ARF1 and ARF4 regulate recycling endosomal morphology and retrograde transport from endosomes to the Golgi apparatus (original) (raw)
The ARF1+ARF4 and ARF1+ARF3 pairs are both required for integrity of recycling endosomes but are involved in distinct transport pathways: the former pair regulates retrograde transport from endosomes to the TGN, whereas the latter is required for the transferrin recycling pathway from endosomes to the plasma membrane.
Abstract
Small GTPases of the ADP-ribosylation factor (ARF) family, except for ARF6, mainly localize to the Golgi apparatus, where they trigger formation of coated carrier vesicles. We recently showed that class I ARFs (ARF1 and ARF3) localize to recycling endosomes, as well as to the Golgi, and are redundantly required for recycling of endocytosed transferrin. On the other hand, the roles of class II ARFs (ARF4 and ARF5) are not yet fully understood, and the complementary or overlapping functions of class I and class II ARFs have been poorly characterized. In this study, we find that simultaneous depletion of ARF1 and ARF4 induces extensive tubulation of recycling endosomes. Moreover, the depletion of ARF1 and ARF4 inhibits retrograde transport of TGN38 and mannose-6-phosphate receptor from early/recycling endosomes to the _trans_-Golgi network (TGN) but does not affect the endocytic/recycling pathway of transferrin receptor or inhibit retrograde transport of CD4-furin from late endosomes to the TGN. These observations indicate that the ARF1+ARF4 and ARF1+ARF3 pairs are both required for integrity of recycling endosomes but are involved in distinct transport pathways: the former pair regulates retrograde transport from endosomes to the TGN, whereas the latter is required for the transferrin recycling pathway from endosomes to the plasma membrane.
INTRODUCTION
Membrane traffic between subcellular compartments is mediated by vesicular and tubular intermediates. Members of the ADP-ribosylation factor (ARF) family of small GTPases are key regulators of transport carrier formation; ARFs trigger budding of coated carrier vesicles by recruiting coat protein complexes and regulating phospholipid metabolism (D'Souza-Schorey and Chavrier, 2006). As with other GTPases, binding of GTP to ARFs induces a conformational change that allows them to interact with a variety of effector proteins, including coat proteins such as the COPI complex, clathrin adaptor protein complexes, and GGAs, as well as lipid-modifying enzymes, such as phospholipase D, phosphatidylinositol 4-kinase, and phosphatidylinositol 4-phosphate 5-kinase (Shin and Nakayama, 2004). Conversion of GDP-bound, inactive forms of ARFs to GTP-bound, active forms is catalyzed by guanine-nucleotide exchange factors (GEFs). In mammals, there are several subfamilies of ARF-GEFs, including the brefeldin A–sensitive Gea/GBF1 and BIG1/BIG2 subfamilies, which localize mainly to the _cis_-Golgi and _trans_-Golgi network (TGN), respectively (Jackson and Casanova, 2000; Shin and Nakayama, 2004).
ARFs can be divided into three classes based on amino acid sequence similarity: class I includes ARF1 and ARF3 (humans lack ARF2); class II comprises ARF4 and ARF5; and the sole member of class III is ARF6 (Tsuchiya et al., 1991). Class I and II ARFs localize primarily to the Golgi apparatus and function in the Golgi and compartments along the secretory pathway, whereas ARF6 functions mainly at the plasma membrane (D'Souza-Schorey and Chavrier, 2006). Class I and class II ARFs share 96 and 90% of their amino acids, respectively, with other proteins of their class, and the two classes share 81% amino acid identity. Because of their high similarity, pairs of class I and II ARFs often play redundant roles (Volpicelli-Daley et al., 2005), but the specific and/or complementary functions of the ARF pairs are largely unknown.
ARF1 and ARF6 are the most thoroughly characterized ARFs. ARF1 is localized mainly to the Golgi apparatus, where it is responsible for formation of COPI-coated vesicles, whereas ARF6 is localized mainly to the plasma membrane and regulates the endocytic and recycling pathways and cytoskeletal remodeling (D'Souza-Schorey and Chavrier, 2006). On the other hand, ARF4 and ARF5 are localized to the endoplasmic reticulum (ER)–Golgi intermediate compartment (ERGIC) as well as to the Golgi (Chun et al., 2008) and are involved in transport between the ERGIC and the Golgi (Volpicelli-Daley et al., 2005). ARF1 and ARF4 are also localized to the ERGIC as well as the Golgi (Chun et al., 2008; Ben-Tekaya et al., 2010), are activated by GBF1 (Szul et al., 2007), and play a role in tubule formation from the ERGIC (Ben-Tekaya et al., 2010).
In addition to their conventional roles at the Golgi, class I and class II ARFs have a variety of functions. We and others showed that BIG2, which activates ARF1 and ARF3, localizes to recycling endosomes, as well as to the TGN, and is required for maintaining the structural integrity of recycling endosomes (Shin et al., 2004; Shen et al., 2006; Ishizaki et al., 2008). We also showed that ARF1 and ARF3 are localized to recycling endosomes and that simultaneous depletion of these ARFs induces tubulation of these compartments and inhibits the transferrin (Tfn) recycling pathway but not the endocytic pathway (Kondo et al., 2012). On the other hand, ARF4 is associated with the TGN and plays a role in transport of ciliary cargoes (Mazelova et al., 2009), and ARF5 mediates integrin internalization (Moravec et al., 2012). A recent study described a functional ARF cascade in which ARF4 and ARF5 activated by GBF1 in turn promote recruitment of BIG1 and BIG2 to the TGN after recruitment of AP-1 (Lowery et al., 2013). Thus it is likely that the cellular functions of ARFs are not confined to their principal locations.
In this study, we unexpectedly found that simultaneous depletion of ARF1 and ARF4 induces drastic tubulation of recycling endosomes and inhibits retrograde transport of TGN38 and mannose 6-phosphate receptor (MPR) from those compartments to the TGN but does not affect the Tfn recycling pathway from those compartments to the plasma membrane.
RESULTS
Combinatorial depletion of class I and class II ARFs
To understand potentially redundant functions of ARFs in membrane traffic (Volpicelli-Daley et al., 2005), we used small interfering RNAs (siRNAs) to systematically deplete HeLa cells of Golgi-localizing ARFs, either singly or in pairwise combinations, and then examined the resulting morphological alterations of the Golgi apparatus and endosomes, as revealed by staining with antibodies against GM130 (for the _cis_-Golgi), golgin-245 (for the TGN), and transferrin receptor (TfnR; for endosomes). Efficient and specific depletion of each ARF was confirmed by immunoblot analysis (Supplemental Figure S1). No single knockdown of any ARF induced significant morphological changes in these compartments (data not shown). As we previously reported, simultaneous knockdown of ARF1 and ARF3 induces tubulation of TfnR-positive endosomes (Figure 1B′′; Kondo et al., 2012). In addition, we observed extensive tubulation of TfnR-positive endosomes in cells depleted of both ARF1 and ARF4 (Figure 1D′′, enlarged images). This observation was unexpected because a previous study (Ben-Tekaya et al., 2010) showed that double knockdown of ARF1+4 induces formation of tubular structures from the ERGIC (also see Supplemental Figure S2, staining for ERGIC-53). Furthermore, the Golgi structure revealed by staining for GM130 and golgin-245 exhibited a restricted juxtanuclear localization in cells depleted of ARF1+4 (Figure 1, D and D′), in contrast to the reticular and perinuclear Golgi morphology observed in the control cells (Figure 1, A and A′). No other combinatorial knockdown (ARF1+5, ARF3+4, or ARF3+5) significantly changed the structure of the Golgi or TfnR-positive endosomes, although depletion of ARF4+5 resulted in a slightly more compact Golgi shape (Figure 1, C and C′).
FIGURE 1:

Combinatorial depletion of class I and class II ARFs. HeLa cells were treated with siRNAs against LacZ (control; A), ARF1 and ARF3 (B), ARF4 and ARF5 (C), ARF1 and ARF4 (D), ARF1 and ARF5 (E), ARF3 and ARF4 (F), or ARF3 and ARF5 (G). The cells were stained for GM130 (A–G), golgin-245 (A′–G′), or TfnR (A′′–G′′). The boxed areas in D′′ are enlarged. Bar, 20 μm.
To confirm that the endosomal tubulation observed in ARF1+4 double-knockdown cells resulted from specific ARF depletion rather than an off-target effect, we performed two sets of experiments. When we treated HeLa cells with another set of siRNA pools targeting ARF1 and ARF4 (siARF1′ and siARF4′, respectively), we also observed tubulation of TfnR-positive compartments (Supplemental Figure S3C). In rescue experiments, exogenous expression of C-terminally enhanced green fluorescent protein (EGFP)–tagged ARF1 or ARF4 in double-knockdown cells restored the tubular TfnR-positive structures to normal punctate ones (Figure 2, A, b and c, and B). By contrast, the tubular phenotype was not rescued by exogenous expression of EGFP, ARF3-EGFP, or ARF5-EGFP (Figure 2, A, a, d, and e, and B). These observations demonstrate that the tubulation of TfnR-positive endosomes observed in cells simultaneously treated with siRNAs against ARF1 and ARF4 results from specific depletion of these ARF proteins. Moreover, these findings indicate that ARF1 and ARF4 play redundant roles in maintaining the integrity of TfnR-positive endosomes. Although ARF1 and ARF3 also are indispensable for the integrity of recycling endosomes (Volpicelli-Daley et al., 2005; Kondo et al., 2012), ARF3 cannot rescue the tubulation induced by simultaneous depletion of ARF1 and ARF4, suggesting that the mechanism underlying the tubulation induced by ARF1+4 depletion is distinct from that of ARF1+3 depletion (see later discussion). Subsequently we focused on the roles of ARF1 and ARF4 in membrane trafficking via endosomes because ARF4 functions on endosomes have not been previously described.
FIGURE 2:

Alteration of endosomal structures by depletion of ARF1 and ARF4 is restored by exogenous expression of either ARF1 or ARF4. (A) HeLa cells treated with siRNAs against ARF1 and ARF4 were transfected with an expression plasmid for EGFP, ARF1-EGFP, ARF4-EGFP, ARF3-EGFP, or ARF5-EGFP. Cells were then stained for TfnR. Each image represents data from two independent experiments. Bar, 20 μm. (B) The number of cells in which TfnR-containing structures were tubulated (white), partially tubulated (gray), or not tubulated (black) in A was counted and normalized against the total number of counted cells (>20 transfected cells were counted).
Depletion of ARF1+4 induces tubulation of Rab11- and Rab4-positive endosomes
We next examined the distribution of Rab11, which localizes to recycling endosomes, in cells depleted of ARF1, ARF4, or ARF1+4 (Figure 3). Rab11- and TfnR-positive structures were extensively tubulated in cells depleted of ARF1+4 but not in cells depleted of ARF1 or ARF4 alone (Figure 3, A–A′′′ and B–B′′′, and enlarged images). The alternative set of siRNA pools against ARF1 and ARF4 (siARF1′ and siARF4′) also induced tubulation of TfnR- and Rab11-positive compartments (Supplemental Figure S3C). The tubular structures positive for TfnR and Rab11 were extensively colocalized (Supplemental Figure S3C), indicating that these structures were generated from recycling endosomal compartments.
FIGURE 3:

Redistribution of some proteins in cells depleted of ARF1 and ARF4. HeLa cells were treated with siRNAs against LacZ (control; A–E), ARF1 (A′–E′), ARF4 (A′′–E′′), or both ARF1 and ARF4 (A′′′–E′′′). Cells were then stained for Rab11 (A–A′′′), TfnR (B–B′′′), CI-MPR (C–C′′′), TGN46 (D–D′′′), or AP-1 (E–E′′′). The boxed areas in A–C are enlarged in the insets, and those in A′′′, B′′′, and C′′′ are enlarged on the right. Bar, 20 μm.
We next asked whether other endosomal Rab proteins localize to the tubular structures induced by depletion of ARF1+4. In addition to Rab11, Rab4, which localizes to early/recycling endosomes, was also observed on the tubular structures (Figure 4). By contrast, early endosomal Rab5 was not associated with the tubules. Consistent with this, punctate structures positive for EEA1, an early endosomal marker, were not changed by depletion of ARF1+4 (Supplemental Figure S3C). Thus simultaneous depletion of ARF1 and ARF4 induces tubules from Rab11- and Rab4-positive recycling endosomes. We previously showed that simultaneous depletion of ARF1 and ARF3 also induces tubulation of Rab11- and Rab4-positive endosomes (Kondo et al., 2012). As noted earlier, however, the tubules derived from recycling endosomes observed in cells depleted of ARF1+3 and of ARF1+4 are likely to be generated by distinct mechanisms.
FIGURE 4:
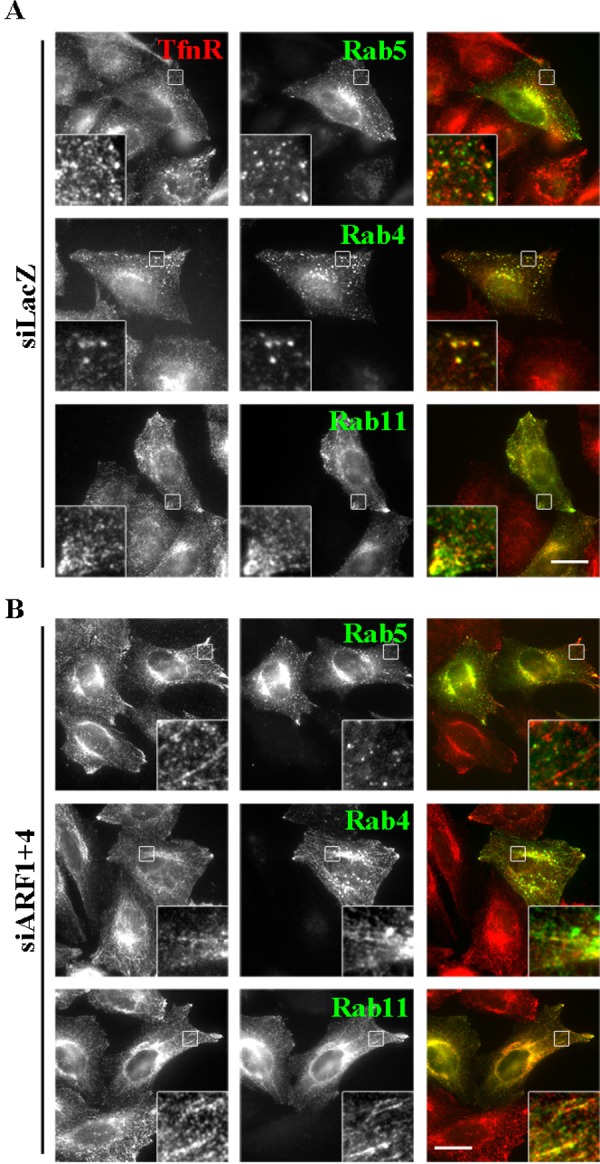
Rab4- and Rab11-positive, but not Rab5-positive, endosomes are tubulated in cells depleted of ARF1 and ARF4. HeLa cells treated with siRNAs against LacZ (A) or ARF1 and ARF4 (B) were transfected with expression plasmids encoding EGFP-Rab5, EGFP-Rab4, or EGFP-Rab11 and then immunostained for TfnR. Bar, 20 μm.
Depletion of ARF1+4 does not inhibit endocytosis or recycling of Tfn
We next examined the dynamics of the Tfn/TfnR-positive tubular structures by time-lapse imaging. When cells stably expressing TfnR-EGFP were incubated at 37°C for 30 min with Alexa Fluor 555–conjugated Tfn, endocytosed Tfn was extensively colocalized with TfnR-EGFP in both control and ARF1+4–depleted cells (Figure 5A and Supplemental Videos S1 and S2), indicating that ARF1+4 depletion does not affect endocytosis of Tfn or its accessibility to TfnR-positive structures, despite the drastic morphological changes. In control cells, a number of vesicular and short tubular structures positive for both Tfn and TfnR could be observed moving around the cytoplasm (Figure 5A, left, and Supplemental Video S1). On the other hand, in the double-knockdown cells, endocytosed Tfn and TfnR were observed in prominent tubular structures (Figure 5A, right, and Supplemental Video S2), and Tfn traveled along the tubules. Thus the Tfn-TfnR–positive tubular compartments seem to be dynamic structures along which endocytosed Tfn moves actively.
FIGURE 5:

Depletion of ARF1 and ARF4 does not affect the endocytic/recycling pathway of Tfn. (A) HeLa cells stably expressing TfnR-EGFP were transfected with siRNAs against LacZ or ARF1 and ARF4, incubated at 37°C for 30 min with Alexa Fluor 555–conjugated Tfn, and then subjected to time-lapse recording. Representative frames from Supplemental Videos S1 and S2 are shown. (B, C) HeLa cells treated with siRNAs against LacZ (B) or both ARF1 and ARF4 (C) were serum starved for 3 h and then incubated at 4°C for 50 min with Alexa Fluor 555–conjugated Tfn at 37°C for the indicated times. Bars, 20 μm. (D) Pixel intensity of Alexa Fluor 555–Tfn estimated at the indicated times. Data are shown as the ratio of the mean of cellular Tfn intensities between cells depleted of ARF1+4 and controls at each time point. The graph is representative of three independent experiments, and >50 cells were analyzed.
Given that ARF1+4 depletion altered the morphology of recycling endosomes, we examined whether this double knockdown affects the endocytic/recycling pathway of Tfn. To this end, we incubated control and double-knockdown cells at 4°C for 50 min with Alexa Fluor 488–conjugated Tfn, washed them to remove unbound Tfn, and then incubated them at 37°C for various time periods in the presence of holo-Tfn to allow internalization of Tfn. Levels of surface-bound Tfn were slightly higher in ARF1+4–depleted cells than in control cells (compare Figure 5, B and C, 0 min, and D). Internalization of Tfn was not significantly affected by knockdown of ARF1+4 (Figure 5, B and C, 5 min). Intracellular Tfn signals were markedly decreased after 30-min incubation at 37°C due to release of internalized Tfn to the extracellular medium in both control and double-knockdown cells (Figure 5, B and C, 30 min). For an unknown reason, the initial signals of surface-bound Tfn were higher in double-knockdown cells than in control cells. Therefore we plotted ratios of the intensity of Tfn signals in the siARF1+4 cells versus the intensity in siLacZ-treated cells and found that the ratios were not significantly changed at any time point (Figure 5D). These observations are consistent with a previous report showing that knockdown of ARF1 and ARF4 by shRNAs did not inhibit endocytosis or recycling of Tfn (Volpicelli-Daley et al., 2005). Taken together, these findings indicate that although ARF1 and ARF4 participate redundantly in maintaining TfnR-positive endosomal morphology, they are dispensable for Tfn endocytosis and recycling. This contrasts with observations in ARF1+3 knockdown cells, in which the double knockdown not only induces tubulation of TfnR- and Rab11-positive endosomes but also inhibits recycling of endocytosed Tfn (Kondo et al., 2012; also see Supplemental Figure S4).
Depletion of ARF1+4 inhibits retrograde transport of TGN38/46 from endosomes to the TGN
Although the Golgi morphology was somewhat altered in cells depleted of ARF1+4, GM130 and golgin-245 were still associated with Golgi membranes (Figure 1), indicating that the Golgi apparatus itself remains intact under these conditions. In striking contrast, levels of TGN46 in the Golgi region were dramatically reduced in the double-knockdown cells (Figure 3, D–D′′′). In addition, the double knockdown of ARF1+4, but neither single knockdown, redistributed cation-independent MPR (CI-MPR) from the perinuclear Golgi region to tubular structures at the cell periphery (Figure 3, C–C′′′). Similarly, we observed reduced levels of TGN46 at the TGN and redistribution of CI-MPR in cells treated with another set of siRNA pools against ARF1 and ARF4 (Supplemental Figure S3C). After arriving at the plasma membrane, CI-MPR and TGN46 are delivered to the TGN via early/recycling endosomes (Maxfield and McGraw, 2004). Therefore ARF1 and ARF4 may play redundant roles in trafficking of these transmembrane proteins from endosomes to the TGN. In addition, levels of the AP-1 γ-subunit were significantly reduced in the TGN and peripheral endosomes in cells depleted of ARF1+4 (Figure 3, E–E′′′), but the total cellular level of this protein was not altered (Supplemental Figure S5), suggesting that ARF1 and ARF4 participate in recruitment of the AP-1 complex to TGN and endosomal membranes (see Discussion).
Given the drastic reduction in TGN46 at the TGN (Figure 3D′′′), retrograde transport of TGN46 to the TGN might be inhibited in the double-knockdown cells. We therefore examined the retrograde transport of rat TGN38, an orthologue of human TGN46, which traverses through early/recycling endosomes en route to the TGN (Ghosh et al., 1998; Mallet and Maxfield, 1999; Maxfield and McGraw, 2004; Kondo et al., 2012). To this end, we performed an antibody-uptake experiment using a cell line stably expressing FLAG-tagged TGN38, as described previously (Ishizaki et al., 2008). Cells treated with siRNAs against LacZ (as a control) or against ARF1 and ARF4 were incubated with anti-FLAG antibody for 50 min at 19.5°C to allow accumulation of internalized anti-FLAG together with FLAG-TGN38, mainly in early endosomes. After an acid wash to remove cell surface–bound antibody, internalization was allowed to proceed at 37°C for various time periods. In both control and double-knockdown cells, anti-FLAG antibody was observed mainly in peripheral punctate structures at 0 min (Figure 6, A and B), indicating that internalization of FLAG-TGN38 was not affected in double-knockdown cells. In control cells, anti-FLAG was partially colocalized with golgin-97 after 30 min and extensively colocalized after 60 or 120 min (Figure 6A), indicating efficient retrograde transport of FLAG-TGN38 to the TGN. In striking contrast, in the double-knockdown cells, the majority of internalized anti-FLAG remained in cytoplasmic puncta, with a fraction in tubular structures, even after 60 or 120 min (Figure 6B). Semiquantitative analysis of the retrograde transport of FLAG-TGN38 to the TGN revealed significant inhibition of transport in double-knockdown cells (Figure 6C). Depletion of neither ARF1 nor ARF4 alone inhibited FLAG-TGN38 transport (data not shown). These results indicate that ARF1 and ARF4 redundantly regulate the retrograde transport of TGN38/46 from endosomes to the TGN. As we reported previously, depletion of ARF1+3 did not affect retrograde transport of TGN38, although it did induce extensive tubulation of endosomes (Kondo et al., 2012), similar to the effect of depleting ARF1+4.
FIGURE 6:

ARF1 and ARF4 are required for retrograde transport of FLAG-TGN38 from endosomes to the Golgi complex. Cells stably expressing FLAG-TGN38 were treated with siRNAs against LacZ (A) or both ARF1 and ARF4 (B). Cells were incubated at 19.5°C for 50 min with anti-FLAG antibodies to allow the antibody to accumulate in early endosomes (0 min) and then chased at 37°C for indicated times. Cells were then immunostained for FLAG and golgin-97. Arrows indicate tubular structures. Bars, 20 μm. (C) Golgi arrival of internalized FLAG-TGN38 was estimated by counting cells in which FLAG-TGN38 was colocalized with golgin-97. Gray bars indicate cells in which internalized FLAG-TGN38 was observed in a distinct ribbon-like structure that extensively colocalized with golgin-97; examples are marked with asterisks. Black bars indicate cells in which internalized FLAG-TGN38 was not colocalized with golgin-97. Results are means ± SD from two independent experiments.
To determine where the internalized FLAG-TGN38 was accumulating in the double-knockdown cells, we compared the trafficking of internalized anti-FLAG antibody with that of Alexa Fluor 555–conjugated Tfn and Alexa Fluor 488–conjugated EGF, which after endocytosis are transported from early endosomes to recycling endosomes and late endosomes, respectively. Endocytosis or lysosomal degradation of EGF and endocytosis of Tfn were unaffected in cells depleted of ARF1+4 (data not shown and Figure 5). In both control and double-knockdown cells, anti-FLAG was extensively colocalized with both internalized Tfn and EGF, mostly in early endosomes, after 0 or 10 min at 37°C (Figure 7, A–D, insets). After longer incubations, anti-FLAG in control cells was segregated from the Tfn- and EGF-positive endosomes and delivered to the TGN (Figure 7, A and B, 30 and 60 min, insets). By contrast, in the double-knockdown cells, anti-FLAG was extensively colocalized with EGF, although some antibody remained partially colocalized with internalized Tfn in tubular structures even after longer incubations (Figure 7, C and D, 30 and 60 min, insets). These observations suggest that in the double-knockdown cells, internalized FLAG-TGN38 was at least partially missorted to EGF-positive late endosomes instead of being transported to the TGN.
FIGURE 7:

Internalized FLAG-TGN38 is retained in endosomal structures in cells depleted of ARF1 and ARF4. Cells stably expressing FLAG-TGN38 were treated with siRNAs against LacZ (A, B) or both ARF1 and ARF4 (C, D). The cells were incubated with anti-FLAG antibody as described in Figure 6 in the presence of Alexa Fluor 555–conjugated Tfn (A, C) or Alexa Fluor 488–conjugated EGF (B, D) for the indicated time periods. Cells were then immunostained for FLAG. Bars, 20 μm.
Depletion of ARF1+4 inhibits retrograde transport of MPR from endosomes to the TGN
As shown in Figure 3, C–C′′′, knockdown of ARF1+4 results in redistribution of CI-MPR. Therefore we next asked whether the double knockdown would affect retrograde transport of MPR from the plasma membrane to the TGN via early/recycling endosomes (Medigeshi and Schu, 2003; Lin et al., 2004; Saint-Pol et al., 2004). To this end, we performed an antibody-uptake experiment using cells stably expressing EGFP-tagged cation-dependent MPR (CD-MPR). Cells treated with siRNAs against either LacZ or ARF1 and ARF4 were incubated for 50 min at 19.5°C with anti-GFP antibody. After an acid wash, internalization of the antibody was allowed to proceed at 37°C for various time periods. In both control and double-knockdown cells at 0 min, we observed anti-GFP in peripheral punctate structures, colocalized with TfnR (Figure 8, A–D), indicating that internalization of EGFP–CD-MPR was not affected in double-knockdown cells. In control cells, anti-GFP was gradually transported to the TGN over time: it was colocalized well with golgin-245 after 30 min (Figure 8A), and after 120 min, the majority of anti-GFP internalized was colocalized with golgin-245 (Figure 8A) and segregated from TfnR (Figure 8B). On the other hand, in cells depleted of ARF1+4, we observed anti-GFP in tubular TfnR-positive endosomes after 10 min, and some anti-GFP was retained in these tubular structures after 120 min (Figure 8D). At this time point, anti-GFP was also observed in close proximity to the plasma membrane, suggesting that EGFP–CD-MPR might be recycled back to the plasma membrane rather than transported retrogradely to the TGN. Semiquantitative analysis revealed that colocalization of internalized anti-GFP was significantly lower in double-knockdown cells than in control cells (Figure 8E). Together, these observations indicate that ARF1 and ARF4 redundantly regulate the retrograde transport of MPR as well as TGN38/46 from recycling endosomes to the TGN. We also investigated whether depletion of ARF1+3 inhibits the retrograde transport of EGFP–CD-MPR, but we did not detect any difference in endosome-to-Golgi transport of EGFP–CD-MPR between control and ARF1+3–knockdown cells (Supplemental Figure S6). Thus the ARF1+3 and ARF1+4 pairs are both required for recycling endosomal integrity, but it is likely that these pairs regulate distinct transport pathways from endosomes, with the former regulating the recycling pathway to the plasma membrane and the latter regulating the retrograde transport pathway to the TGN.
FIGURE 8:

ARF1 and ARF4 are required for retrograde transport of EGFP-CD-MPR from endosomes to the Golgi complex. Cells stably expressing EGFP-CD-MPR were treated with siRNAs against LacZ (A, B) or both ARF1 and ARF4 (C, D). Cells were incubated at 19.5°C for 50 min with anti-GFP antibody to allow the antibody to accumulate in early endosomes and then chased at 37°C for indicated time periods. The cells were then immunostained for GFP and either golgin-245 (A, C) or TfnR (B, D). Images were acquired using an A1R-MP confocal microscope. Bars, 20 μm. (E) Colocalization of internalized EGFP-CD-MPR with golgin-245 was quantified using MetaMorph software. The graph is representative of three independent experiments.
Depletion of ARF1+4 does not inhibit retrograde transport of CD4-furin through late endosomes
Next we examined whether ARF1 and ARF4 are involved in another retrograde pathway, from late endosomes to the TGN. To this end, we used a cell line stably expressing a CD4-furin chimera (Mallet and Maxfield, 1999; Maxfield and McGraw, 2004; Schapiro et al., 2004; Ishizaki et al., 2008) and analyzed retrograde transport of CD4-furin by following extracellularly applied anti-CD4 antibody. Cells treated with siRNAs against either LacZ or ARF1 and ARF4 were incubated for 50 min at 4°C with anti-CD4 antibody. After washing with phosphate-buffered saline (PBS), internalization was allowed to proceed at 37°C for the indicated time periods (Figure 9). Immunofluorescence and subsequent semiquantitative analysis did not reveal any significant differences between control and double-knockdown cells over the course of incubation (Figure 9, A–C). After 120 min, the majority of CD4-furin reached golgin-97–positive Golgi structures (Figure 9C) in both control and double-knockdown cells. Therefore it is likely that the ARF1+4 pair does not participate in the retrograde transport of CD4-furin from late endosomes to the TGN.
FIGURE 9:

ARF1 and ARF4 are not required for retrograde transport of CD4-furin from endosomes to the Golgi complex. Cells stably expressing CD4-furin were treated with siRNAs against LacZ (A) or both ARF1 and ARF4 (B). Cells were then incubated at 4°C for 60 min with anti-CD4 antibodies to allow the antibody to bind cell surface (A, B; 0 min) and then chased at 37°C for indicated times. Cells were immunostained for CD4 and golgin-97. Bars, 20 μm. (C) Golgi arrival of internalized CD4-furin was estimated as described in Figure 6. Gray bars indicate cells in which internalized CD4-furin was observed in a distinct ribbon-like structure that extensively colocalized with golgin-97; examples are marked with asterisks. Black bars indicate cells in which the internalized CD4-furin was not colocalized with golgin-97. Results are means ± SD from two independent experiments.
DISCUSSION
We showed that ARF1 and ARF4 play redundant roles in maintaining the integrity of recycling endosomal compartments, which are positive for Rab11 and Rab4 but not for Rab5. The tubular structures induced by ARF1+4 depletion did not include early endosomes (Figure 4 and Supplemental Figure S3C) or late endosomes (Figure 7). Moreover, we found that ARF1 and ARF4 are required for the retrograde transport of TGN38/46 and MPR from recycling endosomes to the TGN but not that of CD4-furin from late endosomes to the TGN or for endocytosis of TGN38, MPR, Tfn, or EGF from the plasma membrane.
Previous studies showed that exogenously expressed ARF1 and ARF4 are mainly localized to the ERGIC and the Golgi (Kawamoto et al., 2002; Chun et al., 2008; Ben-Tekaya et al., 2010), and their depletion induces formation of tubules from the ERGIC (Ben-Tekaya et al., 2010; Supplemental Figure S2). Although class I and II ARFs localize mainly to the Golgi apparatus, ARF proteins have been proposed to serve various functions at other cellular compartments. Some examples follow: 1) Class I ARFs localize to recycling endosomes and regulate the Tfn recycling pathway (Kondo et al., 2012); BIG2, which is an ARF-GEF for ARF1 and ARF3, localizes to recycling endosomes (Shin et al., 2004). 2) ARF1 can be activated at the plasma membrane, where it regulates dynamin-independent endocytosis (Cohen et al., 2007; Kumari and Mayor, 2008). 3) Class II ARFs and ARF6 share effector proteins, the arfophilins/Rab11-FIPs, with Rab11 (Shin et al., 1999; Meyers and Prekeris, 2002; Shiba et al., 2006), although a functional cross-talk between class II ARFs and Rab11 is not clear. 4) ARF5 is activated by BRAGs, a subfamily of ARF-GEFs, and mediates integrin internalization (Moravec et al., 2012). 5) ARF4 plays an important role in formation of the post-TGN carriers targeted to primary cilia (Mazelova et al., 2009) and has been identified in primary cilia by proteomic analysis (Ishikawa et al., 2012). Here we extended the known repertoire of ARFs in other cellular compartments by elucidating a redundant function of ARF1 and ARF4 in endosome-to-Golgi transport.
Tubulation of recycling endosomes is not induced by depletion of ARF1, ARF3, or ARF4 alone but is induced by pairwise depletion of ARF1+4 or ARF1+3. However, the tubules induced by depletion of ARF1+4 appear to be more extensive than those induced by depletion of ARF1+3 (Figure 1) and cannot be rescued by exogenous expression of ARF3 (Figure 2). These observations indicate that depletion of ARF1+4 and that of ARF1+3 induce tubules from recycling endosomes by distinct mechanisms. Intriguingly, ARF1+4 and ARF1+3 knockdowns affect distinct transport pathways from recycling endosomes: ARF1+4 depletion inhibits the retrograde transport of TGN38 and MPR to the TGN (Figures 6 and 8) but not Tfn recycling (Volpicelli-Daley et al., 2005; Figure 5), whereas ARF1+3 depletion inhibits the Tfn recycling pathway but not the retrograde pathway from recycling endosomes (Kondo et al., 2012; Supplemental Figure S4). On the basis of these results, we conclude that the ARF1+4 and ARF1+3 pairs participate in distinct transport pathways from recycling endosomes, probably by functioning in distinct membrane microdomains: on one hand, ARF1 regulates, redundantly with ARF4, retrograde transport from recycling endosomes to the TGN; on the other hand, redundantly with ARF3, it regulates recycling to the plasma membrane.
How do ARF1 and ARF4 regulate retrograde transport from endosomes? It is possible that the ARF1+ARF4 pair is a primary regulator of recruitment of the AP-1 adaptor complex to TGN and endosomal membranes (Figure 3E′′′). Because AP-1 is required for endosome-to-TGN transport of MPR (Meyer et al., 2000), ARF1 and ARF4 may be involved in retrograde transport through recruitment of the AP-1 complex. The defect in MPR retrograde transport induced by knockdown of ARF1+4, however, was not phenocopied by knockdown of AP-1 (Seaman, 2007; data not shown). Thus a direct functional link between AP-1 and ARF1+ARF4 in retrograde trafficking remains to be elucidated. Because depletion of ARF1+4 dissociates COPI from the Golgi (Volpicelli-Daley et al., 2005), we asked whether knockdown of COPI as well as that of GBF1 phenocopied that of ARF1+4. Both knockdowns, however, failed to induce tubulation of TfnR-positive endosomes (Guo et al., 2008; Saitoh et al., 2009; Supplemental Figure S7). Retromer is also required for the retrograde transport of MPR and TGN38 (Seaman, 2004; Lieu and Gleeson, 2010). We examined whether ARF1+4 knockdown affected the localization of Vps35, a component of the retromer complex, but did not find any changes relative to control cells (Supplemental Figure S2), suggesting that the ARF1+4 pair does not participate in recruitment of the retromer complex. EpsinR, which interacts with AP-1 and clathrin, is required for retrograde transport of TGN46 and CI-MPR (Hirst et al., 2003; Saint-Pol et al., 2004). In addition, brefeldin A causes redistribution of epsinR from membranes into the cytosol, suggesting that epsinR membrane association requires ARF proteins (Hirst et al., 2003). Thus epsinR is another candidate downstream effector of ARF1 and ARF4.
It remains important to determine how distinct combinations of ARFs sort cargoes targeted to distinct organelles and regulate separate transport processes on the same compartments. ARFs may act within distinct membrane microdomains along with distinct effector proteins. Therefore identification of specific downstream effectors for ARF1+ARF4 and ARF1+ARF3 may allow us to understand the mechanisms underlying the regulation of the intricate processes of trafficking from recycling endosomes.
MATERIALS AND METHODS
Plasmids
An expression vector for C-terminally EGFP-tagged ARF4 was constructed by subcloning the entire coding sequence of human ARF4 into the pCAG vector. Construction of expression vectors for C-terminally HA- or EGFP-tagged ARFs was described previously (Takatsu et al., 2002; Kondo et al., 2012; Makyio et al., 2012). Construction of expression vectors for N-terminally EGFP-tagged Rab11a and Rab4 was described previously (Yamamoto et al., 2010; Takahashi et al., 2011). The EGFP-Rab5 construct was kindly provided by Marino Zerial (Max Planck Institute, Dresden, Germany).
Antibodies and reagents
Sources of antibodies used in this study were as follows: polyclonal rabbit anti-GBF1 was prepared as described previously (Kawamoto et al., 2002); polyclonal rabbit anti-TGN46 (Kain et al., 1998) was a kind gift from Minoru Fukuda (Sanford-Burnham Medical Research Institute, La Jolla, CA); polyclonal rabbit anti-golgin-97 (Yoshimura et al., 2004) was a kind gift from Nobuhiro Nakamura (Kyoto Sangyo University, Kyoto, Japan); polyclonal rabbit anti-ARF6 (Honda et al., 1999) was a kind gift from Yasunori Kanaho (Tsukuba University, Tsukuba, Japan); polyclonal rabbit anti-ARF4 was from Protein Tech (Chicago, IL); polyclonal rabbit anti-GFP was from Molecular Probes (Eugene, OR); monoclonal mouse antibodies against GM130, golgin-245, EEA1, CD4, Rab11, ARF3, and GFP (JL-8, for immunoblotting) were from BD Biosciences (San Diego, CA); monoclonal mouse antibodies against γ-adaptin (100.3) and FLAG (M2) were from Sigma-Aldrich (St. Louis, MO); monoclonal mouse anti-CI-MPR was from Thermo Scientific (Waltham, MA); monoclonal mouse anti-TfnR was from Invitrogen (Carlsbad, CA); monoclonal mouse anti-ERGIC53 was from Alexis (San Diego, CA); monoclonal mouse anti-ARF1 was from Enzo Life Sciences (Plymouth Meeting, PA); monoclonal mouse anti-ARF5 was from Abnova (Taipei City, Taiwan); monoclonal mouse anti-ARF6 was from Santa Cruz Biotechnology (Santa Cruz, CA); monoclonal mouse anti–β-actin and anti–β-tubulin were from Millipore (Billerica, MA); monoclonal rat anti-HA was from Roche Applied Sciences (Indianapolis, IN); polyclonal goat anti-VPS35 was from Imgenex (San Diego, CA); Alexa Fluor–conjugated secondary antibodies were from Molecular Probes; and horseradish peroxidase–conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Alexa Fluor–conjugated Tfn and EGF were purchased from Molecular Probes.
Cell culture, RNA interference, immunofluorescence, and time-lapse imaging analysis
Culture of HeLa cells and transfection of expression plasmids were performed as described previously (Shin et al., 2004, 2005). HeLa cell lines stably expressing FLAG-TGN38, CD4-furin, and TfnR-EGFP were established as described previously (Ishizaki et al., 2008; Takahashi et al., 2012). A HeLa cell line stably expressing EGFP-CD-MPR was established by transfection of HeLa cells with pCIpreEGFP-CDMPR, which was constructed by replacing the CI-MPR portion of pCIpreEGFP-CIMPRtail (Waguri et al., 2003) with the coding sequence of mature rat CD-MPR and subsequent selection in the presence of G418 sulfate. Knockdowns of ARFs, β-COP, or GBF1 were performed as described previously (Ishizaki et al., 2006, 2008; Saitoh et al., 2009; Takashima et al., 2011; Kondo et al., 2012). Briefly, pools of siRNAs targeted against the 3′-untranslated region of human ARF1 or ARF3, as described previously (Kondo et al., 2012), ARF4 (PCR amplified using the primer pair 5′-TAAAGAAATTCTCCTTGGGAAAAAA-3′ and 5′-TAACAAAAGCAACATGCAACATAA-3′), or the partial coding and 3′-untranslated regions of human ARF5 (PCR amplified using the primer pair 5′-ACCTCTGCCACCCAAGGCAC-3′ and 5′-CAAACCACAGCAAATTTATTACTC-3′) were prepared using a BLOCK-iT RNAi TOPO transcription kit and a BLOCK-iT Dicer RNAi kit (Invitrogen). For siARF4′, siGENOME SMARTpool (Dharmacon, Lafayette, CO) was purchased. Cells were transfected with the siRNAs using Lipofectamine 2000 (Invitrogen) and incubated for 24 h. The transfected cells were then split and seeded onto culture dishes. After 24 h, the cells were transfected again with the siRNAs and incubated for 48 h. The transfected cells were then transferred to new culture dishes containing coverslips, incubated for another 24 h, and processed for immunofluorescence and immunoblot analyses as described previously (Shin et al., 1997; Ishizaki et al., 2008; Nishimoto-Morita et al., 2009; Takahashi et al., 2012). In the case of simultaneous knockdown of ARF1 and ARF4, cells were directly seeded onto coverslips or culture dishes and incubated for 24 h. The cells were transfected with siRNAs using Lipofectamine 2000, incubated for another 48 h, and processed for immunofluorescence and immunoblot analyses. For time-lapse recording, the TfnR-EGFP expressing cells (Kondo et al., 2012; Takatsu et al., 2013) were treated with siRNAs as described, incubated in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–buffered MEM, placed on a microscope stage prewarmed to 37°C, and then observed using an A1R-MP confocal microscope (Nikon, Melville, NY). Images were acquired sequentially every 2 s.
Tfn and antibody-uptake experiments
Assay for internalization and recycling of Tfn was carried out according to previously described protocols (Shin et al., 2004). HeLa cells were transfected with a pool of siRNAs against either LacZ (as a control) or ARFs as described. At 72 or 120 h posttransfection, cells were serum starved for 3 h in MEM containing 0.2% bovine serum albumin and then incubated on ice for 50 min with Alexa Fluor 555–conjugated Tfn. After washing out of fluorescent Tfn with PBS on ice, cells were incubated in medium containing unlabeled holo-Tfn at 37°C for the indicated times and then processed for immunofluorescence analysis. Immunofluorescence analysis was performed using an Axiovert 200 MAT microscope (Carl Zeiss, Jena, Germany) for epifluorescence images or an A1R-MP confocal microscope for confocal images. The intensity of fluorescent Tfn was quantitated using the MetaMorph imaging software (Molecular Devices, Sunnyvale, CA).
Uptake of anti-GFP, anti-FLAG, or anti-CD4 antibody in cells stably expressing EGFP-CD-MPR, FLAG-TGN38, or CD4-furin, respectively, was assayed as described previously (Takahashi et al., 1995; Shin et al., 2004, 2005; Ishizaki et al., 2008) with some modifications. Briefly, HeLa cells stably expressing EGFP-CD-MPR, FLAG-TGN38, or CD4-furin were mock treated with a pool of siRNAs against LacZ or a control siRNA pool (Dharmacon) or treated with a pool of siRNAs against ARFs or siGENOME SMARTpool against ARF4. Cells expressing FLAG-TGN38 and CD4-furin were incubated with 15 mM sodium butyrate for 16 h before antibody uptake. The cells were then incubated for 60 min with polyclonal anti-GFP or monoclonal anti-FLAG (M2) antibody at 19.5°C or monoclonal anti-CD4 (Leu3a) at 4°C, as appropriate; subjected to an acid wash (0.5% acetic acid, pH 3.0, 50 mM NaCl) in the anti-GFP and anti-FLAG uptake assay; and finally incubated at 37°C for the indicated periods of time. Colocalization of internalized anti-GFP with golgin-245 in the EGFP-CD-MPR–expressing cells was quantified using MetaMorph imaging software.
Supplementary Material
Supplemental Materials
Acknowledgments
We thank Chikako Yagi for technical support. This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Special Coordination Fund for Promoting Science and Technology; the Targeted Proteins Research Program; the Takeda Science Foundation; and the Inamori Foundation.
Abbreviations used:
ARF
ADP-ribosylation factor
CD-MPR
cation-dependent MPR
CI-MPR
cation-independent MPR
EGFP
enhanced green fluorescent protein
ER
endoplasmic reticulum
ERGIC
ER-Golgi intermediate compartment
MPR
mannose-6-phosphate receptor
siRNA
small interfering RNA
Tfn
transferrin
TfnR
transferrin receptor
TGN
_trans_-Golgi network
Footnotes
*These authors contributed equally to this work.
REFERENCES
- Ben-Tekaya H, Kahn RA, Hauri H-P. ADP ribosylation factors 1 and 4 and group VIA phospholipase A2 regulate morphology and intraorganellar traffic in the endoplasmic reticulum-Golgi intermediate compartment. Mol Biol Cell. 2010;21:4130–4140. doi: 10.1091/mbc.E10-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Shapovalova Z, Dejgaard SY, Presley JF, Melancon P. Characterization of class I and II ADP-ribosylation factors (Arfs) in live cells: GDP-bound class II Arfs associate with the ER-Golgi intermediate compartment independently of GBF1. Mol Biol Cell. 2008;19:3488–3500. doi: 10.1091/mbc.E08-04-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LA, Honda A, Varnai P, Brown FD, Balla T, Donaldson JG. Active Arf6 recruits ARNO/cytohesin GEFs to the PM by binding their PH domains. Mol Biol Cell. 2007;18:2244–2253. doi: 10.1091/mbc.E06-11-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- Ghosh RN, Mallet WG, Soe TT, McGraw TE, Maxfield FR. An endocytosed TGN38 chimeric protein is delivered to the TGN after trafficking through the endocytic recycling compartment in CHO cells. J Cell Biol. 1998;142:923–936. doi: 10.1083/jcb.142.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Punj V, Sengupta D, Linstedt AD. Coat-tether interaction in Golgi organization. Mol Biol Cell. 2008;19:2830–2843. doi: 10.1091/mbc.E07-12-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J, Motley A, Harasaki K, Peak Chew SY, Robinson MS. EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol Biol Cell. 2003;14:625–641. doi: 10.1091/mbc.E02-09-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda A, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 1999;99:521–532. doi: 10.1016/s0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Thompson J, Yates JR 3rd, Marshall WF. Proteomic analysis of mammalian primary cilia. Curr Biol. 2012;22:414–419. doi: 10.1016/j.cub.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki R, Shin HW, Iguchi-Ariga SM, Ariga H, Nakayama K. AMY-1 (associate of Myc-1) localization to the trans-Golgi network through interacting with BIG2, a guanine-nucleotide exchange factor for ADP-ribosylation factors. Genes Cells. 2006;11:949–959. doi: 10.1111/j.1365-2443.2006.00991.x. [DOI] [PubMed] [Google Scholar]
- Ishizaki R, Shin H-W, Mitsuhashi H, Nakayama K. Redundant roles of BIG2 and BIG1, guanine-nucleotide exchange factors for ADP-ribosylation factors in membrane traffic between the trans-Golgi network and endosomes. Mol Biol Cell. 2008;19:2650–2660. doi: 10.1091/mbc.E07-10-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CL, Casanova JE. Turning on ARF: the Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000;10:60–67. doi: 10.1016/s0962-8924(99)01699-2. [DOI] [PubMed] [Google Scholar]
- Kain R, Angata K, Kerjaschki D, Fukuda M. Molecular cloning and expression of a novel human trans-Golgi network glycoprotein, TGN51, that contains multiple tyrosine-containing motifs. J Biol Chem. 1998;273:981–988. doi: 10.1074/jbc.273.2.981. [DOI] [PubMed] [Google Scholar]
- Kawamoto K, Yoshida Y, Tamaki H, Torii S, Shinotsuka C, Yamashina S, Nakayama K. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic. 2002;3:483–495. doi: 10.1034/j.1600-0854.2002.30705.x. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Hanai A, Nakai W, Katoh Y, Nakayama K, Shin H-W. ARF1 and ARF3 are required for the integrity of recycling endosomes and the recycling pathway. Cell Struct Funct. 2012;37:141–154. doi: 10.1247/csf.12015. [DOI] [PubMed] [Google Scholar]
- Kumari S, Mayor S. ARF1 is directly involved in dynamin-independent endocytosis. Nat Cell Biol. 2008;10:30–41. doi: 10.1038/ncb1666. [DOI] [PubMed] [Google Scholar]
- Lieu ZZ, Gleeson PA. Identification of different itineraries and retromer components for endosome-to-Golgi transport of TGN38 and Shiga toxin. Eur J Cell Biol. 2010;89:379–393. doi: 10.1016/j.ejcb.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Lin SX, Mallet WG, Huang AY, Maxfield FR. Endocytosed cation-independent mannose 6-phosphate receptor traffics via the endocytic recycling compartment en route to the trans-Golgi network and a subpopulation of late endosomes. Mol Biol Cell. 2004;15:721–733. doi: 10.1091/mbc.E03-07-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery J, Szul T, Styers M, Holloway Z, Oorschot V, Klumperman J, Sztul E. The Sec7 guanine nucleotide exchange factor GBF1 regulates membrane recruitment of BIG1 and BIG2 to the trans-Golgi network (TGN) J Biol Chem. 2013;288:11532–11545. doi: 10.1074/jbc.M112.438481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makyio H, et al. Structural basis for Arf6-MKLP1 complex formation on the Flemming body responsible for cytokinesis. EMBO J. 2012;31:2590–2603. doi: 10.1038/emboj.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet WG, Maxfield FR. Chimeric forms of furin and TGN38 are transported with the plasma membrane in the trans-Golgi network via distinct endosomal pathways. J Cell Biol. 1999;146:345–359. doi: 10.1083/jcb.146.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28:183–192. doi: 10.1038/emboj.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medigeshi GR, Schu P. Characterization of the in vitro retrograde transport of MPR46. Traffic. 2003;4:802–811. doi: 10.1034/j.1600-0854.2003.00136.x. [DOI] [PubMed] [Google Scholar]
- Meyer C, Zizioli D, Lausmann S, Eskelinen EL, Hamann J, Saftig P, von Figura K, Schu P. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 2000;19:2193–2203. doi: 10.1093/emboj/19.10.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JM, Prekeris R. Formation of mutually exclusive Rab11 complexes with members of the family of Rab11-interacting proteins regulates Rab11 endocytic targeting and function. J Biol Chem. 2002;277:49003–49010. doi: 10.1074/jbc.M205728200. [DOI] [PubMed] [Google Scholar]
- Moravec R, Conger KK, D'Souza R, Allison AB, Casanova JE. BRAG2/GEP100/IQSec1 interacts with clathrin and regulates a5b1 integrin endocytosis through activation of ADP ribosylation factor 5 (Arf5) J Biol Chem. 2012;287:31138–31147. doi: 10.1074/jbc.M112.383117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto-Morita K, Shin H-W, Mitsuhashi H, Kitamura M, Zhang Q, Johannes L, Nakayama K. Differential effects of depletion of ARL1 and ARFRP1 on membrane trafficking between the trans-Golgi network and endosomes. J Biol Chem. 2009;284:10583–10592. doi: 10.1074/jbc.M900847200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Pol A, et al. Clathrin adaptor epsinR is required for retrograde sorting on early endosomal membranes. Dev Cell. 2004;6:525–538. doi: 10.1016/s1534-5807(04)00100-5. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Shin H-W, Yamada A, Waguri S, Nakayama K. Three homologous ArfGAPs participate in coat protein I-mediated transport. J Biol Chem. 2009;284:13948–13957. doi: 10.1074/jbc.M900749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro FB, Soe TT, Mallet WG, Maxfield FR. Role of cytoplasmic domain serines in intracellular trafficking of furin. Mol Biol Cell. 2004;15:2884–2894. doi: 10.1091/mbc.E03-09-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MNJ. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MNJ. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J Cell Sci. 2007;120:2378–2389. doi: 10.1242/jcs.009654. [DOI] [PubMed] [Google Scholar]
- Shen X, Xu K-F, Fan Q, Pacheco-Rodriguez G, Moss J, Vaughan M. Association of brefeldin A-inhibited guanine nucleotide-exchange protein 2 (BIG2) with recycling endosomes during transferrin uptake. Proc Natl Acad Sci USA. 2006;103:2635–2640. doi: 10.1073/pnas.0510599103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba T, Koga H, Shin H-W, Kawasaki M, Kato R, Nakayama K, Wakatsuki S. Structural basis for Rab11-dependent membrane recruitment of a family of Rab11-interacting protein 3 (FIP3)/Arfophilin-1. Proc Natl Acad Sci USA. 2006;103:15416–15421. doi: 10.1073/pnas.0605357103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H-W, Kobayashi H, Kitamura M, Waguri S, Suganuma T, Uchiyama Y, Nakayama K. Roles of ARFRP1 (ADP-ribosylation factor-related protein 1) in post-Golgi membrane trafficking. J Cell Sci. 2005;118:4039–4048. doi: 10.1242/jcs.02524. [DOI] [PubMed] [Google Scholar]
- Shin H-W, Morinaga N, Noda M, Nakayama K. BIG2, a guanine nucleotide exchange factor for ADP-ribosylation factors: its localization to recycling endosomes and implication in the endosome integrity. Mol Biol Cell. 2004;15:5283–5294. doi: 10.1091/mbc.E04-05-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H-W, Nakayama K. Guanine nucleotide-exchange factors for Arf GTPases: their diverse functions in membrane traffic. J Biochem. 2004;136:761–767. doi: 10.1093/jb/mvh185. [DOI] [PubMed] [Google Scholar]
- Shin HW, Shinotsuka C, Torii S, Murakami K, Nakayama K. Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p. J Biochem. 1997;122:525–530. doi: 10.1093/oxfordjournals.jbchem.a021784. [DOI] [PubMed] [Google Scholar]
- Shin O-H, Ross AH, Mihai I, Exton JH. Identification of arfophilin, a target protein for GTP-bound class II ADP-ribosylation factors. J Biol Chem. 1999;274:36609–36615. doi: 10.1074/jbc.274.51.36609. [DOI] [PubMed] [Google Scholar]
- Szul T, Grabski R, Lyons S, Morohashi Y, Shestopal S, Lowe M, Sztul E. Dissecting the role of the ARF guanine nucleotide exchange factor GBF1 in Golgi biogenesis and protein trafficking. J Cell Sci. 2007;120:3929–3940. doi: 10.1242/jcs.010769. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kubo K, Waguri S, Yabashi A, Shin H-W, Katoh Y, Nakayama K. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J Cell Sci. 2012;125:4049–4057. doi: 10.1242/jcs.102913. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Nakagawa T, Banno T, Watanabe T, Murakami K, Nakayama K. Localization of furin to the trans-Golgi network and recycling from the cell surface involves Ser and Tyr residues within the cytoplasmic domain. J Biol Chem. 1995;270:28397–28401. doi: 10.1074/jbc.270.47.28397. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Takei T, Koga H, Takatsu H, Shin H-W, Nakayama K. Distinct roles of Rab11 and Arf6 in the regulation of Rab11-FIP3/arfophilin-1 localization in mitotic cells. Genes Cells. 2011;16:938–950. doi: 10.1111/j.1365-2443.2011.01538.x. [DOI] [PubMed] [Google Scholar]
- Takashima K, Saitoh A, Hirose S, Nakai W, Kondo Y, Takasu Y, Kakeya H, Shin HW, Nakayama K. GBF1-Arf-COPI-ArfGAP-mediated Golgi-to-ER transport involved in regulation of lipid homeostasis. Cell Struct Funct. 2011;36:223–235. doi: 10.1247/csf.11035. [DOI] [PubMed] [Google Scholar]
- Takatsu H, Katoh Y, Ueda T, Waguri S, Murayama T, Takahashi S, Shin H-W, Nakayama K. Mitosis-coupled, microtubule-dependent clustering of endosomal vesicles around centrosomes. Cell Struct Funct. 2013;38:31–41. doi: 10.1247/csf.12028. [DOI] [PubMed] [Google Scholar]
- Takatsu H, Yoshino K, Toda K, Nakayama K. GGA proteins associate with Golgi membranes through interaction between their GGAH domains and ADP-ribosylation factors. Biochem J. 2002;365:369–378. doi: 10.1042/BJ20020428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya M, Price SR, Tsai S-C, Moss J, Vaughan M. Molecular identification of ADP-ribosylation factor mRNAs and their expression in mammalian cells. J Biol Chem. 1991;266:2772–2777. [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Li Y, Zhang CJ, Kahn RA. Isoform-selective effects of the depletion of ADP-ribosylation factors 1–5 on membrane traffic. Mol Biol Cell. 2005;16:4495–4508. doi: 10.1091/mbc.E04-12-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waguri S, Dewitte F, Le Borgne R, Rouille Y, Uchiyama Y, Dubremetz JF, Hoflack B. Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol Biol Cell. 2003;14:142–155. doi: 10.1091/mbc.E02-06-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Koga H, Katoh Y, Takahashi S, Nakayama K, Shin HW. Functional cross-talk between Rab14 and Rab4 through a dual effector, RUFY1/Rabip4. Mol Biol Cell. 2010;21:2746–2755. doi: 10.1091/mbc.E10-01-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura S, Yamamoto A, Misumi Y, Sohda M, Barr FA, Fujii G, Shakoori A, Ohno H, Mihara K, Nakamura N. Dynamics of Golgi matrix proteins after the blockage of ER to Golgi transport. J Biochem. 2004;135:201–216. doi: 10.1093/jb/mvh024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials