed with the indicated antibodies. (E) Mammary gland lysates from (D) were precipitated with the anti-flag (M2) antibody, blotted with the indicated antibodies or assessed for in vitro kinase activity against RB (bottom). (F) IHC staining for total cyclin D1 at day 10 of lactation of MMTV-D1 (left panels) and MMTV-D1T286A (right panels) mammary glands. (G) Photomicrographs of palpable mammary tumors (arrows) in transgenic animals.
To determine whether disruption of cyclin D1 phosphorylation abrogates cyclin D1 nuclear export in mammary epithelium, we assessed localization of transgenic cyclin D1 protein by IHC during lactation, a period of increased p-286 cyclin D1 (Fig. 1A). During lactation, transgenic wild-type cyclin D1 exhibited cytoplasmic and nuclear staining (Fig. 2F, left panels). In contrast, cyclin D1T286A was predominantly nuclear during lactation, with a greater nuclear intensity than seen with transgenic wild-type D1, indicating that p-286 induces cyclin D1 nuclear export in the lactating mammary gland (Fig. 2F, right panels). Strikingly, MMTV-D1T286A mice first showed signs of hyperplasia and dysplasia during first lactation at 4.5 months of age compared to both non-transgenic and MMTV-D1 mice (data not shown). These results support the notion that phosphorylation of thr-286 triggers nuclear to cytoplasmic shuttling of cyclin D1 in mammary epithelial cells.
Abrogation of cyclin D1 phosphorylation accelerates mammary carcinogenesis
Long-term cohorts of multiparous females from each genotype were established for the assessment of tumor formation over a 24-month period. Females were allowed to undergo at least two pregnancies to maximize expression of the transgenes. Disease manifestation was characterized as the development of palpable tumors that were subsequently confirmed to be mammary carcinomas via histological analysis. MMTV-D1T286A animals developed palpable mammary adenocarcinomas significantly earlier than MMTV-D1 mice (estimated median: 18 months for D1T286A, estimated 95%CI: 16.2-19.8 months vs. 22 months for D1, estimated 95%CI: 20.5-23.5 months; log rank test: p=0.003, Fig. 3A and Table 1). Both transgenic lines developed tumors with approximately the same penetrance. Because tumor penetrance was less than 100% and estimated medians had to be calculated, as an independent assessment of tumor latency, we also determined the mean age of onset for tumor formation from either transgenic line. In contrast to the median, mean age of onset of palpable tumors was 15.8 months for MMTV-D1T286A mice compared to 19.9 months for MMTV-D1 (student’s t-test: p=0.004, Fig. 3B) confirming that disruption of cyclin D1 localization and proteolysis accelerates mammary carcinogenesis.
Figure 3. Perturbation of cyclin D1 localization and proteolysis accelerates mammary carcinogenesis.
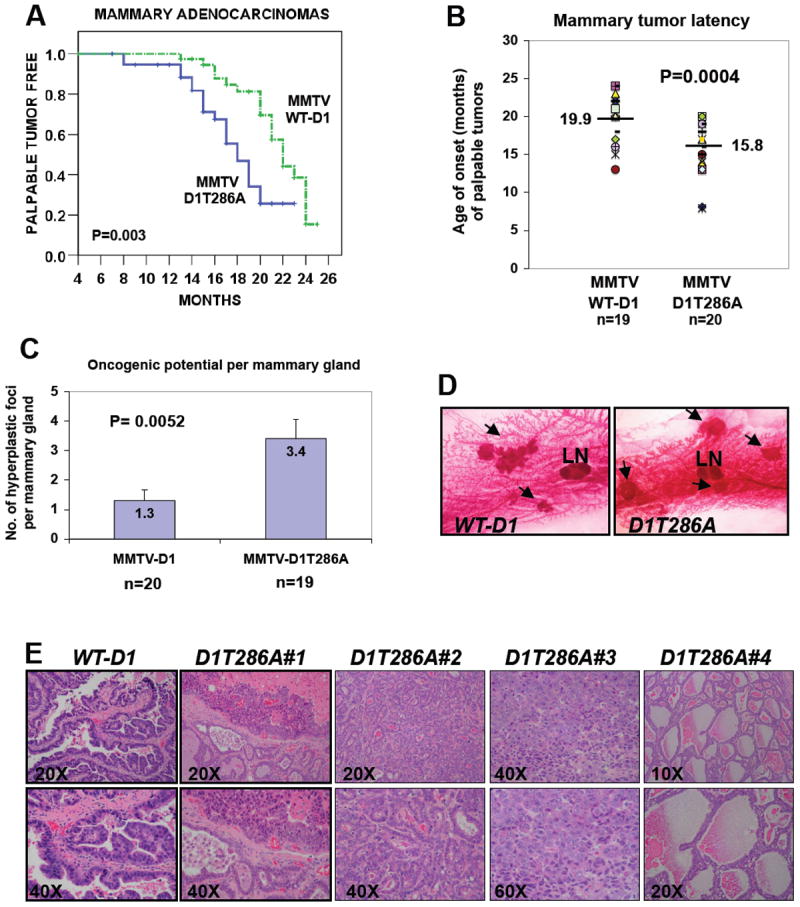
(A) Kaplan-Meier curves for the development of palpable mammary adenocarcinomas in multiparous MMTV-D1 (n=40) and MMTV-D1T286A (n=39) female mice (Log rank Mantel-Cox test: p=0.003). (B) The latency of palpable mammary carcinomas is decreased in MMTV-D1T286A mice (Student’s t-test: p=0.0004). Each marker represents the age of onset of the first palpable mammary carcinoma per mouse. Horizontal bars represent means. (C) One #4 mammary gland per multiparous mouse was harvested at 12 months of age and assessed for hyperplastic foci formation by whole-mount staining. Columns represent means (Student’s t-test: p=0.0052). (D) Representative whole-mount staining of hyperplastic foci (arrows). The intra-mammary lymph node (LN) is included as a size reference. (E, WT-D1) Papillary adenocarcinoma in MMTV-D1 mice. (E, D1T286A#1) MMTV-D1T286A tumor displaying adenocarcinoma of both glandular and solid type with focal necrosis. (E, D1T286A#2) Glandular adenocarcinoma in MMTV-D1T286A mice. (E, D1T286A#3) Adenocarcinoma of the solid type in MMTV-D1T286A mice displaying high nuclear/cytoplasmic ratio, nuclear pleomorphism and hyperchromatism. (E, D1T286A#4) Glandular adenocarcinoma, secretory type, in MMTV-D1T286A mice.
Table 1.
Characterization of mammary carcinomas in MMTV-D1 and MMTV-D1T286A mice.
During the course of our studies, we noted that MMTV-D1T286A mice routinely developed multi-focal tumors (Fig. 2G). Strikingly, the tumors from a single MMTV-D1T286A mouse frequently exhibited distinct histological phenotypes supporting the notion that they reflect independent events (Fig. 3E and data not shown). To directly assess the oncogenic potential of cyclin D1 and D1T286A per mammary gland, we harvested mammary glands from a cohort of approximately 20 multiparous females from each genotype at 12 months of age and assessed hyperplastic foci formation by whole mount analysis. Abrogation of cyclin D1 thr-286 phosphorylation increased the number of multifocal hyperplastic lesions from 1.3 to 3.4 hyperplastic foci per mammary gland (p=0.0004, Fig. 3C and 3D). Together with the acceleration of tumor formation, these results suggest that abrogation of p-286 increases the oncogenic potential of cyclin D1 in mammary epithelium.
Disruption of p-286 alters the histological spectrum of tumors induced by cyclin D1
MMTV-D1 mice typically develop mammary adenocarcinomas of the papillary histological type (Wang et al., 1994). Consistent with this, our MMTV-D1 mice also developed predominantly papillary adenocarcinomas (Fig. 3E and Table 1). In contrast, MMTV-D1T286A mice predominantly developed secretory glandular type adenocarcinomas (Fig. 3E and Table 1). In addition, mice of both genotypes developed a small proportion of acinar and solid types in different ratios (Cardiff et al., 2000) (Fig. 3E and Table 1). In general, tumors of both genotypes appeared aggressive as measured by increased nuclear to cytoplasmic ratio, nuclear pleomorphism, hyperchromatism and layers of disorganized epithelium (Fig. 3E). Both MMTV-D1 and MMTV-D1T286A animals also developed adenosquamous carcinomas, unusual squamous and inflammatory nodules and focal necrotic regions (data not shown). Thus overexpression of both cyclin D1 and D1T286A trigger neoplastic growth. However, the histological ratio of tumors are different demonstrating that perturbation of cyclin D1 nuclear export/proteolysis alters the histological tumor spectrum.
Characterization of MMTV-D1 and MMTV-D1T286A mammary tumors
Most carcinomas of either genotype uniformly expressed cytokeratin 8 (Fig. 4A, tumor A) with occasional tumors also exhibiting expression of cytokeratin 14 (Fig. 4A, tumor B and tumor C). As cytokeratins 8 and 14 are markers of luminal and myoepithelial cells respectively, this implies that cyclin D1 can target a mammary progenitor cell population that gives rise to tumors with both luminal and myoepithelial cell characteristics. Since most human breast carcinomas that overexpress cyclin D1 also express the estrogen receptor, we also determined whether tumors retained expression of estrogen receptor alpha. This analysis revealed that 37.5% D1 and 50% D1T286A tumors retained ERα suggesting that a subset of tumors remained estrogen–dependent (Fig. 4A, tumor D and E). Nuclear staining of uterine epithelial cells and omission of primary antibody served as positive and negative controls respectively (Fig. 4A and data not shown).
Figure 4. Characterization of MMTV-D1 and MMTV-D1T286A mammary tumors.
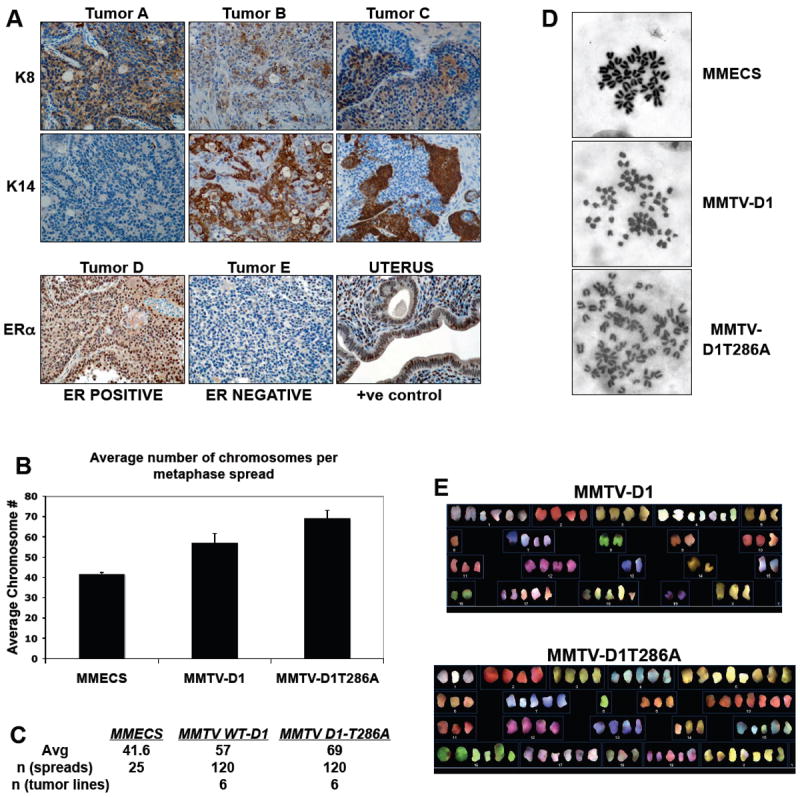
(A) Representative IHC staining for cytokeratin 8 (K8), cytokeratin 14 (K14) and estrogen receptor alpha (ER) in MMTV-D1 and D1T286A tumors. A subset of MMTV-D1 and MMTV-D1T286A display positive staining for cytokeratin 8 (tumor A), cytokeratin 14 (tumors B and C) and ERα (tumor D). (B and C) Average number of chromosomes per metaphase spread in MMTV-D1 and MMTV-D1T286A tumor cells. Metaphase spreads were prepared from early passage tumor derived cell lines. Early passage normal mouse mammary epithelial cells (MECs) served as control. (D) Representative metaphase spreads stained with Giemsa. (E) Representative spectral karyotype (SKY) analysis of MMTV-D1 and MMTV-D1T286A tumor cells.
MMTV-D1T286A tumor cells are aneuploid
Because loss of RB or the CDK4 inhibitor, p16INK4A, induces genetic instability and aneuploidy (Hernando et al., 2004; McDermott et al., 2006), we determined whether nuclear cyclin D1 overexpression by itself induces genomic instability in mammary epithelium. Early passage primary tumor derived cell lines were established and analyzed by staining of metaphase spreads with Giemsa or by spectral karyotyping (SKY). MMTV-D1T286A tumor cells exhibited increased aneuploidy relative to MMTV-D1 tumors cells (Fig. 4B, 4C, 4D). Early passage normal mouse mammary epithelial cells served as controls. Strikingly, no chromosomal translocations were detected by spectral karyotyping (Fig. 4E) suggesting that overexpression of cyclin D1 and D1T286A promotes aneuploidy through random chromosomal gains. These results suggest that overexpression of cyclin D1 promotes genetic instability and that abrogation of cyclin D1 phosphorylation enhances genetic instability of mammary epithelial cells.
The increase in mitotic index and aneuploidy observed in MMTV-D1T286A tumors suggested the potential deregulation of the DNA replication machinery in MMTV-D1T286A tumors. We thus determined whether the expression levels of components of the DNA replication machinery were altered in mammary tumors. Analysis of MMTV-D1 and MMTV-D1T286A tumor tissue extracts for levels of the DNA replication factors revealed overexpression of CDT1 and MCM3 (Fig. 5A; MCM3 is shown as a control). Strikingly, overexpression of CDT1 was largely restricted to tumors expressing D1T286A (2/14, ~14%, of MMTV-D1 vs. 9/11, 81%, of MMTV-D1T286A) (Fig. 5A and Fig. 5B). No changes in CDT1 mRNA levels were observed in the MMTV-D1T286A tumors indicating that CDT1 overexpression likely reflects altered post-translation regulation of CDT1 (Fig. 5C). As CDT1 overexpression promotes re-replication (Vaziri et al., 2003), our results suggest that inhibition of cyclin D1 phosphorylation may promote genomic instability in part through CDT1-dependent re-replication.
Figure 5. CDT1 is overexpressed and p53 is inactivated preferentially in MMTV-D1T286A mammary tumors.
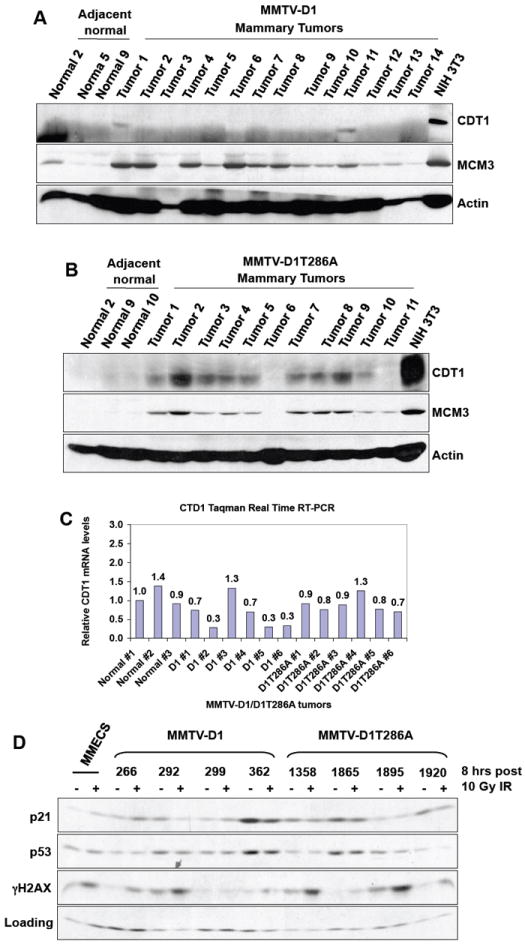
(A) Lysates were prepared from MMTV-D1 tumors and blotted with either CDT1 or MCM3 specific antibodies. NIH3T3 fibroblasts served as positive control and actin served as a loading control. (B) Lysates were prepared from MMTV-D1T286A tumors and blotted as in (A). (C) Total RNA was prepared from MMTV-D1/D1T286A tumors and CDT1 mRNA levels were measured by Taqman Real-Time RT PCR. (D) Mammary tumor derived cell lines were treated with 10Gy of ionizing radiation and allowed to recover for 8 hours. Lysates were prepared and probed with the indicated antibodies.
In other systems, DNA re-replication induced by CDT1 is p53 dependent (Vaziri et al., 2003). For this reason, we assessed whether the p53 pathway was intact in MMTV-D1T286A mammary tumor cells. MMTV-D1T286A tumor derived MECs were irradiated with 10Gy of gamma irradiation. After 8 hours post irradiation, four out of four (100%) MMTV-D1T286A tumor derived MMECs failed to induce the expression of p21 (Fig. 5D), while 2 out of 4 (50%) MMTV-D1 MMECs were able to induce p21 protein after irradiation (Fig. 5D, lines 266, 299). SKP1 levels served as a loading control. Phosphorylation of p53 at ser-18 and of H2AX following irradiation revealed that upstream signaling cascades were intact following DNA damage (Fig. 5D and data not shown). These results indicate that the p53 pathway is disrupted in MMTV-D1T286A tumors thereby eliminating a critical sensor of CDT1-dependent replicative effects.
Combined pharmacologic inhibition of CDK4 and the Estrogen Receptor alpha prevents cancer cell proliferation
As a subset of MMTV-D1 and MMTV-D1T286A tumor cells retained ERα, we determined whether proliferation was estrogen-dependent. Tumor-derived cell lines were treated with 4-hydroxy-tamoxifen and cell cycle distributions were determined through propidium iodide staining and BrdU incorporation. Treatment of two different MMTV-D1T286A tumor-derived cells with increasing concentrations of 4OH-tamoxifen linearly inhibited their proliferation as measured by the percentage of cells in S phase (Fig. 6A and data not shown). Similarly proliferation of primary tumor cells was inhibited in media containing charcoal-dextran stripped serum devoid of any estrogens indicating that MMTV-D1/D1T286A tumor cells were dependent on estrogen for proliferation (data not shown).
Figure 6. Combined pharmacologic inhibition of CDK4/6 and the estrogen receptor has an additive effect on breast cancer proliferation.
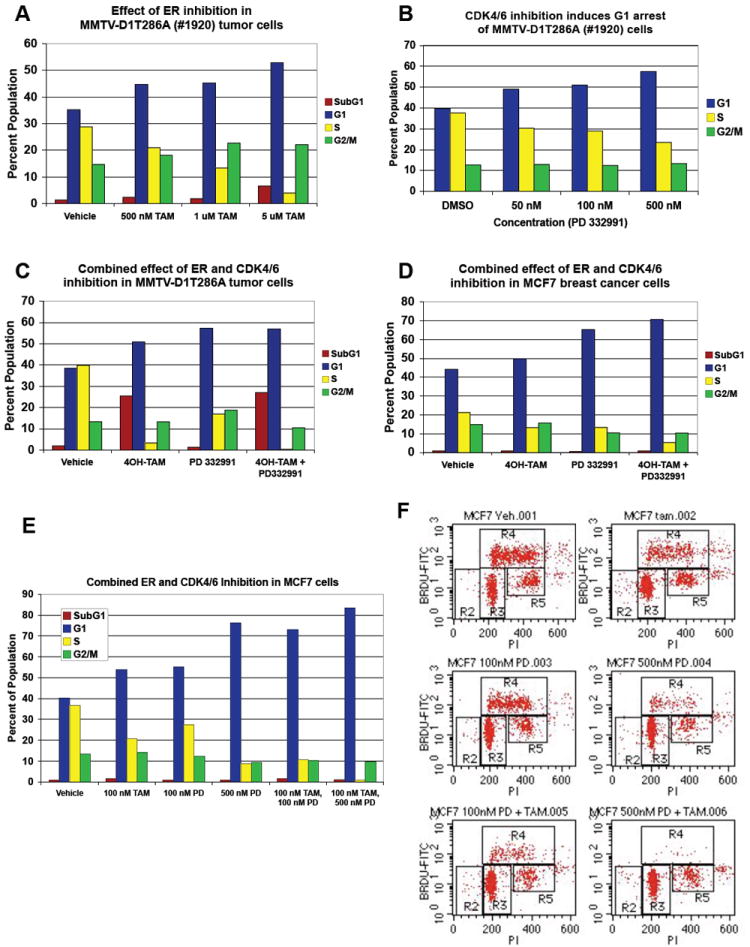
(A) MMTV-D1T286A mammary tumor cells were treated with increasing doses of 4-hydroxy tamoxifen (TAM) for 48 hours. One hour prior to harvesting, cells were labeled with BrdU and cell cycle profiles were assessed by PI-BrdU FACS. (B) MMTV-D1T286A mammary tumor cells were treated with increasing doses of the CDK4/6 inhibitor (PD 0332991) for 48 hours. Cell cycle profiles were assessed as in (A). (C) MMTV-D1T286A mammary tumor cells were treated with both 500 nM of PD 0332991 and 10 μM of 4-OH tamoxifen for 48 hours. Cell cycle profiles were assessed as in (A). (D) Human ERα positive MCF7 breast cancer cells were treated with both 500 nM of PD 0332991 and 100 nM of 4-OH tamoxifen for 48 hours. Cell cycle profiles were assessed as in (A). Representative of three independent experiments are shown.
We next determined whether MMTV-D1 (data not shown) and MMTV-D1T286A (Fig. 6B) cells remained dependent on cyclin D-dependent kinases for growth. Cells were treated with PD 0332991, a specific CDK4/6 inhibitor that is currently in phase I clinical trials for cancer (Fry et al., 2001; Fry et al., 2004). Treatment of MMTV-D1 or MMTV-D1T286A mammary cancer cells with increasing concentrations of PD 0332991 for 48 hours inhibited the percentage of cells in S phase in a dose-dependent manner as measured by PI-BrdU FACS in 5 out of 6 different primary tumor cell lines (Fig. 6B and data not shown). Strikingly, treatment of mammary cancer cells with both 4-OH tamoxifen and PD 0332991 further in inhibited the percentage of cells in S phase indicating that combined pharmacologic inhibition of CDK4/6 and the estrogen receptor has an additive effect on tumor cell proliferation (Fig. 6C). Similar results were obtained by treating the estrogen receptor positive, human breast cancer cell line, MCF7 (Fig. 6D). These results indicate that combined estrogen blockade and CDK4/6 inhibition may be an effective treatment of estrogen receptor positive human breast cancers.
MMTV-D1T286A tumors differentially express genes involved in DNA replication and DNA damage checkpoints
To gain additional mechanistic insight into why inhibition of cyclin D1 phosphorylation accelerates mammary carcinogenesis, we performed a tumor mRNA microarray to compare the expression profiles of MMTV-D1 and MMTV-D1T286A tumors. While on a global scale MMTV-D1 and MMTV-D1T286A tumors were not significantly different from one another (data not shown), we identified several genes involved in distinct biological processes that were differentially expressed in MMTV-D1T286A tumors compared to MMTV-D1 tumors and normal mammary glands (Fig. 7 and data not shown). Because of the genetic instability and overexpression of Cdt1 seen in MMTV-D1T286A tumors, we performed cluster analysis of genes involved in DNA replication and DNA damage checkpoints (Fig. 7 and data not shown). Differential expression of a subset of these genes in MMTV-D1T286A tumors, specifcally Claspin, CDC7, E2F8, DNA ligase 1 and Rpa1, were confirmed by Taqman Real time RT-PCR (Fig. 7). These results suggest that disruption of the normal cytoplasmic localization of cyclin D1 induces DNA replication stress and DNA damage during initiation and progression of mammary carcinogenesis.
Figure 7. DNA replication and DNA damage response genes are differentially expressed in MMTV-D1T286A tumors.
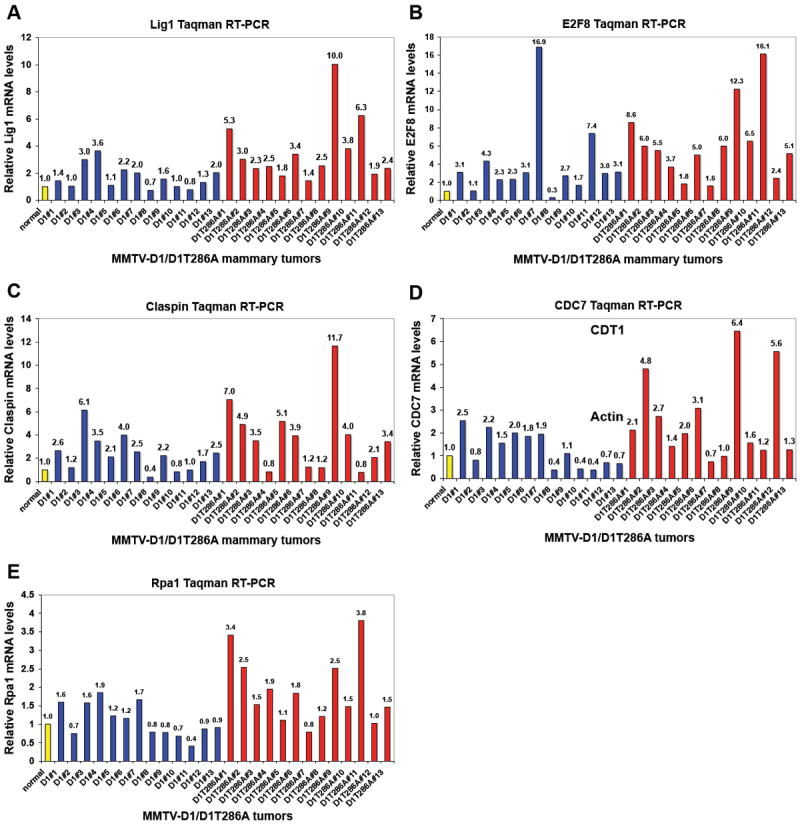
Taqman Real Time RT-PCR analysis of (A) DNA Ligase 1, (B) E2F8, (C) Claspin, (D) CDC7 and (E) Rpa1 in thirteen different MMTV-D1 (blue) and MMTV-D1T286A (red) mammary tumors. mRNA levels were normalized to actin levels and normal mammary glands (yellow).
DISCUSSION
Site-specific phosphorylation has been implicated in the regulation of cyclin D1 degradation and localization in cultured cells. Herein, we demonstrate that, in the mammary gland, p-286 directs cyclin D1 nuclear export and ubiquitination of cyclin D1 by SCFFBX4-αB crystallin. During stages of high epithelial proliferation, cyclin D1 was predominantly nuclear, with weak p-286 staining apparent. In contrast, during lactation, cyclin D1 redistributed to the cytoplasm and exhibited increased in p-286 staining. While IHC revealed an increase in p-286, total cyclin D1 levels actually decreased, suggesting thr-286 phosphorylation triggered cyclin D1 proteolysis. Consistent with these conclusions, cyclin D1T286A remained nuclear through both developmental stages. Furthermore, cyclin D1 was ubiquitinated in a p-286-dependent manner by mammary gland extracts, and ubiquitination towards cyclin D1 was mediated by the SCFFBX4-αB crystallin complex. These results support the notion that the weaker tumor phenotype observed with overexpression of wild-type cyclin D1 may reflect its ability to undergo phosphorylation-dependent nuclear to cytoplasmic shuttling and subsequent FBX4-mediated cytoplasmic proteolysis in mammary epithelial cells.
The concept that decreased proteolysis of cyclin D1 might contribute to its oncogenicity was first inferred from analysis of cyclin D1 in primary breast carcinoma. Here, cyclin D1 is overexpressed in nearly 50% of primary cancers. Paradoxically, gene rearrangements are observed in only approximately 10-30% of these tumors; and it was proposed that decreased proteolysis contributes to the remaining fraction of cancers (Buckley et al., 1993; Butt et al., 2005; Utsumi et al., 2000). Indeed, our work provides direct support for the notion that disruption of cyclin D1 phosphorylation and thus ubiquitin-dependent proteolysis increases the neoplastic potential of cyclin D1 in the mammary gland. First, mice expressing cyclin D1T286A in the mammary epithelium develop mammary adenocarcinomas significantly earlier than mice expressing wild-type cyclin D1. Second, abrogation of cyclin D1 phosphorylation increases the number of multifocal hyperplastic foci lesions per mammary gland. Furthermore, MMTV-D1T286A mice develop mammary tumors with altered tumor histologies and greater genetic instability than MMTV-D1. In addition, tumor mRNA array profiles revealed that D1T286A tumors differentially expressed several genes involved in DNA replication and DNA damage checkpoints compared to D1 tumors suggesting that nuclear, stable cyclin D1 complexes induces genetic instability via perturbations in DNA replication and DNA damage checkpoints. Collectively, these results suggest that disruption of cyclin D1 localization and proteolysis unmasks the neoplastic potential of cyclin D1 in the mammary epithelium.
An important remaining question pertains to whether it is the disruption of cyclin D1 nuclear export or proteolysis that further unmasks the neoplastic potential of cyclin D1 in the mammary gland. Unlike Eμ-D1 mice which do not develop lymphoma (Bodrug et al., 1994; Wang et al., 1994), MMTV-D1 mice develop mammary adenocarcinomas; this result demonstrates that increased cyclin D1 protein levels partly contributes to the oncogenicity of cyclin D1 in mammary epithelium. Consistent with this, loss of specificity components of the cyclin D1 E3 ubiquitin ligase, FBX4 and αB crystallin, occurs in human breast cancers with simultaneous cyclin D1 stabilization again implicating impaired cyclin D1 proteolysis in human breast cancinogenesis (Lin et al., 2006). Critically however, disrupting both nuclear export and proteolysis in MMTV-D1T286A mice further accelerates mammary tumor formation suggesting that altered nuclear export also contributes to the oncogenicity of cyclin D1. Future experiments, in which cyclin D1 nuclear export and proteolysis are uncoupled through targeted deletion of components of the E3 ligase or mutation of ubiquitin-acceptor lysines in cyclin D1, are needed to distinguish the precise contribution of cyclin D1 localization and proteolysis to mammary tumor formation.
It is also important to point out that cyclin D1 may acquire a nuclear gain of function during S phase, due to inhibition of phosphorylation-dependent nuclear export and proteolysis, which could also contribute to the development of additional hits during mammary tumor initiation. Indeed we have observed that MMTV-D1T286A tumors specifically overexpress the DNA replication factor CDT1 suggesting that CDT1 stabilization and DNA re-replication may represent nuclear target. CDT1 is a nuclear DNA replication licensing factor that is targeted for proteasomal degradation by two ubiquitin ligases: SCFSKP2 during S phase and CUL4-DDB1-Cdt2 during both S phase and DNA damage (Arias & Walter, 2006; Jin et al., 2006; Nishitani et al., 2006; Nishitani et al., 2001). Since CDT1 mRNA levels were unchanged in MMTV-D1T286A tumors, we hypothesize that cyclin D1T286A increases the stability of CDT1 protein by a yet unknown mechanism in mammary tumors. Overexpression of both CDT1 and CDC6 can cause DNA re-replication in a p53 dependent manner (Vaziri et al., 2003). Future experiments are warranted to determine whether cyclin D1T286A collaborates with CDT1 in DNA re-replication and mammary tumor formation.
Cyclin D1 is a key mediator of estrogen-dependent proliferation in human breast cancer cells, and human breast cancers that overexpress cyclin D1 are usually estrogen receptor positive (Buckley et al., 1993; Butt et al., 2005). Despite initial responses to the anti-estrogen tamoxifen, human breast cancer patients usually become resistant to anti-estrogen therapies (Robertson et al., 2005). Here we provide evidence that combined pharmacological inhibition of cyclin D dependent kinases CDK4/6, with PD 0332991, and anti-estrogen therapy may be an effective therapeutic option for estrogen receptor positive breast cancer. In addition, the fact that up to 50% of MMTV-D1T286A mice develop estrogen receptor positive mammary tumors indicates that our mouse model may provide an effective tool to study targeted therapies against both the estrogen receptor and cyclin D1-dependent kinases for human breast cancer.
Unlike in esophageal and endometrial carcinomas (Benzeno et al., 2006; Moreno-Bueno et al., 2003), cyclin D1 mutations that abrogate thr-286 phosphorylation have not been found in human breast cancers possibly reflecting the small sample size of screening efforts (Moreno-Bueno et al., 2003). However, overexpression of an alternative splice variant of cyclin D1 that lacks thr-286, cyclin D1b, occurs frequently in human breast carcinomas indicating that nuclear cyclin D1 variants contributes to human breast cancer initiation and progression (Lu et al., 2003; Solomon et al., 2003). In conclusion, we demonstrate that disruption of cyclin D1 nuclear export and proteolysis contributes to the oncogenicity of cyclin D1 in mammary epithelium, and we establish a transgenic mouse model to dissect the mechanisms of mammary oncogenesis induced by constitutively nuclear and non-degradable cyclin D1 molecules.
MATERIALS AND METHODS
Construction of transgenic mice
cDNA encoding FLAG-tagged cyclin D1 and D1T286A were sub-cloned into a transfer vector containing the MMTV-LTR promoter and a rabbit β-globin intron with its endogenous poly-A signal. The MMTV-LTR promoter in this study is the 1.2 kb fragment used in Phil Leder’s and Gilbert Smith’s studies (Hennighausen et al., 1994), which shows less variation in expression between lines and is less dependent on genetic background than the 2.5kb promoter (Rowse et al., 1998). The design for the wild-type cyclin D1 construct is identical to that of the D1T286A transgene. A _Bam_HI digest released the linearized transgene for injection into zygotes from FVB mice. Founder mice were identified by Southern blot of genomic DNA and confirmed by PCR with the following primers: 5’-GGAACAGGAATGCACTTTTGGG-3’ and 5’-CTCACAGACCTCCAGCAT-3’. Founders were subsequently bred to FVB mice to generate MMTV-D1 and MMTV-D1T286A transgenic lines.
Cell Purification and Cell Culture conditions
Normal mouse mammary epithelial cells (MECs) and tumor derived cell lines were purified as previously described (Ip & Asch, 2000). Purified MECs were cultured in DMEM/F12 buffered with HEPES (pH 7.6), 10 μg/ml Insulin, 5 ng/ml EGF, 1 mg/ml BSA fraction V (Sigma), 5 μg/ml linoleic acid complex, antibiotics, and 2% adult calf donor serum (Gibco) (Ip & Asch, 2000). PD 0332991 (Pfizer, Inc.) was dissolved in DMSO at 10 mM and stored at -80 °C in aliquots. Four-hydroxy tamoxifen (Sigma) was dissolved in ethanol.
Immunoprecipitation, CDK kinase assay and Western Blotting
Fresh tissue extracts were prepared and kinase assays were performed as previously described (Diehl & Sherr, 1997; Gladden et al., 2006). The following antibodies were used for western blotting: cyclin D1 (D1-17-13G), CDK4 (Santa Cruz, C-22 or H-22), p21 (Santa Cruz, C-19), p27 (BD Transduction Laboratory, clone 57), p53 (PAB421), CDT1 (generous gift of Dr. Dutta, University of Virginia), MCM3 (N-19, Santa Cruz), FBX4 previously described (Lin et al., 2006), αB crystallin (Stressgen, SPA 223), p53 (pAb 421), γH2AX (Cell Signaling).
In vitro ubiquitination with mammary gland extracts
Mammary glands were lysed in EBC buffer (0.5% NP-40, 120 mM NaCl, 0.1mM PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 25 mM glycerol phosphate, 1 mM NaF and 1 mM DTT), sonicated twice and cleared three times. 50 μg of total extracts were incubated with 20 ng of purified cyclin D1/CDK4 complexes at 37°C in a 15 ul ubiquitination mix containing 50 mM Tric-HCl pH=8, 5 mM MgCl2, 1 mM DTT, 2 mM ATP, 60 μg/ml creatine phosphokinase, 10 mM creatine phosphate and 5 μM ubiquitin. Purified cyclin D1/CDK4 complexes were produced in SF9 cells (Cyclin D1 is readily phosphorylated in SF9 cells). Ubiquitinated cyclin D1 derivatives were detected by direct western blotting.
Immunohistochemistry
Tissues were fixed in 10% buffered formalin. Antigen retrieval was performed with an antigen unmasking solution (Vector Laboratories) followed by blocking of endogenous peroxidase activity with 3% peroxide for 10 min. Sections were incubated in Power Block (Biogenex, CA) for 10 min. The following primary antibodies were used: total cyclin D1 (D1-17-13G), phosho-T286 cyclin D1 previously described (Alt et al., 2000), cytokeratin 8 (TROMA-I, University of Iowa), cytokeratin 14 (Covance), Estrogen Receptor alpha (MC-20, Santa Cruz).
Staining of mammary gland whole mounts
No. 4 mammary glands were fixed with 60% ethanol, 30% chloroform and 10% glacial acetic acid overnight, washed in 70% ethanol for 15 min, washed twice in water for 5 min and stained with 0.2% carmine, 0.5% aluminum potassium sulfate overnight at 4°C. Glands were washed in 70%, 90% and 95% ethanol for 15 min each and in 100% ethanol for 6 hrs. Mammary glands were incubated in xylene overnight and mounted with permount.
Metaphase spreads and spectral karyotyping
Metaphase spreads of early passage, mammary cancer cells were stained with giemsa or by SKY as previously described (Bassing CH, 2003).
Statistical analysis
Kaplan-Meier survival analysis was used to determine tumor latencies, and Log-rank test was performed to determine statistically significant differences.
mRNA microarray and Real Time RT-PCR
Total RNA was prepared with TRIzol followed by RNseasy cleanup. cRNA and hybridization to Affymetrix gene chips were performed by the University of Pennsylvania mRNA microarray core facility according to the manufacture’s instructions. Analysis was performed with Spot fire software. Real Time RT-PCR was performed by the ΔΔCt method with primer/probe master mixes purchased from Applied Biosystems. Target mRNA levels were normalized relative to β-actin levels.
Acknowledgments
We thank Hongwei Yu, the Abramson Histology Core Facility and Robert D. Cardiff for histological assistance; John Tobias for assistance with microarray analysis; and members of the Diehl, Kushner, Bassing and Fuchs labs for critical insight. PD 0332991 was kindly provided by Pfizer. The cytokeratin 8 (TROMA-I) antibody was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa. This work was supported by CA11360 (NIH) and a Leukemia & Lymphoma Scholar award (JAD); the Pew foundation in the Biomedical Sciences and the Department of Pathology of the Children’s Hospital of Philadelphia (CHB).
References
- Alt JR, Cleveland JL, Hannink M, Diehl JA. Genes Dev. 2000;14:3102–14. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias EE, Walter JC. Nat Cell Biol. 2006;8:84–90. doi: 10.1038/ncb1346. [DOI] [PubMed] [Google Scholar]
- Bassing CH, S H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Cell. 2003;114:359–70. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- Benzeno S, Diehl JA. J Biol Chem. 2004;279:56061–6. doi: 10.1074/jbc.M411910200. [DOI] [PubMed] [Google Scholar]
- Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, Klein PS, Rustgi A, Diehl JA. Oncogene. 2006;25:6291–303. doi: 10.1038/sj.onc.1209644. [DOI] [PubMed] [Google Scholar]
- Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM. Embo J. 1994;13:2124–30. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL. Oncogene. 1993;8:2127–33. [PubMed] [Google Scholar]
- Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Endocr Relat Cancer. 2005;12(Suppl 1):S47–59. doi: 10.1677/erc.1.00993. [DOI] [PubMed] [Google Scholar]
- Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE. Oncogene. 2000;19:968–88. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Proc Natl Acad Sci U S A. 1998;95:1091–6. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Sherr CJ. Mol Cell Biol. 1997;17:7362–74. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Bedford DC, Harvey PH, Fritsch A, Keller PR, Wu Z, Dobrusin E, Leopold WR, Fattaey A, Garrett MD. J Biol Chem. 2001;276:16617–23. doi: 10.1074/jbc.M008867200. [DOI] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Mol Cancer Ther. 2004;3:1427–38. [PubMed] [Google Scholar]
- Gladden AB, Woolery R, Aggarwal P, Wasik MA, Diehl JA. Oncogene. 2006;25:998–1007. doi: 10.1038/sj.onc.1209147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cell. 1999;98:859–69. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, McKnight R, Burdon T, Baik M, Wall RJ, Smith GH. Cell Growth Differ. 1994;5:607–13. [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Ip MM, Asch BB. Methods in mammary gland biology and breast cancer research. Kluwer Academic/Plenum Publishers; New York: 2000. [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. Mol Cell. 2006;23:709–21. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cancer Cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Mol Cell. 2006;24:355–66. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Gladden AB, Diehl JA. Cancer Res. 2003;63:7056–61. [PubMed] [Google Scholar]
- McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, Tlsty TD. PLoS Biol. 2006;4:e51. doi: 10.1371/journal.pbio.0040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, Hardisson D, Sarrio D, Prat J, Cigudosa JC, Matias-Guiu X, Palacios J. Oncogene. 2003;22:6115–8. doi: 10.1038/sj.onc.1206868. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, Lygerou Z, Nishimoto T. Embo J. 2006;25:1126–36. doi: 10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Taraviras S, Lygerou Z, Nishimoto T. J Biol Chem. 2001;276:44905–11. doi: 10.1074/jbc.M105406200. [DOI] [PubMed] [Google Scholar]
- Robertson JF, Come SE, Jones SE, Beex L, Kaufmann M, Makris A, Nortier JW, Possinger K, Rutqvist LE. Eur J Cancer. 2005;41:346–56. doi: 10.1016/j.ejca.2004.07.035. [DOI] [PubMed] [Google Scholar]
- Rowse GJ, Ritland SR, Gendler SJ. Cancer Res. 1998;58:2675–9. [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cell. 1995;82:621–30. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Solomon DA, Wang Y, Fox SR, Lambeck TC, Giesting S, Lan Z, Senderowicz AM, Knudsen ES. J Biol Chem. 2003;278:30339–47. doi: 10.1074/jbc.M303969200. [DOI] [PubMed] [Google Scholar]
- Utsumi T, Yoshimura N, Maruta M, Takeuchi S, Ando J, Mizoguchi Y, Harada N. Int J Cancer. 2000;89:39–43. doi: 10.1002/(sici)1097-0215(20000120)89:1<39::aid-ijc7>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, Wagle N, Hwang DS, Dutta A. Mol Cell. 2003;11:997–1008. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Nature. 1994;369:669–71. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P. Nature. 2001;411:1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, Sicinski P. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]