VEGF Inhibition, Hypertension, and Renal Toxicity (original) (raw)
. Author manuscript; available in PMC: 2013 Aug 19.
Published in final edited form as: Curr Oncol Rep. 2012 Aug;14(4):285–294. doi: 10.1007/s11912-012-0242-z
Abstract
The use of anti-angiogenic agents as part of the therapeutic armamentarium for advanced stage solid tumors has become standard of care in several instances, particularly for renal cell carcinoma, non-small cell lung carcinoma, colorectal carcinoma, and gastrointestinal stromal tumors. These agents primarily target vascular endothelial growth factor (VEGF) and/or its receptors, and include bevacizumab, a humanized monoclonal antibody against VEGF, as well as tyrosine kinase inhibitors that target several receptor tyrosine kinases (RTK), including VEGF receptors. These therapies, as a general class of angiogenic medications, have been shown to have common adverse vascular effects attributable directly or indirectly to their anti-VEGF effects, including hypertension, renal vascular injury, often manifested by proteinuria and thrombotic microangiopathy, and congestive heart failure. Knowledge of these common side effects and their underlying mechanisms may allow for more accurate and prompt diagnoses, timely clinical interventions, and the development of rational and standard treatments. These measures may minimize patient morbidity and mortality, not only by the treatment of side effects, but also by minimizing the disruption of treatment of the underlying malignancy, as well as improving patient quality of life.
Keywords: Vascular Endothelial Growth Factor, Hypertension, Renal Toxicity, Proteinuria, Podocyturia, Tyrosine Kinase Inhibitors, Bevacizumab, Thrombotic Microangiopathy
Introduction
The concept of inhibiting angiogenesis as a strategy for the treatment of certain malignancies was proposed by Folkman in 1971 [1]. The use of angiogenesis inhibition as a therapeutic modality, either as monotherapy or as part of combination chemotherapy, has become standard treatment for several tumor types, particularly solid tumors, such as metastatic renal cell carcinoma (mRCC), non-small cell lung carcinoma, gastrointestinal stromal tumors (GIST), and colorectal carcinoma. One of the main angiogenic growth factors, and its downstream pathways, that is a major therapeutic target, is vascular endothelial growth factor (VEGF) and its receptors. This review will concentrate largely on the renal and vascular effects of VEGF inhibition.
The current FDA approved medications that target VEGF include, bevacizumab, a humanized monoclonal antibody directed against VEGF-A, through extracellular blockade of ligand-receptor binding, and more recently, the small molecule tyrosine kinase inhibitors (TKI), sunitinib, sorafenib, and pazopanib, which target VEGF receptors (VEGFR1,VEGFR-2, and VEGFR3), platelet derived growth factor receptor-α (PDGFRα), and/or PDGFRβ, FLT-3 (fms-related tyrosine kinase 3), the stem cell factor receptor (KIT), and the product of the RET proto-oncogene [2]. Sorafenib, in addition, inhibits Raf serine/threonine kinase [2]. The number and severity of the potential target and “off-target” toxicities of these anti-angiogenic agents are dependent in large part on their molecular target(s), whether they are used as monotherapy or in combination with other anti-neoplastic medications, tumor type and extent, as well as patient co-morbidities. However, regardless of the specific medication, certain adverse vascular effects to varying extents are shared among these anti-angiogenic agents, as a class, specifically, abnormalities involving the vasculature, hypertension, proteinuria, renal insufficiency, and congestive heart failure [3].
Vascular Endothelial Growth Factor (VEGF)
VEGF is a 45 kDa glycoprotein expressed in multiple organs which plays a key role in maintaining homeostasis and cell survival. Specific cells that express VEGF include progenitor endothelial cells, endothelial cells (EC), podocytes (renal epithelial cells), fibroblasts, macrophages, and certain tumor types [4, 5•]. The gene undergoes alternative splicing, and six VEGF isoforms have been identified, with the most biologically active variant being VEGF165 (VEGF-A) [4, 5•]. VEGF binds 3 tyrosine kinase receptors, VEGFR-1 (fms-like tyrosine kinase [Flt-1]), VEGFR-2 (kinase domain region [KDR] human homologue or Flk-1 murine homologue), and VEGFR-3 (fms-like tyrosine kinase [Flt-4]), which only responds to VEGF-C or VEGF-D, and is found predominantly on lymphatic endothelium [4, 5•]. VEGFR-1 and VEGFR-2 have extracellular domains, and the extracellular domain of Flt-1 has a soluble form (s-Flt-1), capable of binding and inactivating circulating VEGF, as well as membrane bound VEGFR-1 and VEGFR-2 [6]. These two receptors are also expressed on multiple cell types, such as tumor cells, EC progenitors, and mature endothelial cells, including glomerular, preglomerular and peritubular [5•]. Most of the significant VEGF signaling in EC cells is mediated by VEGFR-2 [5•].
VEGF Dysregulation and Renal and Vascular Disease
The role of VEGF as a survival, trophic and proliferative factor for vascular endothelium is well recognized. However, only recently evidence has emerged implicating its role in the regulation of renal vascular endothelium both in health and disease [7]. Anti-VEGF therapy, leading to low free VEGF levels, may cause endothelial dysfunction and glomerular epithelial cell (podocyte) dysregulation, leading to the two main clinical adverse effects, hypertension and proteinuria, respectively (Figure 1).
Figure 1.
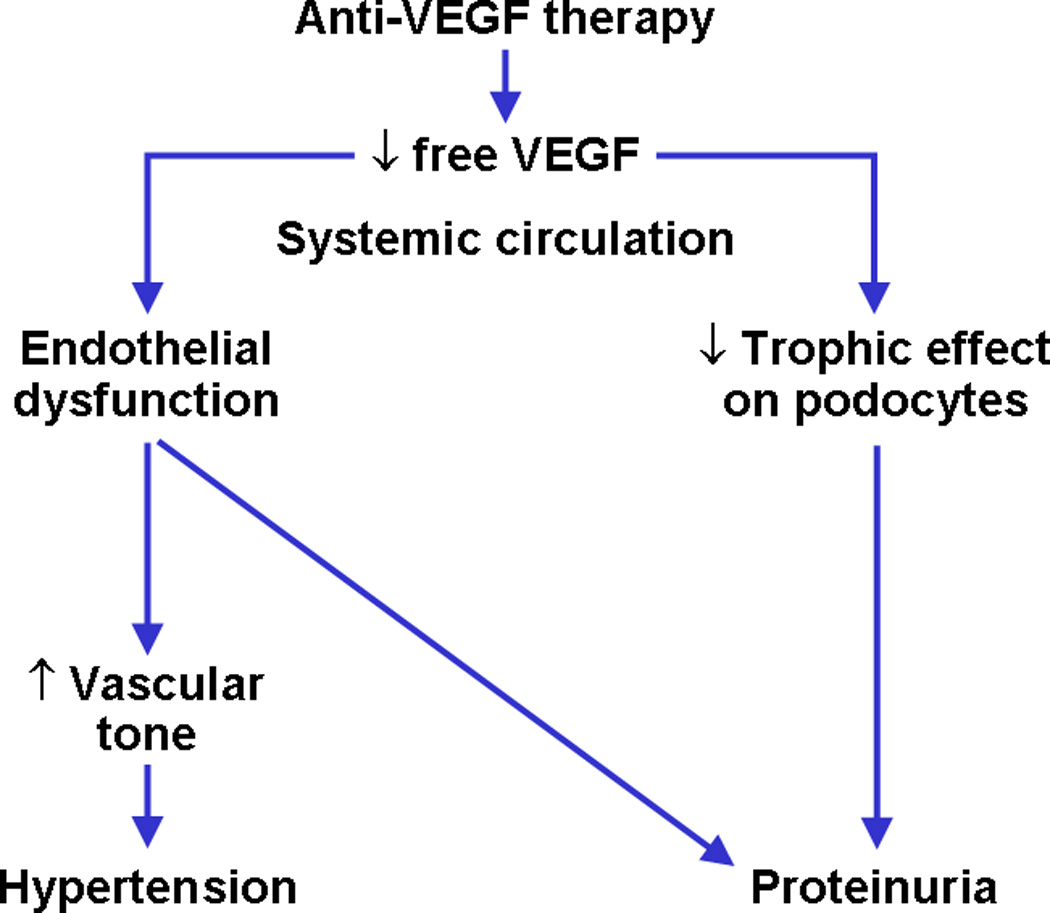
Anti-VEGF therapy, by causing low free VEGF levels, may cause endothelial dysfunction and podocyte dysregulation, leading to hypertension and proteinuria, respectively.
Hypertension
Bevacizumab was the first anti-VEGF drug introduced into clinical practice, and hypertension and proteinuria were described as adverse effects. Zhu, et al reported that the relative risk for hypertension with bevacizumab at low doses (3, 5, or 7.5 mg/kg/dose) was 3.0, compared to 3.5 at a high dose (10 or 15 mg/kg) [8]. Similarly, there appeared to be a dose-dependent increase risk for proteinuria; 1.4 at the lower doses, and 2.2 for the high doses [8]. Reportedly, the systolic pressure is affected more than the diastolic pressure [9, 10], and up to 36% of patients receiving bevacizumab demonstrate increases in blood pressure [11–13].
In a phase II randomized trial in patients with mRCC, those patients treated with bevacizumab at 3mg/kg, the rate of hypertension was only 3%, compared to 36% in those patients treated at the 10 mg/kg dose [13]. Small molecule TKIs also result in dose dependent increases in blood pressure, with reported incidences of 11–43% [14]. The relative potencies of the TKIs also impact the rates of hypertension. The more potent and specific TKIs, axitinib and cediranib (not FDA approved), not surprisingly, have shown higher rates of hypertension than sorafenib and sunitinib at the maximum tolerated dose (MTD) [15, 16]. Similarly, in a phase II study evaluating pazopanib in relapsed or refractory soft tissue sarcomas, during the first month of treatment, virtually all patients developed some degree of hypertension [17].
The accurate determination of the rates of significant hypertension, grades 3 and 4 (Table 1), for anti-VEGF agents has been confounded by several issues. In general, most clinical trials will formally exclude patients with poorly controlled hypertension. In addition, the grading of hypertension was changed from the National Cancer Institute’s, Common Toxicity Criteria of Adverse Events (CTCAE) version 2.0, to CTCAE version 3.0 in 2003, and, more recently, to version 4.0 in 2010 (Table 1) (http://ctep.cancer.gov) [18–20]. These changes in definitions have affected the evaluation and reporting of the adverse blood pressure effects of anti-VEGF medications, and particularly bevacizumab, which has been available for more than 10 years. The most recent classification, version 4.0, is using the blood pressure threshold values that are comparable to those from the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure [21]. This may lead to more consistent reporting of adverse blood pressure outcomes, and, ultimately, to improved understanding and management of anti-VEGF therapy-related hypertension and its complications. With respect to the definitions of the severity of proteinuria, the different classification systems are in agreement, defining proteinuria as grade 1 (urinary protein <1 gr/24 hour urine), grade 2 (1.0–3.4 gr/24 hour urine), or grade 3 (≥ 3.5 gr/24 hour urine). The classifications are not consistent in reporting nephrotic syndrome (grade 4 adverse effect in versions 2.0 and 3.0) and death (grade 5 adverse effect in version 3.0 only).
Table 1.
National Cancer Institute grading systems for HTN as an adverse effect of cancer treatment [18–20]; http://ctep.cancer.gov
| Classification | Category | Definition |
|---|---|---|
| CTCAE Version 4.0 | Grade 1 | Pre-HTN (systolic BP120 – 139 mm Hg or diastolic BP 80 – 89 mm Hg) |
| Grade 2 | Stage 1 hypertension (systolic BP 140 – 159 mm Hg or diastolic BP 90 – 99 mm Hg); medical intervention indicated; recurrent or persistent (≥24 hrs); symptomatic increase by >20 mm Hg (diastolic) or to >140/90 mm Hg if previously WNL; monotherapy indicated Pediatric: recurrent or persistent (≥ 24 hrs) BP>ULN; monotherapy indicated | |
| Grade 3 | Stage 2 hypertension (systolic BP ≥160 mm Hg or diastolic BP ≥100 mm Hg); medical intervention indicated; more than one drug or more intensive therapy than previously used indicated Pediatric: Same as adult | |
| Grade 4 | Life-threatening consequences (e.g., malignant hypertension, transient or permanent neurologic deficit, hypertensive crisis); urgent intervention indicated Pediatric: Same as adult | |
| Grade 5 | Death | |
| CTCAE version 3.0 | Grade 1 | Asymptomatic, transient (<24 hrs) increase by >20 mmHg (diastolic) or to >150/100 if previously WNL; intervention not indicated Pediatric: Asymptomatic, transient (<24 hrs) BP increase >ULN; intervention not indicated |
| Grade 2 | Recurrent or persistent (≥24 hrs) or symptomatic increase by >20 mmHg (diastolic) or to >150/100 if previously WNL; monotherapy may be indicated Pediatric: Recurrent or persistent (≥24 hrs) BP >ULN; monotherapy may be indicated | |
| Grade 3 | Requiring more than one drug or more intensive therapy than previously Pediatric: Same as adult | |
| Grade 4 | Life-threatening consequences (e.g., hypertensive crisis) Pediatric: Same as adult | |
| Grade 5 | Death | |
| Common toxicity criteria: version 2.0 | Grade 0 | None |
| Grade 1 | Asymptomatic, transient increase by 20 mmHg (diastolic) or to 150/100, if previously WNL; not requiring treatment | |
| Grade 2 | Recurrent or persistent or symptomatic increase by 20 mmHg (diastolic) or to 150/100, if previously WNL; not requiring treatment. For pediatric patients, use age and sex appropriate normal values .95th percentile ULN | |
| Grade 3 | Requiring therapy or more intensive therapy than previously | |
| Grade 4 | HTN crisis |
Additional factors that impact the development and/or grade of hypertension while using anti-VEGF therapy include a previous history of hypertension, the concurrent use of more than one anti-VEGF medication, as well as tumor type. It repeatedly has been reported that patients with mRCC treated with anti-angiogenic therapies have higher rates of hypertension than those patients with other tumor types, such as carcinomas of non-small cell lung, hepatocellular, and breast, due to frequent previous nephrectomy and baseline renal insufficiency. However, in a meta-analysis by Wu, et al, the risk of hypertension was similar in those patients treated for mRCC and those being treated for other malignancies [22]. The incidence of hypertension increases with the use of two anti-angiogenic medications concurrently. The combination of bevacizumab and sunitinib and that of bevacizumab and sorafenib in advanced solid tumors, including mRCC, resulted in rates of 92% and 67%, respectively [23, 24]. The first cycle of therapy with an anti-VEGF medication is usually when the majority of blood pressure elevations occur, including in those patients without a history of pre-existing cardiovascular disease [25].
Possible Mechanisms of Hypertension
Inhibition/downregulation of nitric oxide
VEGF is known to stimulate ECs to release NO (nitric oxide) via the upregulation of eNOS (endothelial nitric oxide synthase), as well as prostacyclin (PGI2), resulting in vasodilation, through the activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) downstream pathways [4, 9, 26–29]. This effect of VEGF has been shown to be mediated predominantly through VEGFR-2 (KDR) receptor binding and signaling [5, 30].
This role of VEGF in blood pressure control has been demonstrated in both pre-clinical and clinical studies, in which the infusion of VEGF has been shown to result a drop in blood pressure [30, 31]. The VIVA (VEGF in Ischemia for Vascular Angiogenesis) trial infused recombinant human VEGF, both intravenously and intra-coronary, producing dose-dependent drops in blood pressure, of up to 22% [31]. The antagonism of VEGF by anti-angiogenic therapies is, therefore, considered one of the major contributors to the development of hypertension, through vasoconstriction as a result of decreased NO.
NO also participates in tubuloglomerular feedback, pressure natriuresis and sodium balance, and thus decreased levels may lead to the development of hypertension through sodium retention and direct renal effects [32, 33].
Rarefaction of the microvasculature
Discussion regarding the importance of the microcirculation in the predisposition for and the development of hypertension, both primary and secondary, has been debated, particularly over the past decade. Much of the microvasculature, in the resting state, is closed, allowing for additional vessel recruitment in the event of an increase in metabolic activity. Rarefaction of the microcirculation has been defined as a reduced spatial density of microvascular networks [34]. It can be further divided into functional rarefaction, ie, the abnormal prevalence of existing, but underperfused vessels, versus structural rarefaction, an anatomical absence of microvessels as a result of impaired angiogenesis or capillary apoptosis[34].
One mechanism by which rarefaction has been linked to hypertension is through NO dysregulation[34]. This has been shown in an experimental setting, in which hypertension was induced by pharmacologic inhibition of NO synthesis [33, 35].
The reduction in the microvasculature, whether structural or functional, is postulated to lead to increased systemic vascular resistance and hypertension. The correlation of the development of hypertension with rarefaction in an oncology setting was demonstrated by Steeghs, et al as a side study of a phase I protocol of telatinib, a small molecule TKI of VEGFR-2 and 3, PDGFR, and c-KIT in patients with advanced solid tumors [36]. Several of these patients developed new onset proteinuria, or had worsening of pre-existing proteinuria. Capillary rarefaction was also shown in the finger skin of patients receiving bevacizumab for metastatic colorectal cancer [33]. The degree of rarefaction correlated with the development of hypertension, as well as with the total dose of bevacizumab. However, the cause and effect relationship between rarefaction and hypertension remains unclear.
Neuro-endocrine Mechanisms
The possible contribution of hormonal factors in the development of anti-angiogenesis-induced hypertension continues to be examined. Veronese, et al assessed patients treated with BAY-43-9006 (sorafenib) for three weeks for changes in the following plasma levels: endothelin-1, renin, aldosterone, urotensin II, and catecholamines [37]. Blood pressures rose by 20 mm Hg in 60% of the patients, despite no significant changes in the plasma levels of vasoactive mediators. This suggested that the hypertension is not related to the renin-angiotensin or sympathetic nervous system. Vascular stiffness, as measured by the aortic pulse wave velocity and central aortic augmentation index, was significantly increased [37]. As with rarefaction, however, whether it is a cause or consequence of the hypertension has not been definitively determined. In contrast, Mariette, et al reported a significant reduction in plasma renin levels in patients treated for four weeks with sunitinib [5].
Glomerular Epithelial Cells and Slit Diaphragm
The glomerulus is a highly specialized filtration apparatus with selective permeability that allows free passage of water and solutes, but not protein. It is comprised of three layers that are structurally and functionally distinct. The first layer consists of capillary ECs that are highly fenestrated and allow free passage of albumin. The second is the glomerular basement membrane that is negatively charged and thus repels negatively charged proteins. The third layer is composed of visceral glomerular epithelium, with highly specialized cells situated on the outer aspect of the glomerular basement membrane, also known as podocytes. These cells originate from mesenchyme, in contrast to most other non-renal epithelial cells that are derived from ectoderm [38]. During glomerular development, immature podocytes maintain a high proliferation index [39]. With the acquisition of a mature phenotype, podocytes lose their mitotic activity and develop a highly differentiated cytoarchitecture. Mature podocytes consist of cell bodies, major processes and foot processes. Foot processes are anchored to the glomerular basement membrane. The foot processes interdigitate and connect via specialized cell-to-cell junctions, also known as glomerular slit diaphragms. The slit diaphragm appears to be a modified adherens junction that provides the main size selective filtration barrier in the kidney [40].
The critical role of the slit diaphragm in the normal function of the glomerular filtration barrier has been supported by studies of inherited nephrotic syndromes. In 1998, NPHS1, the gene mutated in the congenital nephrotic syndrome of the Finnish type, was cloned [41].The gene product, nephrin, was localized to podocytes, which likely represent the major structural component of the slit diaphragm. Subsequently, several new proteins that localize either to the slit diaphragm or foot process cytoskeleton have been identified over the past few years, which, through complex interactions, contribute to the maintenance of the structural and functional integrity of the slit diaphragm and podocytes. These include Neph-1, a homologue of nephrin [42], podocin [43], CD2AP, a protein that localizes to the cytoplasmic portion of the slit diaphragm, that may play an important role in protecting the filtration barrier [44], and synaptopodin, which is linked to the formation of foot processes, and thus considered to be a marker of the differentiated podocyte phenotype [45].
Proteinuria
In addition to hypertension, proteinuria is a common adverse side effect attributable to the anti-angiogenic agents. VEGF is crucial to the maintenance of normal renal function, and both over- and under-expression of VEGF may disrupt normal glomerular function. The interaction between VEGF generated by podocytes and VEGFR-2 on glomerular ECs is necessary to maintain the integrity of the glomerular slit diaphragm. It has been theorized that reductions in VEGF lead to down regulation of nephrin expression, subsequently resulting in podocyte injury and proteinuria [46]. Experiments in mice with selective heterozygous or homozygous knock-out of podocyte-specific VEGF have demonstrated the absence of a glomerular filtration barrier in null phenotypes and marked glomerular endothelial cell swelling, bloodless glomeruli, and endotheliosis, (similar to kidney findings in preeclampsia), proteinuria, followed by end-stage kidney failure in haplo-insufficient mice [47]. Eremina, et al further noted that glomerular damage preceded hypertension, suggesting that hypertension per se is not the sole determinant of the development of proteinuria, although there is some association. Yang, et al reported that in patients with mRCC treated with bevacizumab, 54% of the patients who developed grade 2 or 3 hypertension, developed grade 2 or 3 proteinuria, and 16% of patients with no or minimal hypertension (grade 0 or 1) developed proteinuria [13]. In the randomized phase III study evaluating capecitabine alone with capecitabine and bevacizumab in those with previously treated breast cancer, the patients who developed proteinuria were more likely to develop hypertension than those patients who did not have proteinuria (47.1% vs 16.9%, p ≤ 0.001) [12].
Proteinuria related to bevacizumab, as with hypertension, is dose-dependent and has been reported in 41–63% of patients [8]. Despite the high incidence, most cases are asymptomatic and not severe, with nephrotic range proteinuria (> 3.5 g/day) reported in approximately 6.5% [13]. It has been mentioned in the literature that there appears to be a decreased incidence in proteinuria with the TKIs compared to bevacizumab. However, an accurate incidence is unknown given that proteinuria was not evaluated for in the initial studies. Patel, et al reported that 2.8% of 298 patients treated with either sorafenib or sunitinib developed significant proteinuria, with an average of 3.8 g/day, after a median treatment time of 6 months [48]. In a phase I study of RTKI KRN951, 20% of patients developed dose limiting proteinuria, while all but one patient developed hypertension [49].
Patients receiving concurrent intravenous bisphosphonates and/or non-steroidal analgesics while on anti-VEGF treatment have an increased risk for the development or exacerbation of proteinuria [5, 50]. In the phase III metastatic breast cancer study evaluating capecitabine +/- bevacizumab, the incidence of proteinuria was 33.9% in patients receiving bevacizumab and pamidronate vs. 8.5 % (p < 0.026) in the patients receiving just bevacizumab [12].
Renal Pathology
There have been few renal biopsies performed in patients who develop proteinuria+/- hypertension while on therapy. The most probable cause is that few patients have developed frank nephrotic range proteinuria, and also proteinuria, hypertension, and acute renal insufficiency typically resolve or markedly improve after discontinuation of anti-angiogenic therapy. There are usually otherwise few or no other concerning clinical abnormalities.
The renal pathology diagnoses that have been documented to date in these patients include, cryoglobulinemic glomerulonephritis (GN) [51] in a chronic lymphocytic leukemia patient, acute interstitial nephritis attributed to sunitinib and sorafenib [52, 53], collapsing and crescentic GNs thought to reflect the combined toxicity of an intravenous bisphosphonate and anti-angiogenic treatment [12, 54, 55], and focal segmental glomerulosclerosis plus thrombotic microangiopathy in a patient with mRCC on sunitinib [56]. The most common renal pathologic findings, and also the ones most associated with VEGF derangement, are endotheliosis and thrombotic microangiopathy (TMA), indicative of vascular damage (Figure 2) [2, 27, 57, 58]. This pathology is identical to that seen in patients with severe forms of preeclampsia, also believed to be a result of systemic VEGF dysregulation and resulting widespread endothelial damage. In most reported cases in patients who are on anti-VEGF therapy, the TMA appears to be renally localized, rather than part of a systemic process, in which thrombocytopenia, schistocytes on peripheral smear, hemolytic anemia, and rarely, reversible posterior leukoencephalopathy occur. The true rate of renally localized TMA is unknown given the infrequency of renal biopsies. However, there continue to be intermittent reports of systemic organ dysfunction, even with intravitreal injections of anti-VEGF therapy for macular degeneration [59]. Using more than one anti-VEGF concurrently may increase the risk of systemic wide damage. In a phase I dose escalation trial of bevacizumab and sunitinib, 5 of the 12 patients developed systemic TMA [23].The renal biopsy abnormalities, aside from TMA, may or may not be directly related to therapy. Other possibilities would include paraneoplastic nephropathies, “off-target” effects of the medication, or the concurrent presence of unrelated renal disease.
Figure 2.
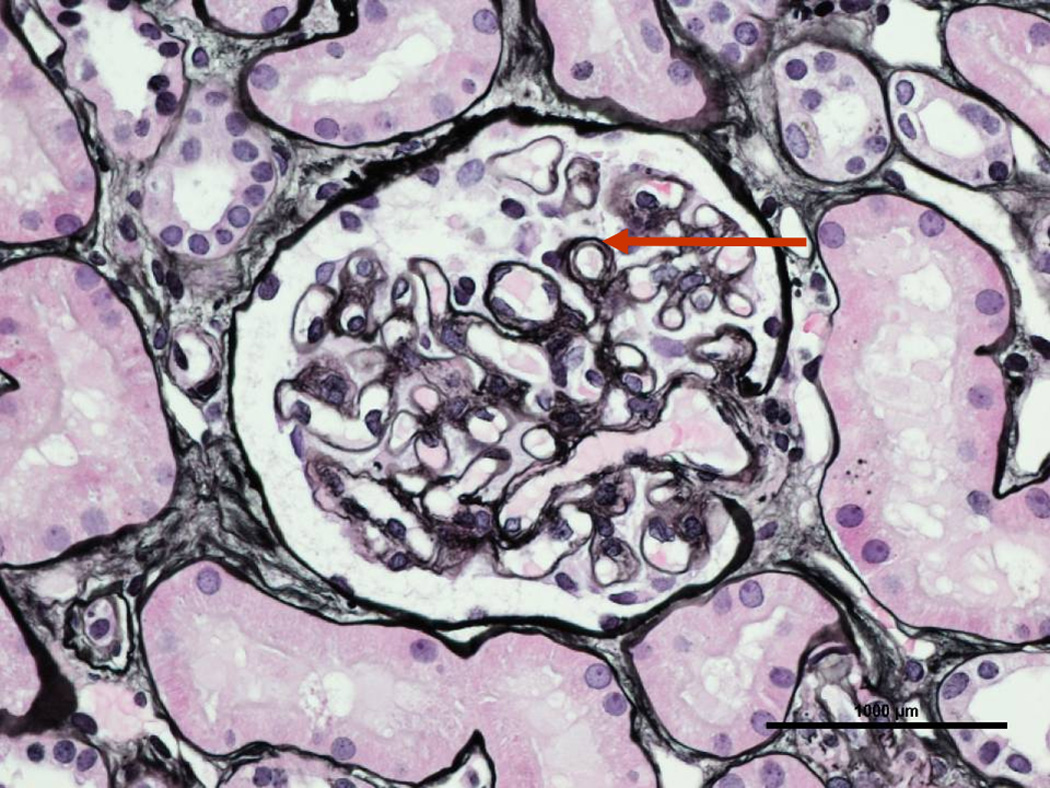
64 year old male with metastatic liver angiosarcoma treated with bevacizumab, who, after 35 months of treatment, developed nephrotic range proteinuria and an active urinary sediment. A renal biopsy (silver stain) showing double contouring (arrow), consistent with thrombotic microangiopathy.
Of concern to clinicians, is the not uncommon discrepancy of minimal non-invasive clinical findings and relatively mild proteinuria, and the severe renal pathology that is obtained from biopsy, suggesting that a renal biopsy should be considered at some point prior to the development of severe proteinuria, in order to maximize the chances of intervening at a time when toxicities may be reversed or at least more easily managed. The additional issue raised is how accurately does the degree of proteinuria, in general, reflect the extent of underlying glomerular damage? Recent publications have suggested that quantifying urinary podocyte excretion, as a measure of podocyte injury, may be a more sensitive indicator of glomerular damage than proteinuria [60]. Muller-Deile et al recently reported for the first time the presence of podocyturia in patients with proteinuria treated with anti-VEGF agents [61••].
Reversibility of Nephrotoxicity and Hypertension
In the majority of cases, proteinuria and hypertension resolve or significantly improve with removal of anti-VEGF therapy. There have been reports, however, of resolution of nephrotic range proteinuria after cessation of treatment, but with limited recovery of actual renal function [62]. Steeghs, et al studied a small group of patients, with either breast or colorectal cancer, being treated with chemotherapy regimens that included bevacizumab, as to whether there was objective evidence of reversibility of capillary density (rarefaction) and/or changes in blood pressure after discontinuation of bevacizumab treatment for 3 months [63••]. There was clear reversibility of capillary regression, with most capillaries growing back within two weeks. This reversal in functional rarefaction, however, did not correlate with significant changes in blood pressure within the 3 month time frame. Renal function was not evaluated.
Medication Toxicities vs. Clinical Benefits
The development or worsening of hypertension during anti-VEGF treatment is not without possible positive aspects. Multiple publications have noted that hypertension may be a surrogate marker of the effectiveness of VEGF blockade [64–68]. Osterlund et al evaluated 101 patients with metastatic colorectal carcinoma treated with bevacizumab. Blood pressures were monitored and graded according to severity. Overall response rate, progression free survival, and overall survival were superior in those who developed hypertension compared to those who did not [69•]. The development of hypertension within 3 months was an independent prognostic factor. Whether dose escalations of anti-VEGF agents to the point of the development of some degree of hypertension as a marker of successful VEGF blockade should be considered, or whether lack of hypertension within a certain period of time after starting therapy should result in a change in therapeutic strategy, remain unknown.
Possible molecular markers to predict the development of hypertension and disease response are being investigated. Associations of certain VEGF and VEGFR-2 single nucleotide polymorphisms (SNPs) with hypertension and clinical outcome have been found in a retrospective, preliminary study of patients with mRCC treated with sunitinib [70]. Certain SNPs were also found to be associated with response and toxicity in patients also with mRCC treated with first-line sunitinib in a multi-center, observational, prospective study [71].
Initial Patient Assessment/Choice of Anti-hypertensive Medications
There are no established guidelines for the treatment of hypertension arising from or worsening as a result of anti-VEGF therapy, as there are few prospectively collected data collected on its management. The results of the first prospective investigation of hypertension management during treatment with cediranib were published in JCO in 2009 [72]. Formal incorporation of hypertension and its management into future clinical studies evaluating anti-VEGF agents should ultimately aid in establishing formal general guidelines. A consensus report by the Investigational Drug Steering Committee of the NCI convened an interdisciplinary panel comprised of cardiovascular toxicity experts to make recommendations to the Cancer Therapy Evaluation Program regarding this particular issue [25]. It has suggested conducting formal cardiovascular risk assessments on patients prior to the start of anti-VEGF treatment and maximizing control of pre-existing hypertension. Optimal blood pressure target goals and monitoring are also addressed.
The choice of agent (s) needs to be made in the context of the patients’ other co-morbidities and medications, as well as the metabolism of the particular anti-angiogenic treatment. In the case of some of the TKIs, they undergo hepatic metabolism via the cytochrome P450 system, especially CYP3A4. As the calcium channel blockers (CCB), verapamil and diltiazem inhibit the CYP3A4 system, resulting in increased plasma concentrations of the TKIs, they are best avoided. This would not be an issue with the dihydropyridine class of CCB, although some take several days before any significant anti-hypertensive effect is seen. Nifedipine has been shown to induce VEGF secretion, and so felodipine or amlodipine are preferred [73]. The use of ace inhibitors (ACEI) or angiotensin II receptor blockers (ARB) has been advocated by some due to several factors. If there is concurrent proteinuria, ACEI and ARBs may confer a renal protective effect.
Angiotensin II is known as a mitogenic agent stimulating growth through epidermal growth factor, transforming growth factor beta, and tyrosine kinase [74]. The potential inhibition of angiotensin II has obvious appeal. In addition, ACEI have been shown to upregulate expression of nephrin in diabetic nephropathy, and also improve endothelial function and capillary density [75], [19, 76]. As one of the mechanisms of action of anti-VEGF therapy is a decrease in NO, with subsequent vasoconstriction and increase in systemic vascular resistance, the use of agents that increase NO are attractive. Dirix, et al reported excellent responses in patients with long acting nitrates [77]. However, the use of nitrates for the control of anti-VEGF induced hypertension has to be weighed against the possible adverse effects, such as tumor progression, with increasing NO levels. Oliver et al, reported that phosphodiesterase inhibitor type 5 (sildenafil) was effective [75, 78]. The use of nebivolol, whose mechanism of action is through reduction in peripheral resistance, has been considered as well [75, 79]. Finally, other antihypertensive agents (such as beta blockers, diuretics, alpha blockers, centrally acting agents, and direct vasodilators) may be required to achieve goal blood pressure [21]. Pre-existing conditions may affect the choice of a specific agent. One notable example is the use of beta blockers for patients with ischemic heart disease.
Special Considerations
Patients that may require more intensive monitoring and earlier intervention are those with nephrectomy and/or renal insufficiency as a result of mRCC. Launay-Vacher et al evaluated the renal function of mRCC patients, on anti-VEGF therapy, by GFR using the aMDRD formula, who had previously undergone nephrectomy [80]. All 73 patients demonstrated declining renal function over time, especially those with pre-existing hypertension. Gupta et al reported that 33% of the mRCC patients they studied had renal insufficiency, as defined by a GFR ≤ 60 mL/minute [81]. Compared to the patients with normal renal function, those with renal insufficiency had larger median increases in blood pressure with bevacizumab and sunitinib. There was no difference with respect to other toxicities, time to progression, response or overall survival [81]. The use of anti-VEGF therapy should be carefully considered in patients with polycystic kidney disease (PKD) [82]. An anti-VEGF-A antibody was administered in a PKD rat model and compared to controls. Those with PKD displayed increased tubular epithelial cell proliferation, increased kidney and cyst growth, as well as severe renal failure associated with glomerular damage [82]. These findings were correlated with low renal VEGF levels and high HIF-1-alpha.
Conclusion
Patients with solid tumors who are currently being treated with anti-VEGF monotherapy or in combination with chemotherapy are typically older, with a higher likelihood of cardiovascular and renovascular co-morbidities. The approved indications for anti-VEGF treatment are largely for incurable metastatic disease, with limited therapeutic options and median survivals. In addition, most, but not all of these medications, have been shown to improve progression free survival, rather than overall survival. The decision as to how to best treat and manage the accompanying toxicities in this setting, while maintaining a reasonable quality of life, should rely on a careful, ongoing evaluation of risks vs. benefits in each individual patient. Future progress in treatment of these patients is critically dependent on development of early markers of vascular injury that would identify those at risk before hypertension and/or proteinuria develop. Early identification may allow for timely dose adjustments and continuation of anti-VEGF therapy, which is commonly the only option for these patients, rather than holding or discontinuing these drugs when severe side effects develop.
Footnotes
Disclosure
No potential conflicts of interest relevant to this article were reported.
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Eremina V, Quaggin SE. Biology of anti-angiogenic therapy-induced thrombotic microangiopathy. Seminars in nephrology. 2010;30:582–590. doi: 10.1016/j.semnephrol.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors a review. Eur J Cancer. 2006;42:3127–3139. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 5.Kappers MH, van Esch JH, Sleijfer S, Danser AH, van den Meiracker AH. Cardiovascular and renal toxicity during angiogenesis inhibition: clinical and mechanistic aspects. J Hypertens. 2009;27:2297–2309. doi: 10.1097/HJH.0b013e3283309b59. [DOI] [PubMed] [Google Scholar]
- 6.Ebos JM, Bocci G, Man S, et al. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Mol Cancer Res. 2004;2:315–326. [PubMed] [Google Scholar]
- 7.Kitamoto Y, Tokunaga H, Miyamoto K, Tomita K. VEGF is an essential molecule for glomerular structuring. Nephrol Dial Transplant. 2002;17(Suppl 9):25–27. doi: 10.1093/ndt/17.suppl_9.25. [DOI] [PubMed] [Google Scholar]
- 8.Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am J Kidney Dis. 2007;49:186–193. doi: 10.1053/j.ajkd.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 9.Sane DC, Anton L, Brosnihan KB. Angiogenic growth factors and hypertension. Angiogenesis. 2004;7:193–201. doi: 10.1007/s10456-004-2699-3. [DOI] [PubMed] [Google Scholar]
- 10.Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007;7:475–485. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 11.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 12.Miller KD, Chap LI, Holmes FA, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–799. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 13.Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mourad JJ, Levy BI. Mechanisms of antiangiogenic-induced arterial hypertension. Curr Hypertens Rep. 2011;13:289–293. doi: 10.1007/s11906-011-0206-y. [DOI] [PubMed] [Google Scholar]
- 15.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–477. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
- 16.Drevs J, Siegert P, Medinger M, et al. Phase I clinical study of AZD2171, an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2007;25:3045–3054. doi: 10.1200/JCO.2006.07.2066. [DOI] [PubMed] [Google Scholar]
- 17.Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043) J Clin Oncol. 2009;27:3126–3132. doi: 10.1200/JCO.2008.21.3223. [DOI] [PubMed] [Google Scholar]
- 18.Trotti A, Byhardt R, Stetz JA, et al. Common toxicity criteria:version 2.0. An improved reference for grading the acute effects of cancer treatment: impact on radiotherapy. Int J Radiat Oncol Biol Phys. 2000;47:13–47. doi: 10.1016/s0360-3016(99)00559-3. [DOI] [PubMed] [Google Scholar]
- 19.Agabiti-Rosei E. Structural and functional changes of the microcirculation in hypertension: influence of pharmacological therapy. Drugs. 2003;63(Spec No 1):19–29. [PubMed] [Google Scholar]
- 20.Trotti A, Colevas AD, Setser A, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13:176–181. doi: 10.1016/S1053-4296(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 21.Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 22.Wu S, Chen JJ, Kudelka A, Lu J, Zhu X. Incidence and risk of hypertension with sorafenib in patients with cancer: a systematic review and meta-analysis. Lancet Oncol. 2008;9:117–123. doi: 10.1016/S1470-2045(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 23.Feldman DR, Baum MS, Ginsberg MS, et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:1432–1439. doi: 10.1200/JCO.2008.19.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azad NS, Posadas EM, Kwitkowski VE, et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J Clin Oncol. 2008;26:3709–3714. doi: 10.1200/JCO.2007.10.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maitland ML, Bakris GL, Black HR, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J Natl Cancer Inst. 2010;102:596–604. doi: 10.1093/jnci/djq091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol. 1998;274:H1054–H1058. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- 27.Horowitz JR, Rivard A, van der Zee R, et al. Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol. 1997;17:2793–2800. doi: 10.1161/01.atv.17.11.2793. [DOI] [PubMed] [Google Scholar]
- 28.Zachary I. VEGF signalling: integration and multi-tasking in endothelial cell biology. Biochem Soc Trans. 2003;31:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]
- 29.Gelinas DS, Bernatchez PN, Rollin S, Bazan NG, Sirois MG. Immediate and delayed VEGF-mediated NO synthesis in endothelial cells: role of PI3K, PKC and PLC pathways. Br J Pharmacol. 2002;137:1021–1030. doi: 10.1038/sj.bjp.0704956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Ogasawara AK, Yang R, et al. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension. 2002;39:1095–1100. doi: 10.1161/01.hyp.0000018588.56950.7a. [DOI] [PubMed] [Google Scholar]
- 31.Henry TD, Annex BH, McKendall GR, et al. The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003;107:1359–1365. doi: 10.1161/01.cir.0000061911.47710.8a. [DOI] [PubMed] [Google Scholar]
- 32.Zou AP, Cowley AW., Jr Role of nitric oxide in the control of renal function and salt sensitivity. Curr Hypertens Rep. 1999;1:178–186. doi: 10.1007/s11906-999-0016-7. [DOI] [PubMed] [Google Scholar]
- 33.Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19:927–934. doi: 10.1093/annonc/mdm550. [DOI] [PubMed] [Google Scholar]
- 34.Feihl F, Liaudet L, Waeber B, Levy BI. Hypertension: a disease of the microcirculation? Hypertension. 2006;48:1012–1017. doi: 10.1161/01.HYP.0000249510.20326.72. [DOI] [PubMed] [Google Scholar]
- 35.Kiefer FN, Misteli H, Kalak N, et al. Inhibition of NO biosynthesis, but not elevated blood pressure, reduces angiogenesis in rat models of secondary hypertension. Blood Press. 2002;11:116–124. doi: 10.1080/08037050211256. [DOI] [PubMed] [Google Scholar]
- 36.Steeghs N, Gelderblom H, Roodt JO, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clin Cancer Res. 2008;14:3470–3476. doi: 10.1158/1078-0432.CCR-07-5050. [DOI] [PubMed] [Google Scholar]
- 37.Veronese ML, Mosenkis A, Flaherty KT, et al. Mechanisms of hypertension associated with BAY 43–9006. J Clin Oncol. 2006;24:1363–1369. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 38.Mundel P, Kriz W. Cell culture of podocytes. Exp Nephrol. 1996;4:263–266. [PubMed] [Google Scholar]
- 39.Nagata M, Yamaguchi Y, Ito K. Loss of mitotic activity and the expression of vimentin in glomerular epithelial cells of developing human kidneys. Anat Embryol (Berl) 1993;187:275–279. doi: 10.1007/BF00195765. [DOI] [PubMed] [Google Scholar]
- 40.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 41.Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 42.Donoviel DB, Freed DD, Vogel H, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol. 2001;21:4829–4836. doi: 10.1128/MCB.21.14.4829-4836.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwarz K, Simons M, Reiser J, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–1629. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shih NY, Li J, Karpitskii V, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286:312–315. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 45.Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. doi: 10.1681/ASN.V10151. [DOI] [PubMed] [Google Scholar]
- 46.Garovic VD, Wagner SJ, Petrovic LM, et al. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol Dial Transplant. 2007;22:1136–1143. doi: 10.1093/ndt/gfl711. [DOI] [PubMed] [Google Scholar]
- 47.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel TV, Morgan JA, Demetri GD, et al. A preeclampsia-like syndrome characterized by reversible hypertension and proteinuria induced by the multitargeted kinase inhibitors sunitinib and sorafenib. J Natl Cancer Inst. 2008;100:282–284. doi: 10.1093/jnci/djm311. [DOI] [PubMed] [Google Scholar]
- 49.Eskens FAP, van Doorn A. An open label phase I dose escalation study of KRN951, a tyrosine kinase inhibitor of vascular endothelial growth factor receptor 2 and 1 in a 4 week on, 2 week off schedule in patients with advanced solid tumors (Abstract) J Clin Oncol. 2006;24:2034. doi: 10.1200/JCO.2008.18.8193. [DOI] [PubMed] [Google Scholar]
- 50.Izzedine H, Massard C, Spano JP, et al. VEGF signalling inhibition-induced proteinuria: Mechanisms, significance and management. Eur J Cancer. 2010;46:439–448. doi: 10.1016/j.ejca.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Nasr SH, Snyder RW, Bhagat G, Markowitz GS. Chronic lymphocytic leukemia and cryoglobulinemic glomerulonephritis. Kidney Int. 2007;71:93. doi: 10.1038/sj.ki.5001891. [DOI] [PubMed] [Google Scholar]
- 52.Khurana A. Allergic interstitial nephritis possibly related to sunitinib use. Am J Geriatr Pharmacother. 2007;5:341–344. doi: 10.1016/j.amjopharm.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 53.Izzedine H, Brocheriou I, Rixe O, Deray G. Interstitial nephritis in a patient taking sorafenib. Nephrol Dial Transplant. 2007;22:2411. doi: 10.1093/ndt/gfm199. [DOI] [PubMed] [Google Scholar]
- 54.Stokes MB, Erazo MC, D'Agati VD. Glomerular disease related to anti- VEGF therapy. Kidney Int. 2008;74:1487–1491. doi: 10.1038/ki.2008.256. [DOI] [PubMed] [Google Scholar]
- 55.Stylianou K, Lioudaki E, Papadimitraki E, et al. Crescentic glomerulonephritis associated with vascular endothelial growth factor (VEGF) inhibitor and bisphosphonate administration. Nephrol Dial Transplant. 2011;26:1742–1745. doi: 10.1093/ndt/gfr093. [DOI] [PubMed] [Google Scholar]
- 56.Costero O, Picazo ML, Zamora P, et al. Inhibition of tyrosine kinases by sunitinib associated with focal segmental glomerulosclerosis lesion in addition to thrombotic microangiopathy. Nephrol Dial Transplant. 2010;25:1001–1003. doi: 10.1093/ndt/gfp666. [DOI] [PubMed] [Google Scholar]
- 57.Bollee G, Patey N, Cazajous G, et al. Thrombotic microangiopathy secondary to VEGF pathway inhibition by sunitinib. Nephrol Dial Transplant. 2009;24:682–685. doi: 10.1093/ndt/gfn657. [DOI] [PubMed] [Google Scholar]
- 58.Izzedine H, Brocheriou I, Deray G, Rixe O. Thrombotic microangiopathy and anti-VEGF agents. Nephrol Dial Transplant. 2007;22:1481–1482. doi: 10.1093/ndt/gfl565. [DOI] [PubMed] [Google Scholar]
- 59.Pelle G, Shweke N, Duong Van Huyen JP, et al. Systemic and kidney toxicity of intraocular administration of vascular endothelial growth factor inhibitors. Am J Kidney Dis. 2011;57:756–759. doi: 10.1053/j.ajkd.2010.11.030. [DOI] [PubMed] [Google Scholar]
- 60.Yu D, Petermann A, Kunter U, et al. Urinary Podocyte Loss Is a More Specific Marker of Ongoing Glomerular Damage than Proteinuria. Journal of the American Society of Nephrology. 2005;16:1733–1741. doi: 10.1681/ASN.2005020159. [DOI] [PubMed] [Google Scholar]
- 61.Müller-Deile J, Bröcker V, Grünwald V, et al. Renal side effects of VEGF-blocking therapy. NDT Plus. 2010;3:172–175. ••First article reporting the presence of podocyturia in patients with proteinuria treated with anti-VEGF therapy.
- 62.Takahashi D, Nagahama K, Tsuura Y, Tanaka H, Tamura T. Sunitinib-induced nephrotic syndrome and irreversible renal dysfunction. Clin Exp Nephrol. 2011 doi: 10.1007/s10157-011-0543-9. [DOI] [PubMed] [Google Scholar]
- 63.Steeghs N, Rabelink TJ, op 't Roodt J, et al. Reversibility of capillary density after discontinuation of bevacizumab treatment. Ann Oncol. 2010;21:1100–1105. doi: 10.1093/annonc/mdp417. •• First study showing that decreased capillary density as a result ot bevacizumab is reversible and that capillary density may represent a marker of treatment efficacy.
- 64.Ravaud A, Sire M. Arterial hypertension and clinical benefit of sunitinib, sorafenib and bevacizumab in first and second-line treatment of metastatic renal cell cancer. Ann Oncol. 2009;20:966–967. doi: 10.1093/annonc/mdp201. author reply 7. [DOI] [PubMed] [Google Scholar]
- 65.Rixe O, Billemont B, Izzedine H. Hypertension as a predictive factor of Sunitinib activity. Ann Oncol. 2007;18:1117. doi: 10.1093/annonc/mdm184. [DOI] [PubMed] [Google Scholar]
- 66.Levy BI. Blood pressure as a potential biomarker of the efficacy angiogenesis inhibitor. Ann Oncol. 2009;20:200–203. doi: 10.1093/annonc/mdp018. [DOI] [PubMed] [Google Scholar]
- 67.Bono P, Elfving H, Utriainen T, et al. Hypertension and clinical benefit of bevacizumab in the treatment of advanced renal cell carcinoma. Ann Oncol. 2009;20:393–394. doi: 10.1093/annonc/mdn729. [DOI] [PubMed] [Google Scholar]
- 68.Mir O, Ropert S, Alexandre J, Goldwasser F. Hypertension as a surrogate marker for the activity of anti-VEGF agents. Ann Oncol. 2009;20:967–970. doi: 10.1093/annonc/mdp206. [DOI] [PubMed] [Google Scholar]
- 69.Osterlund P, Soveri LM, Isoniemi H, et al. Hypertension and overall survival in metastatic colorectal cancer patients treated with bevacizumab-containing chemotherapy. Br J Cancer. 2011;104:599–604. doi: 10.1038/bjc.2011.2. • Study that most clearly hypertension and overall survival in patients treated with bevacizumab.
- 70.Kim JJ, Vaziri SAJ, Rini BI, et al. Association of VEGF and VEGFR2 single nucleotide polymorphisms with hypertension and clinical outcome in metastatic clear cell renal cell carcinoma patients treated with sunitinib. Cancer. 2011 doi: 10.1002/cncr.26491. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Donas J, Esteban E, Leandro-Garcia LJ, et al. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 2011;12:1143–1150. doi: 10.1016/S1470-2045(11)70266-2. [DOI] [PubMed] [Google Scholar]
- 72.Langenberg MH, van Herpen CM, De Bono J, et al. Effective strategies for management of hypertension after vascular endothelial growth factor signaling inhibition therapy: results from a phase II randomized, factorial, double-blind study of Cediranib in patients with advanced solid tumors. J Clin Oncol. 2009;27:6152–6159. doi: 10.1200/JCO.2009.22.2273. [DOI] [PubMed] [Google Scholar]
- 73.Miura S, Fujino M, Matsuo Y, Tanigawa H, Saku K. Nifedipine-induced vascular endothelial growth factor secretion from coronary smooth muscle cells promotes endothelial tube formation via the kinase insert domain-containing receptor/fetal liver kinase-1/NO pathway. Hypertens Res. 2005;28:147–153. doi: 10.1291/hypres.28.147. [DOI] [PubMed] [Google Scholar]
- 74.Molteni A, Heffelfinger S, Moulder JE, Uhal B, Castellani WJ. Potential deployment of angiotensin I converting enzyme inhibitors and of angiotensin II type 1 and type 2 receptor blockers in cancer chemotherapy. Anticancer Agents Med Chem. 2006;6:451–460. doi: 10.2174/187152006778226521. [DOI] [PubMed] [Google Scholar]
- 75.Izzedine H, Ederhy S, Goldwasser F, et al. Management of hypertension in angiogenesis inhibitor-treated patients. Ann Oncol. 2009;20:807–815. doi: 10.1093/annonc/mdn713. [DOI] [PubMed] [Google Scholar]
- 76.Kelly DJ, Aaltonen P, Cox AJ, et al. Expression of the slit-diaphragm protein, nephrin, in experimental diabetic nephropathy: differing effects of antiproteinuric therapies. Nephrol Dial Transplant. 2002;17:1327–1332. doi: 10.1093/ndt/17.7.1327. [DOI] [PubMed] [Google Scholar]
- 77.Dirix LY, Maes H, Sweldens C. Treatment of arterial hypertension (AHT) associated with angiogenesis inhibitors. Ann Oncol. 2007;18:1121–1122. doi: 10.1093/annonc/mdm205. [DOI] [PubMed] [Google Scholar]
- 78.Oliver JJ, Melville VP, Webb DJ. Effect of regular phosphodiesterase type 5 inhibition in hypertension. Hypertension. 2006;48:622–627. doi: 10.1161/01.HYP.0000239816.13007.c9. [DOI] [PubMed] [Google Scholar]
- 79.Porta C, Paglino C, Imarisio I, Bonomi L. Uncovering Pandora's vase: the growing problem of new toxicities from novel anticancer agents. The case of sorafenib and sunitinib. Clin Exp Med. 2007;7:127–134. doi: 10.1007/s10238-007-0145-8. [DOI] [PubMed] [Google Scholar]
- 80.Launay-Vacher V, Ayllon J, Janus N, et al. Evolution of renal function in patients treated with antiangiogenics after nephrectomy for renal cell carcinoma. Urol Oncol. 2011;29:492–494. doi: 10.1016/j.urolonc.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 81.Gupta S, Parsa V, Heilbrun LK, et al. Safety and efficacy of molecularly targeted agents in patients with metastatic kidney cancer with renal dysfunction. Anticancer Drugs. 2011;22:794–800. doi: 10.1097/CAD.0b013e328346af0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Raina S, Honer M, Kramer SD, et al. Anti-VEGF antibody treatment accelerates polycystic kidney disease. Am J Physiol Renal Physiol. 2011;301:F773–F783. doi: 10.1152/ajprenal.00058.2011. [DOI] [PubMed] [Google Scholar]