High-throughput analysis of type I-E CRISPR/Cas spacer acquisition in E. coli (original) (raw)
Abstract
In Escherichia coli, the acquisition of new CRISPR spacers is strongly stimulated by a priming interaction between a spacer in CRISPR RNA and a protospacer in foreign DNA. Priming also leads to a pronounced bias in DNA strand from which new spacers are selected. Here, ca. 200,000 spacers acquired during E. coli type I-E CRISPR/Cas-driven plasmid elimination were analyzed. Analysis of positions of plasmid protospacers from which newly acquired spacers have been derived is inconsistent with spacer acquisition machinery sliding along the target DNA as the primary mechanism responsible for strand bias during primed spacer acquisition. Most protospacers that served as donors of newly acquired spacers during primed spacer acquisition had an AAG protospacer adjacent motif, PAM. Yet, the introduction of multiple AAG sequences in the target DNA had no effect on the choice of protospacers used for adaptation, which again is inconsistent with the sliding mechanism. Despite a strong preference for an AAG PAM during CRISPR adaptation, the AAG (and CTT) triplets do not appear to be avoided in known E. coli phages. Likewise, PAM sequences are not avoided in Streptococcus thermophilus phages, indicating that CRISPR/Cas systems may not have been a strong factor in shaping host-virus interactions.
Keywords: CRISPR adaptation, CRISPR/Cas systems, Escherichia coli, bacteriophage, high-throughput sequencing
Introduction
The interaction of prokaryotes with viruses (phages) and other mobile genetic elements such as plasmids is a process of global significance. In large part it accounts for horizontal gene transfer that underlies, among other things, practically important phenomena such as the spread of antibiotic resistance and acquisition of pathogenicity islands or toxin genes by pathogenic bacteria. Prokaryotes have evolved numerous systems that counter and control horizontal gene transfer by interfering with phage infection and/or plasmid establishment. The best-known are restriction-modification systems. These systems have clearly left a footprint on bacterial genomes and mobile genetic elements, as demonstrated by site avoidance in many genomes.1,2 Another mechanism that can restrict horizontal gene transfer is provided by the CRISPR/Cas systems that are ubiquitous in archaea and common in eubacteria. A CRISPR (clustered regularly interspaced short palindromic repeats) cassette consists of 30–60 bp direct repeats separated by spacers of highly variable sequence.3-5 Together with cas genes,5-7 CRISPR cassettes function as a nucleic acid-based immunity system, excluding viral and plasmid DNA that contains sequences matching spacer sequences.5,8-12 Such sequences are referred to as protospacers.13 Mutations in a conserved protospacer-adjacent motif (PAM14) prevent CRISPR/Cas-mediated immunity (also called “CRISPR interference”) even when there is a perfect match between a spacer and a protospacer.13
CRISPR cassettes are transcribed as long pre-crRNAs that are processed into short crRNAs by one of the Cas proteins (Cas6e in the case of E. coli type I-E CRISPR/Cas system).15,16 Individual crRNAs contain variable central sequences corresponding to CRISPR spacers flanked by common fragments of CRISPR repeat sequences.15,17 crRNAs are bound by Cascade, a complex of several Cas proteins.15,17,18 In E. coli type I-E CRISPR/Cas system, Cascade is composed of Cse1, Cse2, Cas7, Cas5 and Cas6e proteins (subunit composition Cse11 Cse22 Cas76 Cas51 Cas6e1). The Cascade-crRNA complex recognizes double-stranded DNA containing a protospacer matching the crRNA spacer; the presence of PAM strongly increases the strength of the interaction in vitro.19 Upon target recognition, an R-loop containing an extended RNA-DNA heteroduplex involving the entire length of spacer-protospacer is formed. In vivo, target recognition leads to target cleavage.11 In E. coli, CRISPR interference requires Cas3, a large protein with nuclease and helicase activities.20,21 This protein is not part of the Cascade complex but is essential for target cleavage.
For CRISPR immunity to manifest itself, a sequence matching a protospacer must first find its way into a CRISPR cassette and become a spacer in a process, called “CRISPR adaptation.”5,8-12 During selection of bacteriophage insensitive mutants in Streptococcus thermophiles, it was observed that resistant clones contained expanded CRISPR cassettes, with new spacer-repeat units inserted at the leader end of the cassette with spacers derived from the phage.8,13,22 New spacers were responsible for phage resistance, which was ultimately overcome by accumulation of mutations in corresponding protospacers or their PAMs.13,19 Bacteria, in turn, responded by accumulation of additional spacers. Thus, a set of spacers that a particular strain possesses provides a record of prior encounters with mobile genetic elements.
Laboratory strains of E. coli and their viruses are best-understood model organisms and over the years powerful genetic and biochemical tools have been developed for their studies. However, type I-E CRISPR/Cas loci are dormant in E. coli, at least at laboratory conditions, since expression of cas genes is inhibited by H-NS.16,23 When E. coli K12 cas genes are overexpressed and CRISPR cassette is engineered to carry an appropriate spacer, CRISPR interference with viral infection15 and plasmid transformation19 can be observed. Thus, the E. coli type I-E CRISPR/Cas system is functional, at least at the interference stage.
During the adaptation stage (at least) one new spacer-repeat unit must be incorporated into CRISPR cassette in the course of phage infection or plasmid transformation, leading, respectively, to non-productive infection or plasmid loss. Yet, ever since the classical work of Delbruck and Luria,24 selections of phage-resistant E. coli conducted by various laboratories had never resulted in isolation of mutants that had arisen due to CRISPR cassette expansion. We previously developed a robust E. coli type I-E CRISPR/Cas adaptation system25 based on described interference system.15 When cells co-expressing cas genes from plasmids are transformed with compatible plasmids containing protospacers matching a CRISPR spacer, the efficiency of transformation becomes reduced several orders of magnitude due to CRISPR interference.19 Many cells that lose a protospacer-containing plasmid undergo CRISPR cassette expansion and acquire new spacers derived from eliminated plasmid.25 Efficient CRISPR expansion is not observed in cells that have lost plasmids without sequences matching CRISPR spacers.26 The phenomenon of very strong stimulation of spacer acquisition when a prior match between a foreign DNA and CRISPR spacer exists has been referred to as “priming.”25 However, Yosef et al.27 reported efficient spacer acquisition from plasmids without matches to CRISPR spacers in cells overproducing E. coli Cas1 and Cas2 only. Thus, while Cas1 and Cas2 are sufficient for non-primed adaptation (but dispensable for interference15), primed adaptation requires the entire set of Cas proteins.25 Presumably, the recognition of a protospacer by Cascade containing appropriate crRNA stimulates, in the presence of Cas3, Cas1/Cas2-dependent acquisition of additional spacers located in cis.25
Analysis of the distribution of protospacers from which new spacers are derived revealed that the location of the priming protospacer in large part determines the strand from which new spacers are selected.25,26 We previously explained this observation by a sliding hypothesis, where we envisioned that a primed Cascade-crRNA complex moves or slides along a DNA strand, possibly with the help of Cas3 helicase, occasionally selecting sequences that are converted into spacers through the action of Cas1 and Cas2.25 However, the sliding hypothesis was put forward based on analysis of a rather limited number (several dozens) of protospacers.25 Here, we dramatically extend the scope of this analysis. We present extensive data on new spacer acquisition obtained by high-throughput sequencing and we show that albeit the strong bias determined by priming clearly exists, an important prediction of the sliding hypothesis, i.e., existence of a gradient of spacer acquisition efficiency as a function of distance from the priming site, is not fulfilled. We propose an alternative scenario that may explain the strand bias of primed spacer acquisition. Our data also reveal a very strong preference for an AAG PAM during E. coli spacer acquisition. Surprisingly, we do not see evidence of PAM site avoidance in bacteriophage genomes, suggesting that the influence of CRISPR/Cas systems on bacteriophages was less than that of restriction-modification systems.
Results
Experimental set-up
Our CRISPR adaptation system is based on a plasmid-based interference system originally described by Brouns et al.15 E. coli BL21AI cells, devoid of their own cas genes and harboring only one functional CRISPR cassette, CRISPR 2.3,28 were transformed with three compatible plasmids. Two plasmids expressed the entire complement of cas genes from the T7 RNAP promoter,15 while the third plasmid harbored an engineered CRISPR cassette, also under the T7 RNAP control. The CRISPR plasmid is based on a previously described plasmid pWUR477 and carries a spacer corresponding to a fragment of bacteriophage T7 gene. Two additional pT7blue-based plasmids P1 or P2, each carrying a small fragment of the T7 genome containing a sequence matching the T7 spacer in the CRISPR plasmid were created. The T7 fragment is cloned in the same place of the pT7blue vector multiple cloning site, but its orientation is different in P1 and P2. The pT7blue plasmid and its derivatives are compatible with CRISPR and cas plasmids.
The targeted protospacer of pT7blue-based plasmids contains an ATG PAM that is functional in CRISPR interference.20 Indeed, when induced cells carrying cas and CRISPR plasmids were transformed by electroporation with P1 or P2, the number of transformants was four orders of magnitude lower than that observed in control transformation with a pT7blue plasmid without an insert (data not shown). The number of transformants was the same when uninduced cells were transformed. To observe CRISPR adaptation, we essentially followed the procedure described by Datsenko et al.25 Uninduced cells transformed with pT7blue plasmids with or without the protospacer insert were grown overnight in liquid medium supplemented with inducers. The medium also contained antibiotics necessary to maintain the cas and CRISPR plasmids but lacked ampicillin necessary to maintain the pT7blue plasmid. We next prepared genomic DNA from both cultures and amplified a leader-proximal fragment of the CRISPR 2.3 cassette as schematically depicted in Figure 1A. It should be noted that no selection for cells that have lost (or kept) the plasmid was performed. As can be seen from a gel presented in Figure 1B, in control samples only an amplified DNA fragment corresponding to unaltered CRISPR 2.3 cassette was observed (lanes 1 and 3). In case of cultures transformed with protospacer-containing plasmids P1 and P2, an additional band corresponding to CRISPR 2.3 cassette extended by one spacer-repeat unit was observed. This band was estimated to correspond to ~20–40% of material in a band corresponding to unextended cassette. A fragment corresponding to insertion of two spacer-repeat units was also detected but in lower amounts. Overall, the results show that a significant amount of cells undergo CRISPR adaptation at conditions of the experiment, provided that there is a match between CRISPR spacer and plasmid DNA protospacer. No adaptation is detected in the absence of such match. The stimulation of CRISPR adaptation in CRISPR/Cas expressing cells harboring protospacer-containing plasmids is thus due to priming.25,26

Figure 1. Experimental set-up to monitor CRISPR spacer acquisition. (A) At the top, the E. coli BL21 AI CRISPR 2.3 cassette is schematically presented, with repeats indicated as numbered gray rectangles. A leftward arrow indicates CRISPR promoter located in the leader sequence. The primers used to amplify the leader-proximal end of the cassette are shown (thick, not annealed parts of primers correspond to barcodes for high-throughput sequencing). Below, the structures of amplified DNA fragments expected in the absence (middle) or in the presence (bottom) of spacer acquisition are shown. (B) Results of PCR amplification using the primer set shown in (A) of DNA prepared from E. coli BL21 AI cultures transformed with plasmids pT7blue (lanes 1 and 3) or P1 and P2 plasmids (lanes 2 and 4) after an overnight growth at conditions of induction of plasmid-borne CRISPR/Cas components and in the absence of ampicillin needed to maintain pT7blue and its derivatives. The gray arrow indicates a PCR fragment arising from amplification of leader-proximal end of unexpanded CRSIRP 2.3 cassette; black arrow indicates a PCR fragment arising from amplification of CRISPR 2.3 cassette expanded by one spacer-repeat unit. Numbers at the left-hand side of the gel indicate the lengths (in bp) of DNA size markers. (C) Statistics of high-throughput sequencing of PCR amplification products extended by one spacer-repeat unit obtained with P1 and P2 samples.
PCR fragments corresponding to CRISPR 2.3 cassettes extended by one spacer-repeat unit were subjected to MySeq Illumina sequencing. Barcoding allowed simultaneous monitoring of sequences from cultures of cells that have lost P1 and P2 plasmids. The statistics of high-throughput sequencing results is presented in Figure 1C. Approximately half of the total spacers (115,123 in cultures transformed with P1, and 75,755 spacers in cultures transformed with P2) were confidently (less than three mismatches) mapped to the pT7 blue plasmid backbone. Most remaining reads also contained plasmid-derived spacers with larger amounts of mismatches due to low quality sequencing and were excluded from the analysis. Very small number of reads (11 for P1 transformed cultures and 15 for P2 transformed cultures) contained spacers derived from host DNA.
Analysis of PAM preference during primed and not-primed adaptation
Since spacer acquisition in our system is driven by priming, we determined the proportion of spacers that originated from protospacers located at the same strand as the priming protospacer. Indeed, a more than 10-fold bias toward the primed strand was detected in both P1 and P2 cases (Fig. 2A). Presumably, non-primed acquisition also happens in cells transformed with pT7blue vector that lacks the priming protospacer, however the low efficiency of this process does not allow us to detect extended cassettes using the PCR assay used in Figure 1B. Below, we consider P1- and P2-derived spacers arising from the same strand as the priming spacer as resulting from primed acquisition, while those derive from the opposite strand as arising from non-primed acquisition and treat them separately (note that the same strand becomes either primed or non-primed—or vice versa—depending on whether P1 or P2 are considered). Inspection of protospacers, corresponding to newly acquired spacers revealed a preference for an AAG PAM for both primed and non-primed acquisition, though the extent of this preference varied. In the primed strand, 96% of P1 protospacers and 93% of P2 protospacers contained AAG; in the non-primed strand these values were, 43 and 68%, respectively (Fig. 2B). Since latter values are close to those observed during adaptation in cells overproducing Cas1 and Cas2 only,27 if follows that Cascade and/or Cas3 increase the specificity of spacer acquisition machinery with respect to PAM choice.

Figure 2. Analysis of DNA strand and PAM preferences during primed and non-primed spacer acquisition. (A) Percentage of protospacers acquired from primed stand of the pT7blue vector in cells that lost the P1 and P2 plasmids are shown in black, percentage of protospacers from non-primed strand are shown in gray. (B) Percentage of AAG PAM sequences in protospacers acquired in cells that lost the P1 and P2 plasmids. Black bars show percentage of protospacers associated with AAG in the primed strand, gray-in the non-primed strand. (C) The distribution of non-AAG PAMs of primed strand (black bars) and non-primed strand (gray bars) protospacers in cells that lost P1 (“1”) and P2 (“2”) plasmids is presented. Non-AAG PAMs known to be functional in CRISPR interference19 are highlighted by bold typeface. The sum of percentage values for each plasmid/strand equals the total percentage of non-AAG PAMs in Figure 2B. (D) Two examples of imprecise spacer acquisition leading to appearance of non-AAG PAMs during primed acquisition. Spacer sequences are shown in regular typeface; numbers indicate their occurrences. The PAM sequences of corresponding protospacers are shown in bold typeface.
The frequencies of non-AAG PAM sequences corresponding to newly acquired spacers are shown in Figure 2C. Overall, it can be seen that certain non-consensus PAMs were used more often than others but this preference was not maintained for primed and non-primed strand acquisition. Some of non-consensus PAM sequences of primed strand were conjugated with the AAG PAM (i.e., had a sequence of NAA or AGN) and were in fact part of the AAG consensus as illustrated in Figure 2D. The result suggests that after spacer acquisition machinery specifically recognizes an AAG PAM, mistakes that happen during the process of copying and/or excision of the protospacer can lead to incorporation of spacers shifted either downstream or upstream from the “correct” position. Previous analysis of a limited number of spacer acquisition suggested that identity of repeat nucleotide adjacent to a spacer is determined by the last nucleotide of protospacer.25,26,29 Analysis of repeats associated with spacers derived from protospacers associated with non-AAG PAMs in our collection fully supports this observation (data not shown).
Analysis of PAM sequences corresponding to non-primed strand revealed multiple non-AAG sequences, some being used more than others. Since the distribution of non-AAG PAMs is non-random and certain sequences, for example CCC, were never found, the results may reflect intrinsic sequence preferences of spacer acquisition machinery.
The distribution of protospacers in donor DNA is inconsistent with the sliding mechanism of spacer selection
We next analyzed the distribution of protospacers (Fig. 3). On the figure, each strand of the pT7blue plasmid is presented as a horizontal line with the priming protospacer located in the center. Positions of possible PAMs, the AAG sequences, are shown by broken black vertical lines. There are 51 AAG sequences in each strand. The occurrences of newly acquired spacers (63,169 primed and 2,525 non-primed spacers corresponding to plasmid protospacers in the strand shown in the upper part of Fig. 3, and 105,021 primed and 5,170 non-primed spacers derived from the other strand) are shown as red (primed) and blue (non-primed) vertical lines. Inspection of the figure allows several important insights into the spacer acquisition process. First, every potential plasmid protospacer (i.e., a sequence adjacent to an AAG triplet) is actually used as a spacer donor. Second, no bias, or gradient in spacer acquisition efficiency relative to the position of the priming protospacer is detected in either strand, which is in apparent disagreement with the sliding hypothesis. Third, the frequencies of spacers are highly unequal, indicating that some protospacers are clearly preferred as spacer donors. This preference is maintained for both primed and non-primed acquisition (Spearman paired correlation coefficients for primed and non-primed acquisition in each strand equal 0.86 and 0.87). The latter result strongly indicates that in addition to AAG PAM recognition, the spacer acquisition machinery has certain context and/or sequence preferences. However, analysis of overrepresented protospacers (for which CRISPR spacers were observed more than 100 times during priming spacer acquisition) and adjacent areas for sequence commonalities by WebLogo 3 (weblogo.threeplusone.com/create.cgi) failed to reveal any common motif. We also failed to observe a correlation of the frequency of donor protospacer use with its melting temperature, suggesting that general features such as G/C richness do not affect spacer acquisition preference. Likewise, no common properties were observed for underused protospacers (for which CRISPR spacers were observed less than 50 times during priming spacer acquisition).

Figure 3. The distribution of donor protospacers in eliminated plasmids. The location of the ampicillin resistance bla gene, replication origins, and the multiple cloning site (MCS) of the pT7blue plasmid are schematically indicated at the top. Below, the two strands are shown separately as horizontal lines. The location of the priming protospacer is shown by yellow vertical line. The positions of AAG trinucleotides in each strand are shown by punctured vertical lines. For each strand, percentages of spacers corresponding to different protospacers are shown by red and blue vertical lines. Red lines correspond to primed and blue lines- to non-primed acquisition.
To independently test the sliding mechanism of primed spacer acquisition, two pT7blue-based plasmids containing multiple AAG sequences inserted at two different locations were created. The plasmids also carried a priming protospacer in two different orientations (Fig. 4). Multiply repeated AAG sequences should halt the spacer acquisition machinery sliding by presenting multiple preferred PAM sequences. Therefore, we expected that protospacers located downstream of AAG blocks would be chosen less efficiently. The plasmids were transformed into cells expressing crRNA targeting the priming protospacer, cells were cultured in the absence of ampicillin and CRISPR cassettes of individual colonies that lost ampicillin resistance were amplified and newly inserted spacers were identified for several dozens of clones by standard sequencing. Cells that lost pT7Blue-based plasmids with priming protospacer but without AAG blocks were also analyzed as a control. As can be seen from Figure 4, the expectation of the sliding mechanism was not fulfilled: there was no enrichment of donor protospacers at or in front of the AAG sequence blocks and the distribution of protospacer donor did not appear to be significantly different from that seen in cells that lost control plasmids.
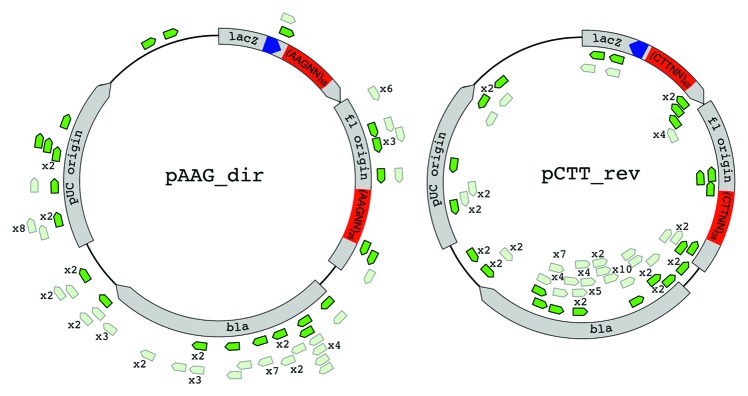
Figure 4. The distribution of donor protospacers acquired during primed spacer acquisition from plasmids containing multiple AAG sequence blocks. Two plasmids with opposing orientations of the priming protospacer (dark blue arrow) are schematically shown, with positions of poly AAG blocks highlighted in red. Protospacers corresponding to spacers acquired by cells that lost each plasmid are shown as green arrows. Protospacers on the outside originate from the coding strand of ampicillin-resistance gene bla. Protospacers on the inside originate from the opposite strand. Protospacers acquired from plasmids that lacked poly AAG tracks are shown in light green. Numbers indicate the number of times identical spacers have been observed.
Discussion
In this work, we analyzed E. coli CRISPR spacers acquired at conditions of overexpression of type I-E CRISPR/Cas system components and revealed by high-throughput sequencing. The results confirmed a strong stimulatory effect of priming on CRISPR spacer acquisition efficiency. The results also confirmed that the spacer acquisition process acquires strand bias only when primed. Importantly, the distribution of protospacers selected during primed adaptation is inconsistent with the simple scanning hypothesis, which posits that spacer selection machinery processively slides along the DNA strand recognized during priming, occasionally recognizing an AAG PAM and initiating the process of spacer acquisition. Such a mechanism should lead to an appearance of a gradient in protospacer selection efficiency as a function of distance from the priming site. This expectation is not fulfilled and no gradient is observed. Nevertheless, certain protospacers are clearly preferred. Since this preference is observed both for primed and non-primed spacer selection, it may reflect the preferences of Cas1/Cas2, which are sufficient for spacer acquisition in the absence of priming.25,27 In the absence of sliding, which mechanism could explain the strong bias of primed spacer acquisition? One hypothesis would be that after the priming interaction with Cascade-crRNA, extended single-stranded regions of only one strand of target DNA are generated (presumably by Cas3) and that protospacers from single-stranded DNA are preferentially selected for acquisition by Cas1/Cas2. Swartz et al.26 have also proposed that spacer acquisition machinery may preferentially use target DNA degradation products that arise during CRISPR interference. A similar model was recently proposed by Sinkunas et al.30 This mechanism, however, is inconsistent with the fact that primed spacer acquisition is induced by the recognition of targets containing escape mutations that render CRISPR interference inactive25 and should, therefore, abolish the generation of Cas3-depedent target degradation products. Moreover, even if spacer acquisition proceeded from single-stranded DNA produced after initial target recognition, a gradient in protospacer usage during acquisition would have been expected. No such gradient is observed experimentally, however. Clearly, additional experiments are needed to determine the molecular basis of primed CRISPR spacer acquisition and its polarity.
If one considers a long-term interaction of bacteriophage with a CRISPR/Cas-carrying bacterial host, then acquisition of the first phage-derived spacer should inevitably lead to primed acquisition of additional spacers. Since primed spacer acquisition in E. coli has a very strong preference for an AAG PAM, underrepresentation of AAG and its complement CTT sequences in phage sequences is expected. A similar site avoidance has been reported for restriction endonuclease recognition sequences in some phages.1,2 To determine if AAG sequences are indeed avoided in E. coli phage genomes, we used a Markov chain-based Z value statistics, which allows one to compare the theoretical expectation for a frequency of a string of nucleotides (“words”) with observed values,31,32 Z scores were calculated for each string of three nucleotides in E. coli bacteriophage genomes retrieved from the ACLAME (aclame.ulb.ac.be/) database. A total of 49 E. coli phages with different life styles and development strategies were analyzed. An avoidance of AAG (and its complement CTT) would have revealed itself in Z scores below -3.32 While strong biases can be expected in phage genomes due to codon usage and non-equal distribution of coding sequences in genomic DNA strands, PAM avoidance should manifest itself equally on both strands. Therefore, simultaneous low Z score values for AAG and CTT can be considered as arising in response to CRISPR/Cas pressure. As can be seen from Table 1, only two phages, phi4795 and Stx2, had Z scores below -3 for both AAG and CTT. In contrast, in phages K1E and RB49, these triplets were overrepresented (Z scores for both AAG and CTT above +3). In some phage genomes, only one triplet was grossly overrepresented, like in T3/T7 or K1F (a Z score for AAG of 12), a reflection of codon bias, since all coding sequences of these phages are located in one DNA strand. In more than half of phage genomes analyzed (26) the Z values for both triplets assumed values between -3 and +3, indicating that they are neither selected nor preferred. To contrast this result, Table 1 also presents Z scores for CCAGG and CCTGG, the recognition sites of E. coli restriction endonuclease EcoRII. As can be seen, both sites (but not their mutated versions that are not recognized by EcoRII, data not shown) are avoided in many phages. A similar result is obtained with several other E. coli restriction endonucleases (data not shown).
Table 1. Avoidance of type I-E CRISPR/Cas PAM consensus sequence and EcoRII sites in E. coli phages.
| Phage | z-score (PAM) | K | z-score (EcoRII) | ||
|---|---|---|---|---|---|
| AAG | CTT | AAG/CTT | CCAGG | CCTGG | |
| phi4795 | -3,68 | -4,63 | 0,92 | -2,07 | -3,2 |
| Stx2 c.b.II | -3,08 | -4,22 | 0,92 | -4,08 | -3,06 |
| P27 | -1,95 | -3,9 | 0,97 | -3,48 | -2,31 |
| lambda | -2,48 | -3,97 | 0,93 | -3,81 | -2,4 |
| WPhi | -2,65 | -4,26 | 0,91 | -3,7 | -4,19 |
| Stx1 с.b. | -2,46 | -3,94 | 0,92 | -3,75 | -2,91 |
| P2 | -2,11 | -4,35 | 0,93 | -3,43 | -3,39 |
| Mu | -2,42 | -6,17 | 0,88 | -3,97 | -3,91 |
| 186 | -2,28 | -6,41 | 0,89 | -4,34 | -4,87 |
| N15 | -2,72 | -3,24 | 0,93 | -1,21 | -2,1 |
| 86 | -2,39 | -4,76 | 0,93 | -4,12 | -4,26 |
| cdtI | -2,83 | -3,89 | 0,96 | -2,73 | -2,63 |
| HK97 | -0,72 | -1,87 | 0,99 | -1,59 | -2,45 |
| P4 | -1,83 | -1,33 | 0,96 | -2,93 | -1,54 |
| HK022 | -0,87 | -2,62 | 0,99 | -1,81 | -1,99 |
| T5 | -2,31 | -2 | 0,99 | -3,88 | -2,85 |
| P1 | -1,42 | 1,11 | 1 | -2,38 | -1,09 |
| RTP | 2,4 | -0,37 | 1 | -1,07 | -1,97 |
| N4 | 2,86 | 2,12 | 1 | -3,54 | -4,79 |
| ID62 | 1,75 | -0,37 | 0,98 | -1,69 | -1,97 |
| If1 | -1,34 | -0,79 | 0,97 | -1,75 | -2,95 |
| alfa3 | 1,14 | -0,58 | 0,94 | -1,63 | -1,87 |
| phiX174 | 1,85 | -0,87 | 0,96 | -1,18 | -1,8 |
| I2–2 | 1,14 | -2,24 | 0,92 | -1,59 | -2,76 |
| Ike | 1,67 | -1,55 | 1 | -1,45 | -1,21 |
| G4 | 2,22 | 0,19 | 0,94 | -0,47 | -2,02 |
| NC1 | 1,95 | -0,67 | 0,98 | -0,65 | -2,28 |
| NC5 | 1,96 | -0,68 | 0,98 | -1,19 | -2,24 |
| NC3 | 1,88 | -1,13 | 0,95 | -0,69 | -1,99 |
| WA13 | 1,61 | 0 | 0,98 | -1,08 | -2,23 |
| ID52 | 1,77 | -0,38 | 0,94 | -1,3 | -2,28 |
| NC29 | 2,28 | -0,51 | 0,99 | -1,64 | -1,83 |
| NC28 | 1,5 | -0,22 | 0,97 | -1,68 | -2,06 |
| ID34 | 1,75 | -0,93 | 0,95 | -0,81 | -2,25 |
| ID1 | 1,76 | -0,55 | 1 | -0,69 | -2 |
| WA5 | 2,16 | 0,04 | 0,93 | -1,35 | -2,66 |
| M13 | -0,49 | -2,58 | 0,9 | -2 | -1,72 |
| phiK | 0,82 | -0,06 | 0,96 | -1,33 | -1,96 |
| phiKT | 7,88 | -3,35 | 1,1 | -5,98 | -7,74 |
| T1 | 3,72 | 2,14 | 1,1 | -2,45 | -0,14 |
| T3 | 12,05 | 0,92 | 1,1 | -5,03 | -6 |
| T7 | 12,08 | 1,45 | 1,1 | -4,39 | -5,73 |
| K1F | 12,42 | 1,58 | 1,1 | -5,78 | -6,27 |
| phiEcoM-GJ1 | 10,64 | -0,98 | 1,1 | 0,52 | -2,14 |
| PRD1 | 3,94 | 1,3 | 1,3 | -3,35 | -3,89 |
| JS98 | 1,85 | 10,93 | 1 | -0,29 | -0,39 |
| RB69 | 0,65 | 8,16 | 1 | -0,2 | -0,22 |
| T4 | -1,18 | 4,55 | 1 | -0,14 | 0,77 |
| K1E | 13,06 | 5,15 | 1,1 | -4,5 | -6,42 |
| RB49 | 3,58 | 7,98 | 1,1 | -6,08 | -5,59 |
Another measure of site abundance statistics, developed by Karlin,33 considers both complementary triplets simultaneously and also takes in account the length of the genome, which can influence Z scores. When this measure assumes values of less than 0.78, the site is considered underrepresented. Based on this criterion, none of E. coli phages analyzed avoid the AAG sequence (Table 1).
The E. coli type I-E CRISPR/Cas system appears to be dormant, at least at laboratory conditions due to strong repression of cas genes transcription by H-NS.16,23 Moreover, almost none of the E. coli phages known have left spacers in E. coli type I-E CRISPR arrays characterized to date. These phages may therefore have never experienced evolutionary pressure from a CRISPR/Cas system, explaining the observed lack of PAM avoidance. As is discussed elsewhere,34 this explanation, however, creates a paradox, for one would have to assume that there must exist a very large number of E. coli phages that did leave their mark on CRISPR and yet escaped detection despite numerous years of studies of E. coli phages by various laboratories worldwide. We repeated the statistical analysis using S. thermophilus phages, since type II-A CRISPR/Cas system of this bacterium is naturally active, numerous phage-derived spacers are known and the PAM sequence (AGAAW) has been established through high-throughput sequencing.35,36 The results showed that none of the S. thermophilus phages, including those that induce CRISPR/Cas adaptation of the host during infection and that left their mark in host CRISPR cassettes show any sign of PAM sequence avoidance (Table 2). Likewise, no evidence of avoidance of the more stringently conserved parts of the consensus, AGAA or GAA, was detected (data not shown).
Table 2. Avoidance of type II-A CRISPR/Cas system PAM consensus sequence in S. thermophilus phages.
| Phage | z-score (PAM) | |||
|---|---|---|---|---|
| AGAAA | AGAAT | ATTCT | TTTCT | |
| ALQ132 | -0,73 | -0,45 | -0,98 | -0,04 |
| 5093 | -0,05 | -0,68 | -1,16 | 0,58 |
| DT1 | 0,09 | -0,81 | -0,90 | 0,11 |
| Abc2 | 0,22 | -0,16 | -1,01 | 0,26 |
| 858 | 0,55 | -1,20 | -0,22 | -1,01 |
| 2972 | 0,34 | -1,13 | -0,79 | -1,01 |
| DT | 0,09 | -0,81 | -0,90 | 0,11 |
| Sfi11 | -0,09 | -0,13 | -0,64 | -0,98 |
| O1205 | 0,75 | -1,74 | -1,25 | -1,87 |
| Sfi19 | -0,59 | -0,90 | -0,92 | 0,70 |
| Sfi21 | -0,13 | -0,66 | -0,45 | -0,04 |
| 7201 | 0,26 | -0,76 | -2,39 | 1,68 |
The results presented above, though tentative, are quite surprising. They suggest that despite their ubiquity, the CRISPR/Cas system may not have had a large impact on phage genomes. On the other hand, these observations are in line with the recently published data that antibiotic resistance plasmids spread among natural isolates in natural Escherichia coli population in spite of CRISPR/Cas system.37
Material and Methods
Bacterial strains and plasmids
E. coli strain BL21AI was used throughout this study. Plasmids pWUR397 (expressing cas3) and pWUR399 (co-expressing _cse1, cse2, cas7, cas5, Cas6_e, cas1, cas2) have been described previously.15 A CRISPR cassette plasmid targeting the T7 phage genome were generated by replacing the EcoRI-BamHI fragment of the previously described pWUR47715 with a synthetic DNA fragment containing a TACTAGGAAGAACCAATAACGCTATGCTCTGG sequence corresponding to T7 phage positions 2932–2963 in gene for protein kinase. The resulting plasmid is named pKP100. P1 and P2 plasmids carry, in opposite orientations, a 285 bp fragment of T7 genome (genome positions 2812–3097) cloned into the _Eco_RV site of the pT7Blue blunt-end vector (Novagen).
The pT7Blue-based plasmids carrying a 209-bp M13 fragment with the g8 protospacer (genome positions 1311–1519) have been described previously.25 The multiple “AAG” blocks were cloned into these plasmids in the orientation corresponding to the priming protospacer. To this end a 60-nucleotide synthetic oligo containing an AAG trinucleotide repeated 14 times and separated by arbitrary mono- or dinucleotides (5′AAGCTAAGTAAGTCAAGTAAGCCAAGTAAGCAAGTAAGTCAAGCAAGTAAGTCAAGTAAG3′) and a complementary oligo were annealed and then blunt-end cloned into pG8_dir and pG8_rev plasmids,25 first at the SmaI and subsequently at the NaeI sites. The molar ratio of insert to plasmid was approximately 10:1, which allowed for obtaining clones with multiple inserts. The orientation of inserts was determined by sequencing. The resulting plasmids pAAG_dir and pCTT_rev contained two AAG blocks in the same orientation as the priming protospacer: 55 AAG sequences in the first block and 28 AAG sequences in the second block for pAAG_dir plasmid and 42 and 28 CTT sequences for plasmid pCTT_rev.
Spacer acquisition experiments
E. coli BL21AI carrying pWUR397, pWUR399 and pKP100 were transformed with P1, P2, or pT7Blue. Transformants were selected on LB agar plates supplemented with 25 μg/ml streptomycin, 25 μg/ml kanamycin, 34 μg/ml chloramphenicol and 150 μg/ml ampicillin. Individual ampicillin-resistant colonies were grown overnight in liquid LB with 25 μg/ml streptomycin, 25 μg/ml kanamycin, 34 μg/ml chloramphenicol, 0.2% arabinose and 1 mM IPTG. Genomic DNA was isolated from overnight cultures with Genomic DNA Purification kit (Thermo Scientific) using the manufacturer’s protocol. CRISPR expansion was monitored by PCR using Illumina barcodes amplicon sequencing primers whose custom parts: GTGGTTTGAGCGATGATAT and AGTTGGTAGATTGTGACTG, matched the CRISPR leader and the first spacer of the CRISPR cassette. To prevent artifacts arising from early acquisition of some spacers, DNA from six independent cultures was pooled. After agarose gel electrophoresis, fragments originating from CRISPR cassettes expanded by a single spacer-repeat unit were purified by QIAquick Gel Extraction Kit.
Amplified fragments were sequenced with MiSeq (Illumina) using MiSeq reagent kit v.2 (2x150). Raw sequencing data were analyzed using ShortRead38 and BioStrings39 packages. The reads were filtered for quality scores of ≥ 20 and reads containing two repeats (with up to two mismatches) were selected. Ninety-nine percent of selected reads contained a sequence of 30–34 bp between the repeats and were considered spacers. Ninety-five percent of spacers had a length of 32 bp and only they were mapped on P1 and P2 plasmids.
To monitor the effects of repeated AAG sequences on spacer acquisition, E. coli BW40119 cells containing genomic cas genes fused to inducible promoters and g8 spacer in CRISPR 2.125 were transformed with pT7Blue-based plasmids carrying the g8 protospacer with or without AAG blogs. Ampicillin-resistant clones were grown overnight in liquid LB supplemented with 1 mM IPTG and 1 mM arabinose (to induce cas genes expression) in the absence of antibiotic. Culture aliquots were spread on LB agar plates containing IPTG and arabinose. Individual ampicillin-sensitive colonies were analyzed for CRISPR expansion by PCR using appropriate primers.25
Analysis of site representation
The analysis of nucleotide word frequencies was performed by statistics based on Markov chains.31,32 Expected count Nexp for an n-letter word (L1L2…Ln) in a sequence was calculated as
Nexp (L1L2….Ln) = Nob(L1L2….Ln-1) × Nob(L2…Ln)/ Nob(L2Ln-1).
A Z score was calculated as
Z(W) = Nob(W)-Nexp(W)/√ Nexp(W),
where Nob stands for observed word count in a sequence. Words with Z scores < -3 were taken as underrepresented and those with z > 3 as overrepresented, correspondingly. The alternative nucleotide word frequencies estimation was performed by methods described previously.33 Briefly, relative abundance measure for three nucleotide word (L1L2L3) was estimated as
K3 = [f*(L1L2L3) × f*(L1) × f*(L2) × f*(L2)]/[(f*(L1L2) × f*(L2L3) × f*(L1LanyL3)],
where f*(A) = f* (T) = [f(A) + f(T)]/2 and f*(C) = f*(G) [f(C) + f(G)]/2 (f being nucleotide frequency). For 2- and 3-nucleotide words f* was counted as an arithmetical mean of the word in the direct and complementary strands. For example, f*(AAG) = f*(CTT) = [f(AAG) + f(CTT)]/2. If K3 value is less than 0.78 or more, then 1.23 the word occurrence is under or overrepresented, correspondingly.
Acknowledgments
This work was supported by an NIH grant GM10407, a Russian Foundation for Basic Research grant and Molecular and Cellular Biology Russian Academy of Sciences grant to K.S. We thank Interlabservice for high-throughput sequencing and Mikhail S. Gelfand, Sergei A. Spirin and Andrei V. Alexeevski for help with statistical analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
References
- 1.Sharp PM. Molecular evolution of bacteriophages: evidence of selection against the recognition sites of host restriction enzymes. Mol Biol Evol. 1986;3:75–83. doi: 10.1093/oxfordjournals.molbev.a040377. [DOI] [PubMed] [Google Scholar]
- 2.Gelfand MS, Koonin EV. Avoidance of palindromic words in bacterial and archaeal genomes: a close connection with restriction enzymes. Nucleic Acids Res. 1997;25:2430–9. doi: 10.1093/nar/25.12.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mojica FJ, Díez-Villaseñor C, Soria E, Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol. 2000;36:244–6. doi: 10.1046/j.1365-2958.2000.01838.x. [DOI] [PubMed] [Google Scholar]
- 4.Jansen R, van Embden JD, Gaastra W, Schouls LM. Identification of a novel family of sequence repeats among prokaryotes. OMICS. 2002;6:23–33. doi: 10.1089/15362310252780816. [DOI] [PubMed] [Google Scholar]
- 5.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–97. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 6.Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–75. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 7.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 9.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–7. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–70. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 11.Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 12.Deveau H, Garneau JE, Moineau S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol. 2010;64:475–93. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 13.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–40. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 15.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–4. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pougach K, Semenova E, Bogdanova E, Datsenko KA, Djordjevic M, Wanner BL, et al. Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol Microbiol. 2010;77:1367–79. doi: 10.1111/j.1365-2958.2010.07265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol. 2011;18:529–36. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 18.Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, et al. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477:486–9. doi: 10.1038/nature10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci USA. 2011;108:10098–103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westra ER, van Erp PB, Künne T, Wong SP, Staals RH, Seegers CL, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011;30:1335–42. doi: 10.1038/emboj.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horvath P, Romero DA, Coûté-Monvoisin AC, Richards M, Deveau H, Moineau S, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190:1401–12. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol Microbiol. 2010;75:1495–512. doi: 10.1111/j.1365-2958.2010.07073.x. [DOI] [PubMed] [Google Scholar]
- 24.Luria SE, Delbrück M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 26.Swarts DC, Mosterd C, van Passel MW, Brouns SJ. CRISPR interference directs strand specific spacer acquisition. PLoS One. 2012;7:e35888. doi: 10.1371/journal.pone.0035888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–76. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Díez-Villaseñor C, Almendros C, García-Martínez J, Mojica FJ. Diversity of CRISPR loci in Escherichia coli. Microbiology. 2010;156:1351–61. doi: 10.1099/mic.0.036046-0. [DOI] [PubMed] [Google Scholar]
- 29.Goren MG, Yosef I, Auster O, Qimron U. Experimental definition of a clustered regularly interspaced short palindromic duplicon in Escherichia coli. J Mol Biol. 2012;423:14–6. doi: 10.1016/j.jmb.2012.06.037. [DOI] [PubMed] [Google Scholar]
- 30.Sinkunas T, Gasiunas G, Waghmare SP, Dickman MJ, Barrangou R, Horvath P, et al. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013;32:385–94. doi: 10.1038/emboj.2012.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schbath S. An efficient statistic to detect over- and under-represented words in DNA sequences. J Comput Biol. 1997;4:189–92. doi: 10.1089/cmb.1997.4.189. [DOI] [PubMed] [Google Scholar]
- 32.Rocha EP, Viari A, Danchin A. Oligonucleotide bias in Bacillus subtilis: general trends and taxonomic comparisons. Nucleic Acids Res. 1998;26:2971–80. doi: 10.1093/nar/26.12.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karlin S, Cardon LR. Computational DNA sequence analysis. Annu Rev Microbiol. 1994;48:619–54. doi: 10.1146/annurev.mi.48.100194.003155. [DOI] [PubMed] [Google Scholar]
- 34.Semenova E, Nagornykh M, Pyatnitskiy M, Artamonova II, Severinov K. Analysis of CRISPR system function in plant pathogen Xanthomonas oryzae. FEMS Microbiol Lett. 2009;296:110–6. doi: 10.1111/j.1574-6968.2009.01626.x. [DOI] [PubMed] [Google Scholar]
- 35.Sun CL, Barrangou R, Thomas BC, Horvath P, Fremaux C, Banfield JF. Phage mutations in response to CRISPR diversification in a bacterial population. Environ Microbiol. 2013;15:463–70. doi: 10.1111/j.1462-2920.2012.02879.x. [DOI] [PubMed] [Google Scholar]
- 36.Paez-Espino D, Morovic W, Sun CL, Thomas BC, Ueda K, Stahl B, et al. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat Commun. 2013;4:1430. doi: 10.1038/ncomms2440. [DOI] [PubMed] [Google Scholar]
- 37.Touchon M, Charpentier S, Pognard D, Picard B, Arlet G, Rocha EP, et al. Antibiotic resistance plasmids spread among natural isolates of Escherichia coli in spite of CRISPR elements. Microbiology. 2012;158:2997–3004. doi: 10.1099/mic.0.060814-0. [DOI] [PubMed] [Google Scholar]
- 38.Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–8. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pages H, Aboyoun P, Gentleman R, DebRoy S. String objects representing biological sequences, and matching algorithms. Biostrings. 2:11. [Google Scholar]