Codon usage between genomes is constrained by genome-wide mutational processes (original) (raw)
Abstract
Analysis of genome-wide codon bias shows that only two parameters effectively differentiate the genome-wide codon bias of 100 eubacterial and archaeal organisms. The first parameter correlates with genome GC content, and the second parameter correlates with context-dependent nucleotide bias. Both of these parameters may be calculated from intergenic sequences. Therefore, genome-wide codon bias in eubacteria and archaea may be predicted from intergenic sequences that are not translated. When these two parameters are calculated for genes from nonmammalian eukaryotic organisms, genes from the same organism again have similar values, and genome-wide codon bias may also be predicted from intergenic sequences. In mammals, genes from the same organism are similar only in the second parameter, because GC content varies widely among isochores. Our results suggest that, in general, genome-wide codon bias is determined primarily by mutational processes that act throughout the genome, and only secondarily by selective forces acting on translated sequences.
Translation of mRNA to protein is universal, and the genetic code describing how the 64 nucleotide triplets (codons) specify 20 amino acids is nearly universal (1). Grantham's genome hypothesis proposes that each species systematically uses certain synonymous codons (codons that code for the same amino acid) in coding sequences (2–4), in other words, that each species has a distinct codon bias. Many studies have since confirmed that, at least in prokaryotes, selective forces acting at the level of translation maintain biased codon usage (5–7). The realization that selection may act on gene sequences in the absence of amino acid changes has had profound implications for the study of the molecular evolution of genes. In particular, analysis of codon bias has helped establish that horizontal gene transfer is a major evolutionary force (8–10).
What causes differences in codon bias and why? Does codon bias exist (i) because it is necessary for efficient and accurate protein expression or (ii) because codons, as DNA sequences, are subject to mutational pressures acting on all the DNA sequences in a given organism? Explanation i is generally termed a selective or selectionist explanation for codon bias. In contrast, explanation ii is referred to as a neutral or mutational explanation. Variation in codon bias among genes from the same organism has been shown to depend on many parameters, including expression level (4, 5, 11), amino acid composition (12–15), gene length (16, 17), mRNA structure (18–20), and protein level noise considerations (21). In most of these cases, evidence exists that selection at different steps during protein expression shapes codon bias. In addition, global forces differentiate the codon bias of genes between different organisms: species-specific codon bias is strongly correlated with overall genome percentage GC content (22, 23), genes from organisms with similar phylogeny or with similar tRNA content have similar codon bias (22), and an organism's optimal growth temperature influences the codon bias of its genes (24). Most of these global forces are thought to be mutational, acting on all DNA sequences, although it has also been argued that growth temperature exerts a selective force on mRNA structure (25) and codon bias (24). Although both selection and mutation are clearly important for establishing codon bias, the relative importance of selection and mutation has been difficult to define in general.
With the recent availability of many complete genome sequences, it has become possible to directly analyze the determinants of genome-wide codon bias. Studying genome-wide codon bias allows us to focus on global forces shaping codon bias. In this article, we examine the relative importance of mutation versus selection in shaping genome-wide codon bias.
Because each of the 20 amino acids, on average, is encoded by three synonymous codons, the space of possible patterns of codon usage is very large. Here we analyze variation in codon bias of genes in archaeal and eubacterial organisms. We introduce a method to quantitatively separate within-genome from between-genome variation in codon bias, and, to analyze global forces shaping codon bias, we focus on between-genome variation. We find, consistent with others, that GC content variation is the most important parameter differentiating codon bias between different organisms. A key finding in our analysis is that a combination of nearest-neighbor nucleotide biases is the next most important parameter differentiating codon bias between different organisms. We demonstrate that genome-wide codon bias in prokaryotic genomes may be predicted with surprising accuracy by using only intergenic sequence statistics, which are unaffected by selective forces acting during protein expression. Furthermore, we find that the codon bias of genes from several nonmammalian eukaryotes is also characterized by genome GC content and nearest-neighbor nucleotide biases. We conclude that genome-wide codon bias can be well characterized by only two parameters, which are determined predominantly by genome-wide mutational forces rather than by coding-region-specific selective forces in all three domains of life.
Materials and Methods
Data Sources. All genome sequences are from GenBank (ftp://ftp.ncbi.nih.gov). A list of genome sequences used is in Data Set 1, which is published as supporting information on the PNAS web site. Some eukaryotic sequences were taken from the RefSeq project (www.ncbi.nlm.nih.gov/RefSeq) on June 5, 2003; only sequences marked “provisional,” “reviewed,” or “validated” were used. Genome sequence processing was done with perl (www.perl.com) with the genome-tools (http://genome-tools.sourceforge.net) (26) and pdl packages (http://pdl.perl.org) by using ad hoc scripts on debian gnu/linux 3.0 (www.debian.org and www.gnu.org). Growth temperature data were taken from the recommended culture conditions of American Type Culture Collection (www.atcc.org) or from ref. 27.
Using the Singular Valve Decomposition (SVD) to Find a Basis for the Space of Codon Vectors. Similar to ref. 22, we represent the codon bias of a gene, i, with a codon vector, ci, with components ci,m(w), where ci,m(w) is the codon frequency of the m(w)th codon (the _w_th codon for amino acid m), normalized for amino acid content (notation used throughout is listed in Table 1). Each c i,m(w) is calculated as 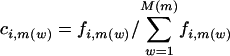 , where f i,m(w) is the number of times the m(w)th codon is used in gene i, and M(m) is the number of synonymous codons that code for amino acid m. The denominator in the calculation of ci,m(w) normalizes for amino acid content in that the sum of the components ci,m(w), which code for the same amino acid, add to 1 regardless of how many times that amino acid is coded for in the gene. Start codons and stop codons are excluded from the calculations of ci,m(w). After excluding stop codons, ci is a 61-dimensional vector. When the genome, g, from which a gene, i, was taken is relevant, it is denoted with a superscript, as in
, where f i,m(w) is the number of times the m(w)th codon is used in gene i, and M(m) is the number of synonymous codons that code for amino acid m. The denominator in the calculation of ci,m(w) normalizes for amino acid content in that the sum of the components ci,m(w), which code for the same amino acid, add to 1 regardless of how many times that amino acid is coded for in the gene. Start codons and stop codons are excluded from the calculations of ci,m(w). After excluding stop codons, ci is a 61-dimensional vector. When the genome, g, from which a gene, i, was taken is relevant, it is denoted with a superscript, as in 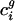 . Because different genomes contain different numbers of genes, we randomly selected N = 400 genes from each of the G = 100 genomes so that each genome had equal weight in the SVD. Altogether, NG = 40,000 genes were selected. We define the mean codon vector for the genes in the study as the mean codon vector of these 40,000 randomly selected genes,
. Because different genomes contain different numbers of genes, we randomly selected N = 400 genes from each of the G = 100 genomes so that each genome had equal weight in the SVD. Altogether, NG = 40,000 genes were selected. We define the mean codon vector for the genes in the study as the mean codon vector of these 40,000 randomly selected genes, 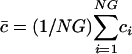 . Let
. Let 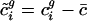 be the mean-centered codon vector for gene i in genome g. Define the matrix
be the mean-centered codon vector for gene i in genome g. Define the matrix
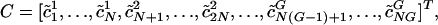 |
[1] |
|---|
where each row is a mean-centered codon vector and the superscript T indicates the transpose of the matrix. Only the 400 randomly selected genes from each genome are used in C.
Table 1. Frequently used symbols.
| m | Index variable for amino acids |
|---|---|
| g | Index variable for genomes |
| m(w) | _w_th codon for amino acid m |
| N | Number of genes selected from each genome for SVD (400) |
| G | Number of prokaryotic genomes used in study (100) |
| ci | Codon vector for gene i |
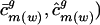 |
Codon vector for gene i from genome g |
| c̄ g | Genome-wide codon bias for genome g; mean of the codon vectors for all genes in g, not just the N used in the SVD |
| c̄ | Mean codon vector for the NG selected genes |
| c̃ i | Mean-centered codon vector for gene i |
| vj | _j_th eigencodon |
| v m(w)j | Component of vj representing codon m(w) |
| σ_j_ | _j_th singular value; global scale factor representing the variance of the selected NG genes in the direction of vj |
| ui,j | Amount of vj, scaled by σ_j_, in c̃ i |
| ū j | Mean usage of σ_j_ vj among the mean-centered codon vectors for all genes in all genomes |
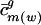 |
Mean of ui,j taken over all genes i from genome g; mean usage of σ_j_ vj among the mean-centered codon vectors for all genes from genome g |
var(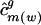 ) ) |
Variance in ui,j among all genes from genome g; variance in usage of σ_j_ vj among the mean-centered codon vectors for all genes from genome g |
| var(uj) | Overall variance in ui,j for all genes, i, among all genomes, g |
| var(uj)within | Within-genome variance in var(uj) |
| var(uj)between | Between-genome variance in var(uj) |
| dg | Vector of intergenic bias parameters for genome g |
We used a thin SVD (28) to decompose C into C = USVT, where UTU = I, VTV = I, S = diag(σ1, σ2,..., σ61), and σ1 ≥ σ2 ≥ · · · ≥σ61 ≥ 0. The matrix V and the singular values σ_j_ are given in Table 3, which is published as supporting information on the PNAS web site. Fig. 1_a_ shows a plot of the σ_j_ for j = 1,..., 61. Because of the normalization for amino acid content, C is not full rank; thus, σ42 = σ43 =... = σ61 = 0 and can be excluded from further consideration. By using the SVD, the mean-centered codon vector for gene i can be written as 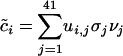 , where ui,j is the component in the i_th row and j_th column of U and vj is the j_th column of V. In other words, each mean-centered codon vector is a weighted sum of the columns of V. More specifically, each column of V, vj, is scaled by two different weights. The first weight, σ_j, is the j_th singular value and can be thought of as a global scale factor. The larger the value of σ_j, the more the codon vectors vary in the direction of vj. The second scalar weight, ui,j, is a gene-specific weight that describes how much vj, scaled globally by σ_j, contributes to c̃i. We refer to the columns of V, {v_1,..., v41}, as eigencodons. Table 2 shows values for v1 and v2. The mean normalized usage of eigencodon vj, i.e., the arithmetic mean of ui,j over all genes (not just the subset used in the SVD) is denoted as ūj and the corresponding variance is denoted as var(uj). The mean and variance of eigencodon vj usage for all genes (not just the subset used in the SVD) within a single genome g are denoted as
, where ui,j is the component in the i_th row and j_th column of U and vj is the j_th column of V. In other words, each mean-centered codon vector is a weighted sum of the columns of V. More specifically, each column of V, vj, is scaled by two different weights. The first weight, σ_j, is the j_th singular value and can be thought of as a global scale factor. The larger the value of σ_j, the more the codon vectors vary in the direction of vj. The second scalar weight, ui,j, is a gene-specific weight that describes how much vj, scaled globally by σ_j, contributes to c̃i. We refer to the columns of V, {v_1,..., v41}, as eigencodons. Table 2 shows values for v1 and v2. The mean normalized usage of eigencodon vj, i.e., the arithmetic mean of ui,j over all genes (not just the subset used in the SVD) is denoted as ūj and the corresponding variance is denoted as var(uj). The mean and variance of eigencodon vj usage for all genes (not just the subset used in the SVD) within a single genome g are denoted as 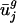 and var(
and var(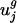 ), respectively.
), respectively.
Fig. 1.

(a) Scree plot of singular values. Singular values (σ_j_) were obtained from a SVD of 400 genes from each of 100 genomes. (b) Contribution of var(uj)between (between-genome variance) to overall variance. Overall variance is scaled to 1 in each dimension. The rest of the overall variance is due to var(uj)within (within-genome variance). In only two dimensions, j = 1 and 2, is var(uj)between the major source of variance.
Table 2. Codon vectors _v_1 and _v_2.
| Codon | _v_1 | _v_2 | Codon | _v_1 | _v_2 | Codon | _v_1 | _v_2 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ala | GCA | –0.088 | 0.077 | Gly | GGA | –0.099 | 0.176 | Pro | CCA | –0.107 | 0.091 |
| GCC | 0.118 | –0.056 | GGC | 0.150 | –0.178 | CCC | 0.076 | 0.030 | |||
| GCG | 0.078 | –0.093 | GGG | 0.014 | 0.022 | CCG | 0.139 | –0.161 | |||
| GCT | –0.108 | 0.071 | GGT | –0.065 | –0.022 | CCT | –0.109 | 0.040 | |||
| Arg | AGA | –0.139 | 0.263 | His | CAC | 0.165 | 0.294 | Ser | AGC | 0.067 | –0.023 |
| AGG | 0.004 | 0.240 | CAT | –0.165 | –0.292 | AGT | –0.062 | 0.002 | |||
| CGA | –0.018 | –0.045 | Ile | ATA | –0.070 | 0.297 | TCA | –0.063 | 0.057 | ||
| CGC | 0.131 | –0.231 | ATC | 0.200 | –0.155 | TCC | 0.064 | 0.006 | |||
| CGG | 0.053 | –0.053 | ATT | –0.130 | –0.138 | TCG | 0.073 | –0.068 | |||
| CGT | –0.031 | –0.176 | Leu | CTA | –0.025 | 0.042 | TCT | –0.079 | 0.026 | ||
| Asn | AAC | 0.187 | 0.134 | CTC | 0.077 | 0.050 | Thr | ACA | –0.106 | 0.084 | |
| AAT | –0.187 | –0.132 | CTG | 0.153 | –0.108 | ACC | 0.144 | –0.100 | |||
| Asp | GAC | 0.174 | 0.064 | CTT | –0.031 | 0.050 | ACG | 0.067 | –0.065 | ||
| GAT | –0.174 | –0.062 | TTA | –0.160 | 0.012 | ACT | –0.105 | 0.080 | |||
| Cys | TGC | 0.196 | –0.162 | TTG | –0.014 | –0.045 | Trp | TGG | 0.000 | 0.000 | |
| TGT | –0.196 | 0.161 | Lys | AAA | –0.181 | –0.079 | Tyr | TAC | 0.167 | 0.240 | |
| Gln | CAA | –0.216 | –0.077 | AAG | 0.181 | 0.089 | TAT | –0.167 | –0.244 | ||
| CAG | 0.217 | 0.051 | Met | ATG | 0.000 | 0.000 | Val | GTA | –0.087 | 0.080 | |
| Glu | GAA | –0.133 | –0.107 | Phe | TTC | 0.212 | 0.064 | GTC | 0.099 | –0.066 | |
| GAG | 0.133 | 0.109 | TTT | –0.211 | –0.070 | GTG | 0.098 | –0.113 | |||
| GTT | –0.110 | 0.100 |
Separation of Within-Genome from Between-Genome Variation. Var(uj) can be decomposed into two parts: (i) variance present within individual genomes [within-genome variance, var(uj)within] and (ii) mean values within genomes that vary from the overall mean [between-genome variance, or var(uj)between]. This can be expressed as:
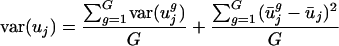 |
[2] |
|---|
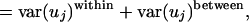 |
[3] |
|---|
where G is the number of genomes considered. For convenience, var(uj)within and var(uj)between will hereafter refer to a fraction of var(uj) [obtained by dividing both sides of the equation above by var(uj)], such that var(uj)within + var(uj)between = 1.
Context-Dependent Intergenic Nucleotide Biases (Intergenic Bias). Context-dependent intergenic nucleotide biases were calculated by using a fixed, second-order Markov model to analyze all intergenic sequences for each of the 100 genomes examined in this study. The frequency of each nucleotide was calculated for each possible combination of nucleotides immediately 5′ and immediately 3′. Taking all intergenic sequences 5′-_N_1_N_2_N_3-3′, where _N_1 and _N_3 are fixed, we calculated the fraction in which _N_2 is G, A, T, or C, which we denote as p(_N_2|_N_1, _N_3). All such intergenic three-nucleotide sequences were included in the calculation, except for the first three and last three nucleotides of each intergenic region. Because Σ_N_2=G,A,T,C p(_N_2|_N_1, _N_3) = 1 for all 16 pairs of _N_1 and _N_3, 64 parameters exist of which 64 - 16 = 48 are linearly independent. In total, this set of 64 nearest-neighbor nucleotide bias parameters calculated from the intergenic sequences of genome g is denoted as dg. For a given organism, g, we will refer to the set of parameters dg as the intergenic bias of that organism.
Least-squares techniques (29) were used to model the average usage of eigencodon v2 in all genes in genome g, denoted 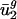 , as a function of dg, the intergenic bias of genome g. We let
, as a function of dg, the intergenic bias of genome g. We let 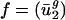 be a vector with G components (one for each genome), and let f̄ be the mean of the components of f. We further let D = [d1... _dG_]T be a matrix with intergenic bias parameters as its rows and let D̃ be a version of D with every column centered about zero. By using a thin SVD, we decomposed D̃ into D̃ = YTXT, where YTY = I, XTX = I, and T = diag(_t_1,..., _t_64), where _t_1 ≥ _t_2 ≥ _t_3 ≥ · · · ≥ _t_64 ≥ 0. The matrix Y and the singular values tj are given in Table 4, which is published as supporting information on the PNAS web site. Because D̃ has rank 48, t_49 = · · · = t_64 = 0, and the first 48 columns of Y form an orthogonal basis for the range of D̃. In a least-squares model, f is approximated as
be a vector with G components (one for each genome), and let f̄ be the mean of the components of f. We further let D = [d1... _dG_]T be a matrix with intergenic bias parameters as its rows and let D̃ be a version of D with every column centered about zero. By using a thin SVD, we decomposed D̃ into D̃ = YTXT, where YTY = I, XTX = I, and T = diag(_t_1,..., _t_64), where _t_1 ≥ _t_2 ≥ _t_3 ≥ · · · ≥ _t_64 ≥ 0. The matrix Y and the singular values tj are given in Table 4, which is published as supporting information on the PNAS web site. Because D̃ has rank 48, t_49 = · · · = t_64 = 0, and the first 48 columns of Y form an orthogonal basis for the range of D̃. In a least-squares model, f is approximated as 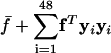 . The larger the (fTyi)2, the greater the amount of variance in f that can be explained by yi. In our case, the yi corresponding to larger singular values, in general, explained more variance than those corresponding to smaller singular values, with y2, y3, and y8 being most critical to the model. To avoid overfitting, we model f using yi for i = 1,..., 8.
. The larger the (fTyi)2, the greater the amount of variance in f that can be explained by yi. In our case, the yi corresponding to larger singular values, in general, explained more variance than those corresponding to smaller singular values, with y2, y3, and y8 being most critical to the model. To avoid overfitting, we model f using yi for i = 1,..., 8.
To determine the quality of the resulting fit, we tested the ability of randomized versions of D to explain f by using models of the same complexity. Specifically, we permuted the entries of D and renormalized the values so that each row satisfied the constraints of a set of intergenic bias parameters. Then D was centered as before and a least-squares fit to f was generated by using the directions corresponding to the eight largest singular values of the centered matrix. The randomization and fitting procedure was repeated 10,000 times. For both the real and randomized data, the quality of the fit was taken to be the fraction of the variance in f explained by the model (the _R_2 statistic).
Results
Codon Bias Varies Between Genomes Primarily Along Two Dimensions. Using a SVD as described above, we decomposed the space of possible codon vectors into 41 orthogonal directions {v1,..., v41}, referred to as eigencodons. The eigencodons are ordered so that gene to gene usage varies most in the direction of eigencodon v1 and least in the v41 direction. Every codon vector can be represented uniquely as a linear combination of the 41 eigencodons. The fraction of var(uj) due to between-genome variance [var(uj)between] is plotted for each eigencodon, vj, in Fig. 1_b._ For j = 1 and 2, between-genome variance accounts for the majority of var(uj) (90% and 62%); for all other vj values, between-genome variance accounts for much less of var(uj) (5–32%). This means that 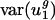 and
and 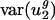 , the within-genome variances, are relatively small for all genomes; thus, for most genes, i, in a given genome, g,
, the within-genome variances, are relatively small for all genomes; thus, for most genes, i, in a given genome, g, 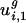 and
and 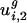 are close to
are close to 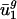 and
and 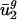 , and
, and 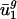 and
and 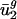 tend to differ for different genomes. In other words,
tend to differ for different genomes. In other words, 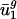 and
and 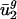 are characteristic values for each genome g because
are characteristic values for each genome g because 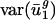 and
and  are small for each genome g. On the other hand, usage of all other eigencodons, vj, for j = 3,..., 41, varies little between genomes compared with its variance among the genes within a genome. Therefore, differences in codon bias between genomes can be reasonably modeled by using only two parameters, average usage of v1 and v2 (
are small for each genome g. On the other hand, usage of all other eigencodons, vj, for j = 3,..., 41, varies little between genomes compared with its variance among the genes within a genome. Therefore, differences in codon bias between genomes can be reasonably modeled by using only two parameters, average usage of v1 and v2 (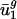 and
and 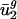 , respectively). Inclusion of additional eigencodons adds little discriminatory power.
, respectively). Inclusion of additional eigencodons adds little discriminatory power.
Genome GC Content Correlates with  . Each component of each eigencodon represents a codon, m(w). Simple inspection of the components of v1 suggests that v1 is related to gene GC content. Nearly all codons ending in G or C contribute positively to v1 (positive vm(w),1) and those ending in A or T contribute negatively (Table 2). Plotting
. Each component of each eigencodon represents a codon, m(w). Simple inspection of the components of v1 suggests that v1 is related to gene GC content. Nearly all codons ending in G or C contribute positively to v1 (positive vm(w),1) and those ending in A or T contribute negatively (Table 2). Plotting 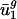 versus genome GC content (Fig. 2_a_) shows a strong positive correlation (_R_2 = 0.961). This correlation also holds for individual genes; plotting ui,1 versus gene GC content (data not shown) gives a squared correlation coefficient of _R_2 = 0.895.
versus genome GC content (Fig. 2_a_) shows a strong positive correlation (_R_2 = 0.961). This correlation also holds for individual genes; plotting ui,1 versus gene GC content (data not shown) gives a squared correlation coefficient of _R_2 = 0.895.
Fig. 2.

(a) Plot of 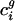 versus genome GC content for each organism. Usage of the first eigencodon correlates with genome GC content (_R_2 = 0.961). (b) Plot of
versus genome GC content for each organism. Usage of the first eigencodon correlates with genome GC content (_R_2 = 0.961). (b) Plot of 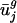 versus intergenic bias. The second eigencodon correlates with a model constructed as a linear combination of intergenic bias parameters (_R_2 = 0.669). In both plots, open boxes are data points for A. thaliana, C. elegans, E. cuniculi, P. falciparum, S. cerevisiae, and S. pombe.
versus intergenic bias. The second eigencodon correlates with a model constructed as a linear combination of intergenic bias parameters (_R_2 = 0.669). In both plots, open boxes are data points for A. thaliana, C. elegans, E. cuniculi, P. falciparum, S. cerevisiae, and S. pombe.
Intergenic Context-Dependent Nucleotide Biases Correlate with 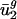 . We created a 64-parameter model, dg (referred to as intergenic bias), from each genome's intergenic regions, which describes nucleotide biases that depend on the identity of immediately adjacent bases (see Materials and Methods). The nearest-neighbor nucleotide biases found in the intergenic regions are in all cases (except those where the context specifies a stop codon) positively correlated with biases in the third codon position of genes from the same organism, when some constraints of the genetic code are corrected for by fixing the first and second codon positions and the first codon position of the following codon (data not shown). This finding is not surprising because it has been shown that dinucleotide biases influence codon bias in several organisms (30, 31). To quantify whether differences in
. We created a 64-parameter model, dg (referred to as intergenic bias), from each genome's intergenic regions, which describes nucleotide biases that depend on the identity of immediately adjacent bases (see Materials and Methods). The nearest-neighbor nucleotide biases found in the intergenic regions are in all cases (except those where the context specifies a stop codon) positively correlated with biases in the third codon position of genes from the same organism, when some constraints of the genetic code are corrected for by fixing the first and second codon positions and the first codon position of the following codon (data not shown). This finding is not surprising because it has been shown that dinucleotide biases influence codon bias in several organisms (30, 31). To quantify whether differences in 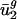 could be explained by intergenic bias, we first constructed a matrix, D = [d1... dG]T, whose rows were the intergenic bias parameters dg. We used a SVD to find the directions of largest variance in D and then used least-squares techniques to model
could be explained by intergenic bias, we first constructed a matrix, D = [d1... dG]T, whose rows were the intergenic bias parameters dg. We used a SVD to find the directions of largest variance in D and then used least-squares techniques to model 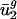 as a linear combination of the eight directions of largest variance (the approximation of
as a linear combination of the eight directions of largest variance (the approximation of  is denoted
is denoted 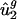 ). As shown in Fig. 2_b_, the resulting model (referred to as the _û_2 model) explained 66.9% of the variance in
). As shown in Fig. 2_b_, the resulting model (referred to as the _û_2 model) explained 66.9% of the variance in 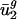 . As a control, we also estimated estimated
. As a control, we also estimated estimated 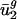 by using 10,000 randomized versions of D. In no case did the resulting models explain >30.3% of the variance in
by using 10,000 randomized versions of D. In no case did the resulting models explain >30.3% of the variance in  (see Materials and Methods for details).
(see Materials and Methods for details).
Eukaryotic Genomes Have Characteristic Values for 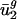 . Several eukaryotic species, namely Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, and Saccharomyces cerevisiae, have been noted to have a “prokaryotic-like” pattern of codon bias (7, 32, 33), in that they obey the genome hypothesis. Others, such as humans and other mammals, do not; GC content varies greatly between regions of the mammalian genome, which are termed isochores (34). Because GC content influences codon bias (35), genes from different isochores have distinct patterns of codon bias.
. Several eukaryotic species, namely Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, and Saccharomyces cerevisiae, have been noted to have a “prokaryotic-like” pattern of codon bias (7, 32, 33), in that they obey the genome hypothesis. Others, such as humans and other mammals, do not; GC content varies greatly between regions of the mammalian genome, which are termed isochores (34). Because GC content influences codon bias (35), genes from different isochores have distinct patterns of codon bias.
As expected, when we expressed the codon bias of A. thaliana, C. elegans, D. melanogaster, and S. cerevisiae genes in terms of the eigencodon basis generated from the SVD of prokaryotic codon vectors, we found that the 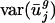 for all eigencodons, vj, is similar to that in prokaryotic organisms. Namely,
for all eigencodons, vj, is similar to that in prokaryotic organisms. Namely, 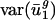 and
and 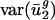 are small, whereas
are small, whereas 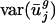 for j = 3,..., 41 are large (Fig. 5, which is published as supporting information on the PNAS web site, and Fig. 3). Expressing the codon bias of Danio rerio, Encephalitozoon cuniculi, Plasmodium falciparum, and Schizosaccharomyces pombe genes in terms of eigencodons also and produced the same pattern. Therefore,
for j = 3,..., 41 are large (Fig. 5, which is published as supporting information on the PNAS web site, and Fig. 3). Expressing the codon bias of Danio rerio, Encephalitozoon cuniculi, Plasmodium falciparum, and Schizosaccharomyces pombe genes in terms of eigencodons also and produced the same pattern. Therefore, 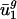 and
and 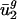 are also characteristic values in these eukaryotic organisms.
are also characteristic values in these eukaryotic organisms.
Fig. 3.

Eukaryotic genomes have low variance in usage of the second eigencodon. Expanded view of box and whisker plots of 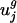 for j = 1,..., 8 for all prokaryotic genomes g, with values for eukaryotic genomes superimposed. A full diagram can be found in Fig. 5. Box and whisker plots are drawn in gray. Asterisks indicate outlying prokaryotic values. Values for eukaryotic organisms are drawn individually with symbols as indicated in the upper left corner. Compared with prokaryotic genomes, many eukaryotic genomes have large variance in the usage of eigencodon v1 but relatively small variance in usage of eigencodon v2. In general, variance is smaller for eukaryotic genomes than for prokaryotic genomes because eukaryotic genes tend to be longer than prokaryotic genes and hence provide less noisy samples of codon bias. Considering only long prokaryotic genes does not change the results qualitatively (see Figs. 7–9, which are published as supporting information on the PNAS web site).
for j = 1,..., 8 for all prokaryotic genomes g, with values for eukaryotic genomes superimposed. A full diagram can be found in Fig. 5. Box and whisker plots are drawn in gray. Asterisks indicate outlying prokaryotic values. Values for eukaryotic organisms are drawn individually with symbols as indicated in the upper left corner. Compared with prokaryotic genomes, many eukaryotic genomes have large variance in the usage of eigencodon v1 but relatively small variance in usage of eigencodon v2. In general, variance is smaller for eukaryotic genomes than for prokaryotic genomes because eukaryotic genes tend to be longer than prokaryotic genes and hence provide less noisy samples of codon bias. Considering only long prokaryotic genes does not change the results qualitatively (see Figs. 7–9, which are published as supporting information on the PNAS web site).
Human genes, on the other hand, have high 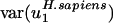 (i.e., they vary greatly in usage of v1), as expected from the differences in GC content between isochores. Rat and mouse genes also have somewhat high
(i.e., they vary greatly in usage of v1), as expected from the differences in GC content between isochores. Rat and mouse genes also have somewhat high 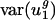 , although much smaller than
, although much smaller than 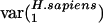 . This finding is consistent with the observation that isochores in humans vary more in GC content than those in rodents (36). Interestingly,
. This finding is consistent with the observation that isochores in humans vary more in GC content than those in rodents (36). Interestingly,  was small for all three mammals examined, similar to
was small for all three mammals examined, similar to 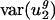 for prokaryotic genomes. Therefore, although
for prokaryotic genomes. Therefore, although  is not characteristic of all mammalian genomes,
is not characteristic of all mammalian genomes, 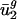 is still characteristic of each of these three mammalian genomes.
is still characteristic of each of these three mammalian genomes.
Codon Usage in Prokaryotes Can Be Estimated from Intergenic Sequences. For any given genome, g, we define genome-wide codon bias (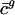 ) as the mean codon vector for all the genes in g (not just the subset used in the SVD). Given the intergenic sequences of any prokaryote, we estimate that organism's genome-wide codon bias in the following manner. First, we calculate the GC content of the intergenic sequences, which is highly correlated with the overall GC content (35) and therefore
) as the mean codon vector for all the genes in g (not just the subset used in the SVD). Given the intergenic sequences of any prokaryote, we estimate that organism's genome-wide codon bias in the following manner. First, we calculate the GC content of the intergenic sequences, which is highly correlated with the overall GC content (35) and therefore 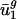 , allowing us to estimate
, allowing us to estimate 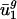 (denoted
(denoted 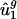 ) by using the following equation:
) by using the following equation: 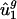 = 0.000359·(intergenic GC content)-0.0143, where GC content is measured in percent. Also from the intergenic sequences, we can calculate intergenic bias parameters, dg. From dg and the _û_2 model, we can compute
= 0.000359·(intergenic GC content)-0.0143, where GC content is measured in percent. Also from the intergenic sequences, we can calculate intergenic bias parameters, dg. From dg and the _û_2 model, we can compute  , an estimate for
, an estimate for 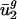 . Then, predicted genome-wide codon bias can be approximated as
. Then, predicted genome-wide codon bias can be approximated as 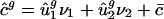 , where c̄ is the average codon vector for the genes used in the SVD. This method predicts the genome-wide codon bias of any individual prokaryotic genome with an average _R_2 of 0.840. As shown in Fig. 4_a_, the components of c̄g correlate with the corresponding components of ĉg; in other words, the average usage of individual codons among all genes within a genome correlates very well with the usage predicted based on intergenic sequence statistics.
, where c̄ is the average codon vector for the genes used in the SVD. This method predicts the genome-wide codon bias of any individual prokaryotic genome with an average _R_2 of 0.840. As shown in Fig. 4_a_, the components of c̄g correlate with the corresponding components of ĉg; in other words, the average usage of individual codons among all genes within a genome correlates very well with the usage predicted based on intergenic sequence statistics.
Fig. 4.

Graph of components of predicted genome-wide codon bias vector, ĉg, based on intergenic nucleotide sequences versus components of actual genome-wide codon bias vector, c̄g. Each point in the plot represents a 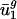 coordinate pair for some organism g and some codon m(w).
coordinate pair for some organism g and some codon m(w). 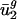 is a component of c̄g, and
is a component of c̄g, and 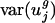 is a component of ĉg. Different organisms and codons are not differentiated in these plots. Stop codons (TAA, TAG, and TGA) and the single codons for methionine (ATG) and tryptophan (TGG) were excluded. (a) Prokaryotes. Overall _R_2 = 0.858. Average for individual genomes is _R_2 = 0.840. (b) Data for the following eukaryotes: A. thaliana, C. elegans, E. cuniculi, P. falciparum, S. cerevisiae, and S. pombe. Overall _R_2 = 0.847. _R_2 values for the individual genomes are given in the text.
is a component of ĉg. Different organisms and codons are not differentiated in these plots. Stop codons (TAA, TAG, and TGA) and the single codons for methionine (ATG) and tryptophan (TGG) were excluded. (a) Prokaryotes. Overall _R_2 = 0.858. Average for individual genomes is _R_2 = 0.840. (b) Data for the following eukaryotes: A. thaliana, C. elegans, E. cuniculi, P. falciparum, S. cerevisiae, and S. pombe. Overall _R_2 = 0.847. _R_2 values for the individual genomes are given in the text.
Prokaryotic Parameters Can Be Used to Effectively Predict Eukaryotic Genome-Wide Codon Bias from Eukaryotic Intergenic Sequences. We then tested whether 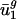 and
and 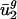 for eukaryotic organisms correlated with organism GC content and intergenic bias, respectively. Not surprisingly, as shown by the open boxes in Fig. 2_a_,GC content for A. thaliana, C. elegans, E. cuniculi, P. falciparum, S. cerevisiae, and S. pombe has a correlation with
for eukaryotic organisms correlated with organism GC content and intergenic bias, respectively. Not surprisingly, as shown by the open boxes in Fig. 2_a_,GC content for A. thaliana, C. elegans, E. cuniculi, P. falciparum, S. cerevisiae, and S. pombe has a correlation with 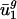 similar to what it has for prokaryotic organisms. As shown by the open boxes in Fig. 2_b_, using the _û_2 model with eukaryotic intergenic bias parameters results in values close to the regression line for prokaryotic data. Based on these results, we can also predict genome-wide codon bias quite well in these eukaryotes based only on their intergenic sequences by using the relationships between intergenic GC content and
similar to what it has for prokaryotic organisms. As shown by the open boxes in Fig. 2_b_, using the _û_2 model with eukaryotic intergenic bias parameters results in values close to the regression line for prokaryotic data. Based on these results, we can also predict genome-wide codon bias quite well in these eukaryotes based only on their intergenic sequences by using the relationships between intergenic GC content and 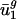 and
and 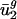 calculated from prokaryotic sequences (Fig. 4_b_). The squared correlation coefficients (_R_2) for the individual organisms were the following: A. thaliana, 0.789; C. elegans, 0.753; E. cuniculi, 0.717; P. falciparum, 0.932; S. cerevisiae, 0.892; and S. pombe, 0.915.
calculated from prokaryotic sequences (Fig. 4_b_). The squared correlation coefficients (_R_2) for the individual organisms were the following: A. thaliana, 0.789; C. elegans, 0.753; E. cuniculi, 0.717; P. falciparum, 0.932; S. cerevisiae, 0.892; and S. pombe, 0.915.
Discussion
Using a SVD, we defined 41 eigencodons, v1,..., v41. Linear combinations of these eigencodons can completely describe the codon bias of any gene. By quantitatively decomposing variation in codon bias into a term for variation within genomes and a term for variation between genomes, we show that between-genome variation accounts for most of the variation in the usage of only two of the 41 eigencodons, v1 and v2. In other words, genes from the same genome are typically more similar to each other in their usage of v1 and v2 than genes from different genomes. Genes from each prokaryotic organism, g, examined thus use characteristic amounts of v1 and v2, denoted 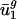 and
and 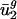 , respectively. Because (i) codon bias varies more in the direction of v1 and v2 than in any other direction, and (ii) between-genome variation [var(uj)between] is large in these two directions,
, respectively. Because (i) codon bias varies more in the direction of v1 and v2 than in any other direction, and (ii) between-genome variation [var(uj)between] is large in these two directions, 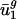 and
and 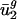 (usage of v1 and v2) are the most important (i.e., most necessary) parameters with respect to our eigencodon basis for describing codon bias in archaeal and eubacterial organisms. In addition, because (i) codon bias varies less in the directions of v3,..., v41 (i.e., σ3,..., σ41 are small) and (ii) between-genome variation in
(usage of v1 and v2) are the most important (i.e., most necessary) parameters with respect to our eigencodon basis for describing codon bias in archaeal and eubacterial organisms. In addition, because (i) codon bias varies less in the directions of v3,..., v41 (i.e., σ3,..., σ41 are small) and (ii) between-genome variation in 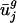 for j = 3,..., 41, is small,
for j = 3,..., 41, is small, 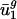 and
and 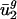 are also largely sufficient for describing genome-wide codon bias. The limitation to only two parameters is somewhat surprising because differences between genomes would be expected to be caused by many global differences between organisms, such as in the replication and transcription machineries, repair systems, and physical and chemical environments.
are also largely sufficient for describing genome-wide codon bias. The limitation to only two parameters is somewhat surprising because differences between genomes would be expected to be caused by many global differences between organisms, such as in the replication and transcription machineries, repair systems, and physical and chemical environments.
Genome GC content, which is determined by directional mutation pressure (ref. 37; although see ref. 38), correlates closely with 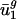 ; thus,
; thus, 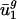 is likely also specified by directional mutation pressure. In this article, we show that
is likely also specified by directional mutation pressure. In this article, we show that 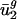 is also determined by mutational pressures acting throughout the genome. Referred to as intergenic bias, the mutational pressures correlated with
is also determined by mutational pressures acting throughout the genome. Referred to as intergenic bias, the mutational pressures correlated with 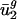 depend on adjacent (nearest-neighbor) nucleotide context. Because
depend on adjacent (nearest-neighbor) nucleotide context. Because 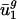 and
and 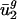 are determined by mutational pressures, they may be predicted from parameters calculated from intergenic sequences. Intergenic sequences can therefore be used in a two-parameter model to predict genome-wide codon bias in eubacterial and archaeal genomes. This two-parameter model is also accurate in predicting codon bias from intergenic sequences in most eukaryotic genomes, confirming that codon bias is largely determined by mutational processes unrelated to protein expression in all three domains of life.
are determined by mutational pressures, they may be predicted from parameters calculated from intergenic sequences. Intergenic sequences can therefore be used in a two-parameter model to predict genome-wide codon bias in eubacterial and archaeal genomes. This two-parameter model is also accurate in predicting codon bias from intergenic sequences in most eukaryotic genomes, confirming that codon bias is largely determined by mutational processes unrelated to protein expression in all three domains of life.
Two observations argue that mutational processes are mostly responsible for differences in codon bias between genomes in the direction of v2. First, usage of v2 varies little among genes within the same genome regardless of effective population size. The effective population size of Escherichia coli has been estimated at 108 to 109 (39), whereas that for mammals such as humans is ∼104 (40). Estimates for the difference in selection coefficients for different synonymous codons range from 10-9 (39) to 10-5 (41). Despite the large range, all estimates are consistent with the notion that selection on synonymous codons may be operative in E. coli (Nes ≥ 1, i.e., effective population size times selective coefficient is large) but not in Homo sapiens (Nes ≤ 0.1) (5, 7, 42). However, although population size varies over more than four orders of magnitude, 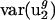 for all genomes studied varies only over a five-fold range. Because H. sapiens has a small effective population size compared with E. coli, one would expect, given the same difference in selective coefficients for different synonymous codons, that selectively maintained codon usage would vary more in H. sapiens. In fact,
for all genomes studied varies only over a five-fold range. Because H. sapiens has a small effective population size compared with E. coli, one would expect, given the same difference in selective coefficients for different synonymous codons, that selectively maintained codon usage would vary more in H. sapiens. In fact, 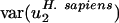 is actually half the value of
is actually half the value of 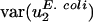 . Thus, the small value of
. Thus, the small value of 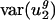 for all organisms is difficult to explain by using selection. Second, intergenic nucleotide biases explain more than two-thirds of the variation in genome-wide average usage of v2 in all archaeal, all eubacterial, and most eukaryotic organisms examined. Thus, most of the variation in
for all organisms is difficult to explain by using selection. Second, intergenic nucleotide biases explain more than two-thirds of the variation in genome-wide average usage of v2 in all archaeal, all eubacterial, and most eukaryotic organisms examined. Thus, most of the variation in 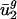 can be explained by properties of sequences in the same genome that are never translated. In principle, some selective process acting during protein expression may cause these correlated changes in intergenic sequences; however, a simpler explanation is that mutational processes affect all the DNA within a given organism. These mutational processes result in correlations between intergenic sequence nucleotide biases and codon bias. Taking GC content as a special case of nucleotide biases (where adjacent nucleotide context is ignored), the preceding statement reduces to the statement that directional mutation pressure influences codon bias by causing qualitatively similar changes in GC content in all DNA within a given organism.
can be explained by properties of sequences in the same genome that are never translated. In principle, some selective process acting during protein expression may cause these correlated changes in intergenic sequences; however, a simpler explanation is that mutational processes affect all the DNA within a given organism. These mutational processes result in correlations between intergenic sequence nucleotide biases and codon bias. Taking GC content as a special case of nucleotide biases (where adjacent nucleotide context is ignored), the preceding statement reduces to the statement that directional mutation pressure influences codon bias by causing qualitatively similar changes in GC content in all DNA within a given organism.
It might be that selection maintains the small value of 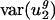 in E. coli and other bacteria, whereas mutation maintains it in H. sapiens and other mammals. More generally, it might be that different mechanisms besides mutation are responsible for maintaining the small value of
in E. coli and other bacteria, whereas mutation maintains it in H. sapiens and other mammals. More generally, it might be that different mechanisms besides mutation are responsible for maintaining the small value of 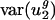 in different organisms. This possibility cannot be completely excluded. However, because of the good correlation of
in different organisms. This possibility cannot be completely excluded. However, because of the good correlation of 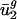 with parameters calculated from intergenic sequences and because usage of v2 measures usage of all codons to some extent, we prefer the simpler explanation that mutation is the primary force maintaining small values of
with parameters calculated from intergenic sequences and because usage of v2 measures usage of all codons to some extent, we prefer the simpler explanation that mutation is the primary force maintaining small values of 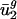 in all organisms.
in all organisms.
Usage of v2 is also correlated with organism optimal growth temperature (Fig. 6, which is published as supporting information on the PNAS web site). Organisms with higher optimal growth temperature tend to have higher values of 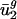 . This result is in agreement with the results of others who note that the second factor in a principal-components analysis or correspondence analysis correlates with the organism's optimal growth temperature (24, 43). The results of ref. 24 demonstrate that selection related to elevated growth temperature plays a role in establishing codon bias in thermophilic organisms, which may be related to the tendency for thermophilic organisms to systematically load RNA sequences with purines (25). However, our results emphasize the importance of mutational (not related to protein expression) forces in determining global trends in codon bias. Specifically, because selection on codon bias or mRNA structure during protein expression cannot explain the correlation we observe with patterns of nearest-neighbor nucleotide bias in intergenic sequences, we conclude that mutational pressures are primarily responsible for the differences in usage of v2 between genomes, as discussed above. The role of selection is instead appropriately ascribed to generating the relatively smaller variation in usage of v2 among highly expressed ribosomal genes and other genes within the same genome (24) and to a minor role in determining overall genome-wide codon bias.
. This result is in agreement with the results of others who note that the second factor in a principal-components analysis or correspondence analysis correlates with the organism's optimal growth temperature (24, 43). The results of ref. 24 demonstrate that selection related to elevated growth temperature plays a role in establishing codon bias in thermophilic organisms, which may be related to the tendency for thermophilic organisms to systematically load RNA sequences with purines (25). However, our results emphasize the importance of mutational (not related to protein expression) forces in determining global trends in codon bias. Specifically, because selection on codon bias or mRNA structure during protein expression cannot explain the correlation we observe with patterns of nearest-neighbor nucleotide bias in intergenic sequences, we conclude that mutational pressures are primarily responsible for the differences in usage of v2 between genomes, as discussed above. The role of selection is instead appropriately ascribed to generating the relatively smaller variation in usage of v2 among highly expressed ribosomal genes and other genes within the same genome (24) and to a minor role in determining overall genome-wide codon bias.
In agreement with our interpretation that mutation is primarily responsible for 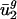 , other studies of the effect of high growth temperature on DNA sequences also point toward a mutational effect on DNA sequences in general and codon bias in particular. A linear combination of dinucleotide abundances calculated over entire genomes correlates well with optimal growth temperature for one mesophilic and several thermophilic archaeal organisms (30). The same result is obtained when coding sequences and intergenic sequences are analyzed separately. Because context-dependent nucleotide biases also influence codon bias (30, 31), one would therefore expect growth temperature to correlate with a mutational effect on codon bias. Furthermore, recent work has shown that patterns of codon bias across many different organisms, both thermophilic and mesophilic, can be explained by a single mutational model dependent on position-specific nucleotide parameters (23). Finally, elevated temperatures result in markedly elevated rates of DNA damage (44), but GC content does not correlate with optimal growth temperature (30), suggesting a role for biases that are not captured by simple GC content.
, other studies of the effect of high growth temperature on DNA sequences also point toward a mutational effect on DNA sequences in general and codon bias in particular. A linear combination of dinucleotide abundances calculated over entire genomes correlates well with optimal growth temperature for one mesophilic and several thermophilic archaeal organisms (30). The same result is obtained when coding sequences and intergenic sequences are analyzed separately. Because context-dependent nucleotide biases also influence codon bias (30, 31), one would therefore expect growth temperature to correlate with a mutational effect on codon bias. Furthermore, recent work has shown that patterns of codon bias across many different organisms, both thermophilic and mesophilic, can be explained by a single mutational model dependent on position-specific nucleotide parameters (23). Finally, elevated temperatures result in markedly elevated rates of DNA damage (44), but GC content does not correlate with optimal growth temperature (30), suggesting a role for biases that are not captured by simple GC content.
Based on our results and those of others, we propose the following interpretation of observed codon bias and the genome hypothesis (in the special case of mammals, the following general statements may not apply to codon bias changes related to isochores). The genome-wide codon bias of each organism is set primarily by mutational forces, which create a point about which the codon bias of individual genes in that organism are clustered. The codon bias of individual genes or subsets of genes is additionally perturbed from the genome-wide average codon bias by selective and other mutational forces acting during translation, but this effect is relatively much smaller. Therefore, in all three domains of life, the “systems of codon usage” referred to by Grantham (which we have called genome-wide codon bias) are coarsely set by mutational pressures and precisely modified by selective pressures.
Supplementary Material
Supporting Information
Acknowledgments
This work was supported by National Institutes of Health Grants 2T32GM07365 to the Medical Scientist Training Program (to S.L.C.), GM51426 (to S.L.C. and L.S.), and HG00044 (to A.K.H.), Department of Energy Grant DE-FG03-01ER63219 (to W.L., A.K.H., L.S., and H.H.M.), and Defense Advanced Research Projects Agency Defense Sciences Office Grant MDA972-00-1-0032 (to W.L., L.S., and H.H.M.).
Abbreviation: SVD, singular valve decomposition.
References
- 1.Osawa, S., Jukes, T. H., Watanabe, K. & Muto, A. (1992) Microbiol. Rev. 56**,** 229-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grantham, R. (1980) Trends Biochem. Sci. 5**,** 327-331. [Google Scholar]
- 3.Grantham, R., Gautier, C., Gouy, M., Mercier, R. & Pave, A. (1980) Nucleic Acids Res. 8**,** r49-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grantham, R., Gautier, C., Gouy, M., Jacobzone, M. & Mercier, R. (1981) Nucleic Acids Res. 9**,** r43-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ikemura, T. (1985) Mol. Biol. Evol. 2**,** 13-34. [DOI] [PubMed] [Google Scholar]
- 6.Sharp, P. M. & Li, W. H. (1987) Mol. Biol. Evol. 4**,** 222-230. [DOI] [PubMed] [Google Scholar]
- 7.Sharp, P. M., Stenico, M., Peden, J. F. & Lloyd, A. T. (1993) Biochem. Soc. Trans. 21**,** 835-841. [DOI] [PubMed] [Google Scholar]
- 8.Doolittle, W. F. (1998) Trends Genet. 14**,** 307-311. [DOI] [PubMed] [Google Scholar]
- 9.Ochman, H., Lawrence, J. G. & Groisman, E. A. (2000) Nature 405**,** 299-304. [DOI] [PubMed] [Google Scholar]
- 10.Woese, C. R. (2002) Proc. Natl. Acad. Sci. USA 99**,** 8742-8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gouy, M. & Gautier, C. (1982) Nucleic Acids Res. 10**,** 7055-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Onofrio, G., Mouchiroud, D., Aissani, B., Gautier, C. & Bernardi, G. (1991) J. Mol. Evol. 32**,** 504-510. [DOI] [PubMed] [Google Scholar]
- 13.Collins, D. W. & Jukes, T. H. (1993) J. Mol. Evol. 36**,** 201-213. [DOI] [PubMed] [Google Scholar]
- 14.Lobry, J. R. & Gautier, C. (1994) Nucleic Acids Res. 22**,** 3174-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Onofrio, G., Jabbari, K., Musto, H. & Bernardi, G. (1999) Gene 238**,** 3-14. [DOI] [PubMed] [Google Scholar]
- 16.Akashi, H. (1994) Genetics 136**,** 927-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eyre-Walker, A. (1996) Mol. Biol. Evol. 13**,** 864-872. [DOI] [PubMed] [Google Scholar]
- 18.Hasegawa, M., Yasunaga, T. & Miyata, T. (1979) Nucleic Acids Res. 7**,** 2073-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gambari, R., Nastruzzi, C. & Barbieri, R. (1990) Biomed. Biochim. Acta 49**,** S88-S93. [PubMed] [Google Scholar]
- 20.Huynen, M. A., Konings, D. A. & Hogeweg, P. (1992) J. Mol. Evol. 34**,** 280-291. [DOI] [PubMed] [Google Scholar]
- 21.Blake, W. J., Kærn, M., Cantor, C. R. & Collins, J. J. (2003) Nature 422**,** 633-637. [DOI] [PubMed] [Google Scholar]
- 22.Kanaya, S., Kinouchi, M., Abe, T., Kudo, Y., Yamada, Y., Nishi, T., Mori, H. & Ikemura, T. (2001) Gene 276**,** 89-99. [DOI] [PubMed] [Google Scholar]
- 23.Knight, R. D., Freeland, S. J. & Landweber, L. F. (2001) Genome Biol. 2**,** RESEARCH0010. [DOI] [PMC free article] [PubMed]
- 24.Lynn, D. J., Singer, G. A. & Hickey, D. A. (2002) Nucleic Acids Res. 30**,** 4272-4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lao, P. J. & Forsdyke, D. R. (2000) Genome Res. 10**,** 228-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, W. & Chen, S. L. (2002) Biotechniques 33**,** 1334-1341. [DOI] [PubMed] [Google Scholar]
- 27.Vieille, C. & Zeikus, G. J. (2001) Microbiol. Mol. Biol. Rev. 65**,** 1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golub, G. H. & Van Loan, C. F. (1996) in Matrix Computations (Johns Hopkins Univ. Press, Baltimore), 3rd Ed., pp. 48-86.
- 29.Weisberg, S. (1985) Applied Linear Regression, Wiley Series in Probability and Mathematical Statistics. Applied Probability and Statistics (Wiley, New York), 2nd Ed.
- 30.Kawashima, T., Amano, N., Koike, H., Makino, S., Higuchi, S., KawashimaOhya, Y., Watanabe, K., Yamazaki, M., Kanehori, K., Kawamoto, T., et al. (2000) Proc. Natl. Acad. Sci. USA 97**,** 14257-14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlin, S., Campbell, A. M. & Mrazek, J. (1998) Annu. Rev. Genet. 32**,** 185-225. [DOI] [PubMed] [Google Scholar]
- 32.Moriyama, E. N. & Powell, J. R. (1997) J. Mol. Evol. 45**,** 514-523. [DOI] [PubMed] [Google Scholar]
- 33.Duret, L. & Mouchiroud, D. (1999) Proc. Natl. Acad. Sci. USA 96**,** 4482-4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernardi, G. (2000) Gene 241**,** 3-17. [DOI] [PubMed] [Google Scholar]
- 35.Muto, A. & Osawa, S. (1987) Proc. Natl. Acad. Sci. USA 84**,** 166-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mouchiroud, D., Gautier, C. & Bernardi, G. (1988) J. Mol. Evol. 27**,** 311-320. [DOI] [PubMed] [Google Scholar]
- 37.Sueoka, N. (1988) Proc. Natl. Acad. Sci. USA 85**,** 2653-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naya, H., Romero, H., Zavala, A., Alvarez, B. & Musto, H. (2002) J. Mol. Evol. 55**,** 260-264. [DOI] [PubMed] [Google Scholar]
- 39.Hartl, D. L., Moriyama, E. N. & Sawyer, S. A. (1994) Genetics 138**,** 227-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shields, D. C., Sharp, P. M., Higgins, D. G. & Wright, F. (1988) Mol. Biol. Evol. 5**,** 704-716. [DOI] [PubMed] [Google Scholar]
- 41.Bulmer, M. (1991) Genetics 129**,** 897-907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulmer, M. (1987) Nature 325**,** 728-730. [DOI] [PubMed] [Google Scholar]
- 43.Lobry, J. R. & Chessel, D. (2003) J. Appl. Genet. 44**,** 235-261. [PubMed] [Google Scholar]
- 44.Lindahl, T. (1993) Nature 362**,** 709-715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information