Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy (original) (raw)
. Author manuscript; available in PMC: 2014 Apr 1.
Abstract
Fragment-based drug design (FBDD) is a promising approach for the generation of lead molecules with enhanced activity and especially drug-like properties against therapeutic targets. Herein, we report the fragment-based drug design, systematic chemical synthesis and pharmacological evaluation of novel scaffolds as potent anticancer agents by utilizing six privileged fragments from known STAT3 inhibitors. Several new molecules such as compounds 5, 12, and 19 that may act as advanced chemical leads have been identified. The most potent compound 5 (HJC0123) has demonstrated to inhibit STAT3 promoter activity, downregulate phosphorylation of STAT3, increase the expression of cleaved caspase-3, inhibit cell cycle progression and promote apoptosis in breast and pancreatic cancer cells with low micromolar to nanomolar IC50 values. Furthermore, compound 5 significantly suppressed estrogen receptor (ER)-negative breast cancer MDA-MB-231 xenograft tumor growth in vivo (p.o.), indicating its great potential as an efficacious and orally bioavailable drug candidate for human cancer therapy.
Keywords: Fragment-based drug design, Anticancer agents, Drug discovery, STAT3, Breast cancer
1. Introduction
STAT3 (signal transducers and activators of transcription 3) is a member of a family of seven transcription factors (STATs 1, 2, 3, 4, 5a, 5b, and 6) that transmit signals from cell surface receptors to the nucleus, and are crucial for the signaling of many cytokines and growth factors that are mediators in the fundamental cellular and biological processes such as the immune response, angiogenesis, cell proliferation, differentiation, and apoptosis [1–7]. It contains four functional domains that contribute to its oligomerization, DNA binding, Src homology 2 (SH2) dimerization, and transactivation, respectively. Once activated by extracellular signaling proteins, STAT3 is phosphorylated at a tyrosine residue 705 (Tyr-705) and phosphorylated STAT3 forms dimers via a reciprocal phosphotyrosines (pTyr)-SH2 domain interaction. Then the dimers translocate to the nucleus where they bind to specific DNA response elements and induce transcription [8]. In contrast to the transient nature of STAT3 activation in normal cells, constitutive STAT3 activity has been observed and reported in many human solid and hematological tumors [9,10]. Aberrant STAT3 activation is found to be correlated with worse prognosis by promoting tumorigenesis, inducing tumor invasion and metastasis, and driving the malignant progression in many carcinomas. Thus, STAT3 is considered to be a promising target for prevention and treatment of cancer, thereby providing the rationale to develop novel anticancer agents targeting the STAT3 signaling pathway [11–18].
Over the past decade, several peptidic and non-peptidic STAT3 inhibitors that directly inhibit the different structural domains of STAT3 protein or indirectly inhibit the upstream components of the STAT3 activation [19] such as PY*LKTK [20], STX-0119 [21], and S31-201 [22] to inhibit STAT3 dimerization, Stattic [23] to inhibit phosphorylation, CAP-1 [24] and IS3 295 [25] to inhibit DNA-binding, niclosamide [26] to inhibit transcriptional function of STAT3, WP1066 [27] and AG490 [28] to inhibit the upstream Janus kinases activity have been explored (Fig. 1). Despite these significant advances, no STAT3 inhibitor drugs have reached the market. The challenges include the lack of membrane permeability and stability, low water-solubility, weak binding affinity, or low specificity of effects [29–38]. Therefore, it remains highly valuable to identify novel potent and efficacious STAT3 inhibitors with new scaffolds.
Fig. 1.

Chemical structures of representative non-peptidic STAT3 inhibitors.
Fragment-based drug design (FBDD) is a promising approach for the generation of lead molecules against therapeutic targets in the past decade. New molecules with enhanced drug-likeness could be identified by chemically merging privileged-fragments from small molecule libraries that bind cooperatively in a given binding pocket [39–42]. These favorable fragments may maintain their original orientation in the protein binding pocket, and thus contribute jointly to the final binding energy. Till now, FBDD has proven to be a successful approach to identify potent inhibitors for specific targets [43,44]. Herein, we describe our efforts for the fragment-based design, synthesis and characterization of new molecules as STAT3 inhibitors for cancer therapy. Compound 5 (HJC0123) was discovered as a potent and orally bioavailable anticancer agent in this endeavor.
2. Results and discussion
2.1. Design
We initiated our studies with a small privileged fragment library containing six fragments selected from representative non-peptidic STAT3 inhibitors including stattic, WP1066, STX-0119, and niclosamide as potential binding pharmacophores to STAT3 (Fig. 2). The selection of the privileged fragment library was governed by data from literature with respect to the most active inhibitors of STAT3 and the drug-like properties such as molecular weight of these fragments, which ranges from 120 to 251 Da. Taking inspiration from the FBDD approach, we designed a number of novel molecules with diversified scaffolds for structure–activity relationship (SAR) studies in an attempt to identify new anticancer compounds with enhanced potency and drug-like properties.
Fig. 2.
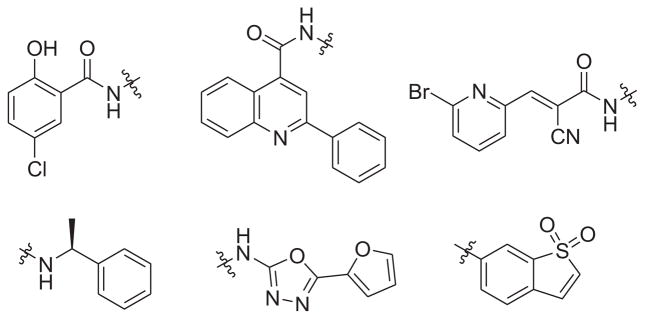
Privileged fragments selected from known STAT3 inhibitors including niclosamide, STX-0119, WP1066, and stattic.
2.2. Chemistry
As shown in Schemes 1 and 2, coupling of 1,1-dioxo-1_H_-1λ6-benzo[_b_]thiophen-6-ylamine (3) with cyanoacetic acid (1) or 2-phenyl-quinoline-4-carboxylic acid (2) in the presence of HBTU and DIPEA in CH2Cl2 generated the corresponding amides 4 and 5 in moderate to high yields. Condensation of L(−)-α-methylbenzylamine (7) with 2-phenylquinoline-4-carboxylic acid (2) or 5-chloro-2-hydroxybenzoic acid (6) in the same fashion provided new compounds 8 and 9, respectively. The synthesis of compound 10 was achieved in moderate yield via Knoevenagel condensation of 6-bromopyridine-2-carbaldehyde with the intermediate 4 in the presence of piperidine as the catalyst. The above mentioned coupling methods failed to give the desired compounds 12 and 13, which were successfully obtained by an alternative protocol in three steps. First, the carboxylic acid 6 was converted to the acid chloride by the treatment with SOCl2 in toluene at 120 °C for 16 h. After concentration, the acid chloride, which contained some ester formed from the hydroxyl group of phenol, was used directly for next step without further purification because the hydrolysis of the ester appendage afterward could lead to the same desired product. 1,1-Dioxo-1_H_-1λ6-benzo[_b_]thiophen-6-ylamine or 5-furan-2-yl-[1,3,4]oxadiazol-2-ylamine was then treated with the above acid chloride in the presence of pyridine as the base. The crude products were subsequently saponified using 1 N LiOH (aq.) in THF to generate the desired compounds 12 and 13 in yield of 39% and 50% (three steps), respectively.
Scheme 1.
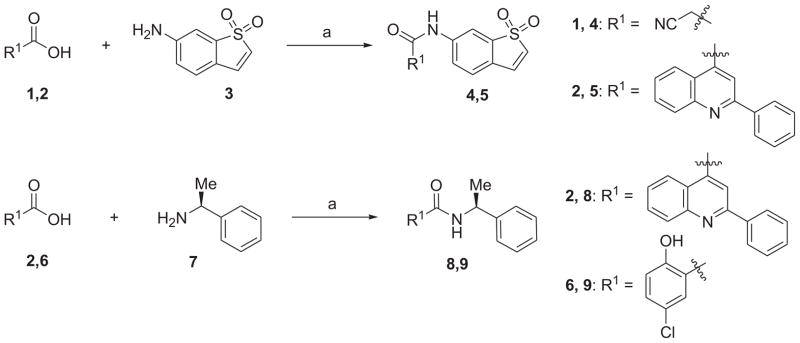
a Reagents and conditions: (a) HBTU, DIPEA, CH2Cl2, rt, 39–94%.
Scheme 2.
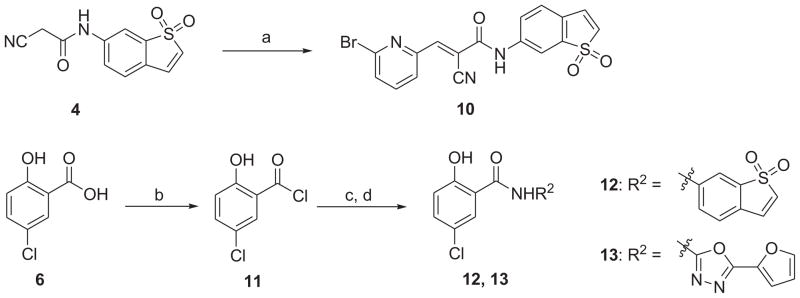
a Reagents and conditions: (a) 6-bromo-pyridine-2-carbaldehyde, piperidine (cat.), EtOH, 90 °C, 72%; (b) SOCl2, toluene, reflux; (c) R2NH2, pyridine, DMF, 0 °C to rt; (d) 1 N LiOH (aq.), THF, H2O, 0 °C to rt, 39–50% (three steps).
2.3. Biology
To explore the SAR, we first evaluated the in vitro anticancer effects of the compounds 4, 5, 8–10 and 12, 13 on the proliferation of human breast cancer cell lines MCF-7 (ER-positive) and MDA-MB-231 (ER-negative and triple-negative), as well as pancreatic cancer cell lines AsPC1 and Panc-1 using MTS assays as described in the Experimental section. The ability of these new scaffolds to inhibit the growth of cancer cells is summarized in Table 1. It is noteworthy that most of the newly synthesized compounds described herein exhibited promising antiproliferative activity with low micromolar to nanomolar IC50 values. Among them, compounds 5, 10, and 12 possessing the 1,1-dioxo-1_H_-1λ6-benzo[_b_] thiophen-6-yl fragment (B1) exhibited a similar or significantly higher potency than the reference compound niclosamide. Interestingly, compound 4 containing the fragment B1 and the moiety A2 instead of the 6-bromopyridin-2-yl fragment (A3) was found inactive with a dramatic loss of antiproliferative activity in comparison with compound 10. Compounds 8 and 9 with (S)-(1-phenylethyl) amide moiety (B2) showed moderate to potent anti-proliferative effects against the tested cancer cells. For example, compound 9 displayed an IC50 value of 0.9 μM against ER-positive breast cancer cell line MCF-7. The salicylic amide compound 13 with the 5-furan-2-yl-[1,3,4]oxadiazol-2-yl moiety (B3) was found inactive, while compound 12 with the fragment B1 instead exhibited significant antiproliferative activity, indicating that fragment B1 rather than B3 is more favorable for the anticancer activity of molecules with the salicylic amide scaffold.
Table 1.
Effects of newly synthesized compounds 4, 5, 8–10 and 12, 13 on proliferation of human breast and pancreatic cancer cell lines.
 |
||||||
|---|---|---|---|---|---|---|
| Compound | A | B | IC50 (μM)a | |||
| Breast cancer ER-Positive | Breast cancer ER-Negative | Pancreatic cancer | ||||
| MCF-7 | MDA-MB-231 | AsPC1 | Panc-1 | |||
| 4 | A2 | B1 | >10b | >10 | NDc | ND |
| 5 | A1 | B1 | 0.1 | 0.29 | 1.25 | 0.26 |
| 8 | A1 | B2 | 2.24 | 86.0 | >10 | >10 |
| 9 | A4 | B2 | 0.9 | 8.88 | 7.54 | 8.44 |
| 10 | A3 | B1 | 3.31 | 1.53 | 1.54 | 1.64 |
| 12 | A4 | B1 | 0.91 | 1.64 | 1.92 | 2.34 |
| 13 | A4 | B3 | >10 | >10 | >10 | >10 |
| Niclosamide | 1.06 | 0.79 | 1.47 | 1.73 |
Among the active new scaffolds discussed above, compound 5 notably exhibited remarkable potency against all the tested cancer cells. Since STX-0119 with the 2-phenylquinoline-4-carboxylic acid amide fragment (A1) [21,45] and stattic with the 1,1-dioxo-1_H_-1λ6-benzo[_b_]thiophen-6-yl fragment (B1) [23] have been reported as the STAT3-SH2 domain inhibitors, the molecular docking studies of compound 5 with both A1 and B1 fragments were performed to investigate the possible conformations and the required spatial relationship between the scaffold and STAT3-SH2 domain Ref. [46] using AutoDock Vina [47] docking approach. Examination of the predicted binding model for 5 complexed with STAT3 revealed that the 2-phenyl group on the quinoline ring could fit effectively into the hydrophobic cleft around Ile634 (Fig. 3). To further verify our findings, chemical optimization of 5 was carried out to gain insights on this potential series. Additional five compounds (19–23) with different hydrophobic groups were prepared (Scheme 3), analogously to the synthesis of 4 and 5 described above, and their anti-cancer activities were evaluated in the same fashion (Table 2).
Fig. 3.

Predicted binding mode for compound 5. (A) Surface of the electrostatic map. (B) Residues of STAT3. Compound 5 is shown in small sticks and in pink color. Hydrogen bonds are indicated by dashed lines. The figures were generated using Pymol. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Scheme 3.

a Reagents and conditions: (a) HBTU, DIPEA, CH2Cl2, rt, 39–56%.
Table 2.
Effects of newly synthesized compounds 19–23 on proliferation of human breast and pancreatic cancer cell lines.
The obtained SAR results suggest that the hydrophobic substituent at C2 of the quinoline or pyridine framework is crucial for targeting STAT3. For example, compounds 20, 21 and 23 without the 2-Ph moiety displayed moderate to low antiproliferative activity against the tested cancer cells. Instead, compound 22 with 2-Ph group in contrast with compound 21 regained antiproliferative activity with low micromolar IC50 values. Compound 19 with an additional phenyl substituent at the 6-position of pyridine ring exhibited further enhanced anticancer activity when compared with compound 22, indicating the important role of hydrophobic substituents on the quinoline and pyridine fragments.
Through the SAR studies, compound 5 has been identified with most desirable antiproliferative activity and physicochemical parameters (see Table S1 in the Supporting information), and was subjected to further biological characterization. Cellular morphological change in MDA-MB-231 breast cancer cells treating with compound 5 for 48 h was examined under light microscopy. As shown in Fig. S1, compound 5 significantly inhibited cell proliferation and induced apoptosis accompanying cellular morphological changes in a dose-dependent manner.
To determine whether compound 5 acts as a potent small-molecule inhibitor of STAT3 activation, we measured the effect of 5 on promoter activity using the cell-based transient transfection and dual luciferase reporter assays. The STAT3 promoter activity in MDA-MB-231 cells was determined after transient transfecting with pSTAT3-Luc vector. As shown in Fig. 4, treatment with 5 μM of compound 5 inhibited the STAT3 promoter activity in MDA-MB-231 cells by approximately 65% compared with control.
Fig. 4.
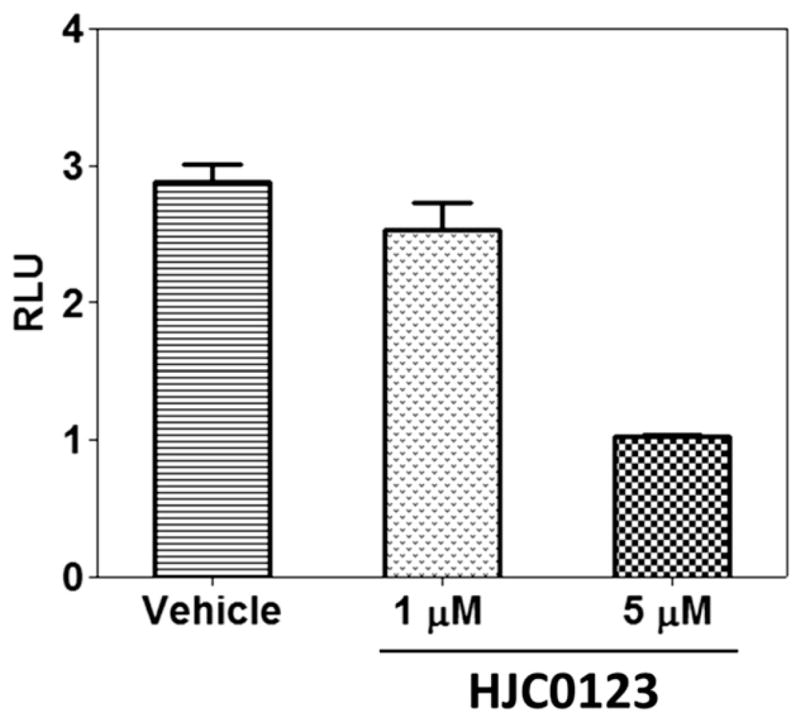
Compound 5 (HJC0123) inhibited the STAT3 mediated luciferase reporter activity in MDA-MB-231 cells. STAT3 promoter activity was measured using dual luciferase assay with a STAT3 reporter. Promoter activity obtained from DMSO-treated MDA-MB-231 cells was used as control. Error bars represent standard deviation of triplicate wells. Representative experiment from at least 3 independent experiments is shown. RLU: relative luciferase unit.
To further investigate the inhibitory activity of compound 5 against the STAT3 pathway, we examined STAT3 phosphorylation and expression of the known STAT3 target genes in MDA-MB-231 cell line. The cells were treated with different doses of compound 5 for 24 h and 48 h, and levels of total STAT3 and phosphorylated STAT3 at Tyr-705 were then examined by Western blot. It was found that the total STAT3 expression in these cells was reduced after the treatment with compound 5 (Fig. 5). Similarly, phos-phorylated STAT3 at Tyr-705 was suppressed by compound 5. Blocking STAT3 signaling in many different tumor cells leads to growth arrest and apoptosis [7,22,48–50]. To investigate whether compound 5 induces apoptosis, we first determined its ability in inducing cleaved caspase 3 in MDA-MB-231 breast cancer cells. As shown in Fig. 5, compound 5 induced a higher cleaved caspase-3 level in MDA-MB-231 cells, suggesting that compound 5 promoted the apoptosis of cancer cells. We further confirmed these results using annexin V-based measurement using flow cytometry. As depicted in Fig. S2, compound 5 activated apoptosis in MDA-MB-231 breast cancer cells in a dose-dependent manner. Further flow cytometry analysis revealed that compound 5 arrested MDA-MB-231 cells at S phase in a dose-dependent manner (Fig. S3). These results suggested that compound 5 was conferred the ability to arrest cells. The more extensive mechanism studies on compound 5 including STAT3 upstream targets and related signaling pathways are undergoing, and the results will be reported somewhere else in due course.
Fig. 5.
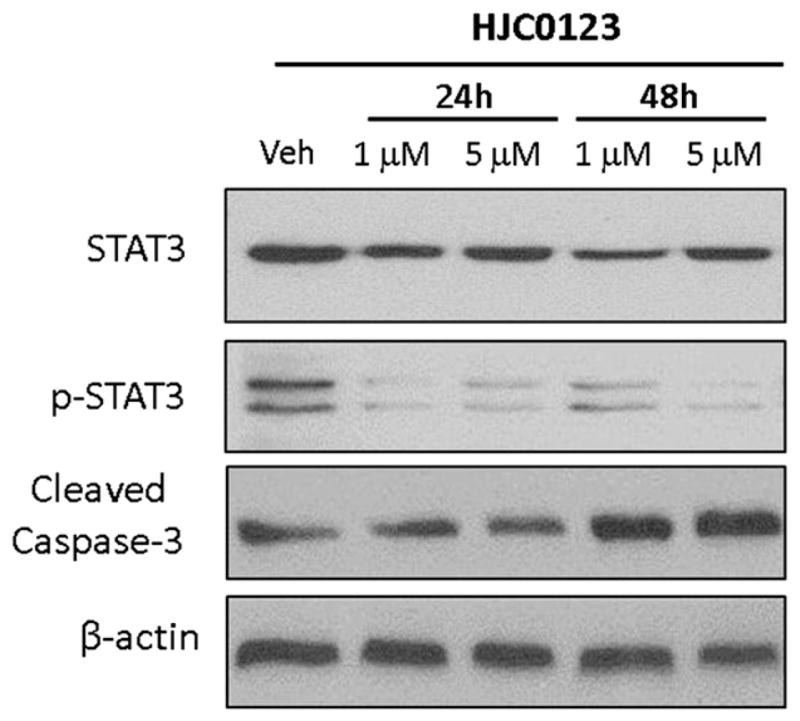
Western blot analysis of biochemical markers for apoptosis induction and inhibition of STAT3 activity by compound 5 (HJC0123) in the MDA-MB-231 cell line. Cells were treated with compound 5 for 24 h and 48 h, and levels of STAT3, pSTAT3, cleaved caspase-3 were probed by specific antibodies. β-Actin was used as the loading control.
Compound 5 was next evaluated for its antitumor activity in inhibition of tumor growth in the MDA-MB-231 xenograft model. MDA-MB-231 xenograft tumors were developed in immunodefficient nude mice and tumor volume was measured daily in oral gavage (p.o.) group. We treated the MDA-MB-231 xenograft mice through oral administration of compound 5 (50 mg/kg) and found that the growth of xenograft tumors in mice was significantly suppressed by compound 5 (Fig. 6). Notably, compound 5 did not show significant signs of toxicity even at the dose of 150 mg/kg (data not shown). These results have demonstrated that compound 5 is a potent, efficacious and orally bioavailable anti-cancer drug candidate that is promising for further clinical development.
Fig. 6.

In vivo efficacy of compound 5 (HJC0123) in inhibiting growth of xenograft tumors (Breast cancer MDA-MB-231) in mice (p.o.).
3. Conclusions
Taken together, a fragment-based drug design, systematic chemical synthesis and pharmacological evaluation of novel scaffolds as potent anticancer agents have been carried out by utilizing six privileged fragments from known STAT3 inhibitors. Several new molecules such as compounds 5,12, and 19 that may act as advanced chemical leads have been identified. The most potent compound 5 has demonstrated to inhibit STAT3 promoter activity, down-regulate phospho-STAT3, increase the expression of cleaved caspase-3, inhibit cell cycle progression and promote apoptosis in breast and pancreatic cancer cells with low micromolar to nanomolar IC50 values. Furthermore, compound 5 significantly suppressed ER-negative breast cancer MDA-MB-231 xenograft tumor growth in vivo (p.o.), indicating its great potential as an efficacious and orally bioavailable drug candidate for human cancer therapy. This promising compound has been selected for further preclinical assessment and the results will be reported somewhere else in due course.
4. Experimental
4.1. Chemistry
All commercially available starting materials and solvents were reagent grade, and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70– 230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Brucker-600 (1H, 600 MHz; 13C, 150 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; capillary temperature was 275 °C and the resolution was 60,000; ionization was achieved by positive mode. Melting points were measured on a Thermo Scientific Electrothermal Digital Melting Point Apparatus and uncorrected. Purity of final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 30% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min followed by 30 min of the last-named solvent. All biologically evaluated compounds are >95% pure.
4.1.1. 2-Cyano-N-(1,1-dioxo-1H-1λ6-benzo[b]thiophen-6-yl) acetamide (4)
To a solution of cyanoacetic acid (340 mg, 4.0 mmol) and 1,1-dioxo-1_H_-1λ6-benzo[_b_]thiophen-6-ylamine (362 mg, 2.0 mmol) in 10 mL of CH2Cl2 was added DIPEA (774 mg, 6.0 mmol). HBTU (1.14 g, 3.0 mmol) was added at 0 °C. The resulting mixture was stirred at r.t. for 28 h. The reaction mixture was diluted with CH2Cl2 (80 mL) and washed with water (20 mL). The organic layer was separated and dried with anhydrous Na2SO4. The solution was concentrated to give a crude product, which was purified with silica gel column (EtOAc/hexane = 1/1) to obtain the desired product (400 mg, 81%) as a yellow solid (mp 207–208 °C). HPLC purity 98.6% (tR = 10.93 min). 1H NMR (600 MHz, DMSO-_d_6) δ 10.79 (s,1H), 8.04 (s, 1H), 7.67–7.69 (m, 1H), 7.60 (d, 1H, J = 7.2 Hz), 7.57 (d, 1H, J = 7.8 Hz), 7.30 (d, 1H, J = 7.2 Hz), 3.98 (s, 2H). 13C NMR (150 MHz, DMSO-_d_6) δ 162.0, 140.7,137.3, 132.8, 130.3, 126.7, 126.1, 123.4, 115.6, 111.4, 27.0. HRMS (ESI) calcd for C11H19N2O3S 249.0328 (M + H)+, found 249.0330.
4.1.2. 2-Phenylquinoline-4-carboxylic acid (1,1-dioxo-1H-1λ6-benzo[b]thiophen-6-yl)amide (5)
To a solution of 2-phenylquinoline-4-carboxylic acid (249 mg, 1.0 mmol) and 1,1-dioxo-1_H_-1λ6-benzo[_b_]thiophen-6-ylamine (181 mg, 1.0 mmol) in 10 mL of CH2Cl2 was added DIPEA (387 mg, 3.0 mmol). HBTU (759 mg, 2.0 mmol) was added at 0 °C. The resulting mixture was stirred at r.t. for 48 h. The reaction mixture was diluted with EtOAc (300 mL) and washed with water (50 mL). The organic layer was separated and dried with anhydrous Na2SO4. The solution was concentrated to give a crude product, which was further purified with short silica gel column (EtOAc/hexane = 1/1 to 4/1) to obtain the desired product (160 mg, 39%) as a pale yellow solid (mp 277–278 °C). HPLC purity 99.2% (tR = 32.39 min). 1H NMR (600 MHz, DMSO-_d_6) δ 11.29 (s, 1H), 8.44 (s, 1H), 8.38 (d, 2H, J = 7.2 Hz), 8.34 (s, 1H), 8.20 (dd, 2H, J = 5.4 Hz, 13.8 Hz), 7.98 (d,1H, J = 7.8 Hz), 7.88 (t,1H, J = 7.2 Hz), 7.54–7.70 (m, 6H), 7.34 (d, 1H, J = 6.6 Hz). 13C NMR (150 MHz, DMSO-_d_6) δ 165.8, 155.8, 147.9, 142.2, 141.2, 138.0, 137.2, 132.9, 130.5, 130.4, 130.0, 129.7, 129.0, 127.6, 127.3, 126.6, 126.3, 125.1, 124.2, 123.0, 117.1, 117.1, 112.3, 112.3. HRMS (ESI) calcd for C24H17N2O3S 413.0954 (M + H)+, found 413.0959.
4.1.3. (S)-2-Phenylquinoline-4-carboxylic acid (1-phenylethyl) amide (8)
To a solution of 2-phenyl-quinoline-4-carboxylic acid (249 mg, 1.0 mmol) and L(−)-α-methylbenzylamine (127 mg, 1.05 mmol) in 10 mL of CH2Cl2 was added DIPEA (388 mg, 3.0 mmol). HBTU (569 mg, 1.5 mmol) was added at 0 °C. The resulting mixture was stirred at r.t. for 3 h. The reaction mixture was diluted with CH2Cl2 (80 mL) and washed with water (20 mL). The organic layer was separated and dried with anhydrous Na2SO4. The solution was concentrated to give a crude product, which was purified with silica gel column (EtOAc/hexane = 1/3) to obtain 8 (330 mg, 94%) as a white solid (mp 157–158 °C). HPLC purity 99.8% (tR = 17.22 min). 1H NMR (600 MHz, CDCl3) δ 8.03–8.07 (m, 3H), 7.97 (d, 1H, J = 8.4 Hz), 7.71 (s, 1H), 7.67 (t, 1H, J = 7.2 Hz), 7.28–7.48 (m, 9H), 6.78 (d,1H, J = 7.2 Hz), 5.38–5.43 (m,1H),1.66 (d, 3H, J = 6.6 Hz). 13C NMR (150 MHz, CDCl3) δ 166.9, 156.9, 148.7, 143.0, 142.6, 138.9, 130.3, 130.2, 129.8, 129.0, 127.8, 127.6, 127.4, 126.4, 125.0, 123.4, 116.4, 49.8, 21.9. HRMS (ESI) calcd for C24H21N2O 353.1648 (M + H)+, found 353.1653.
4.1.4. (S)-5-Chloro-2-hydroxy-N-(1-phenylethyl)benzamide (9)
Compound 9 was prepared in 39% yield by a procedure similar to that used to prepare compound 8. The title compound was obtained as a white solid (mp 124–125 °C). HPLC purity 97.1% (tR = 19.75 min). 1H NMR (600 MHz, CDCl3) δ 12.22 (s, 1H), 7.28–7.48 (m, 7H), 6.93 (d, 1H, J = 9.0 Hz), 6.55 (d, 1H, J = 6.6 Hz), 5.29–5.34 (m, 1H), 1.64 (d, 3H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 168.2, 160.4, 142.3, 134.3, 129.1, 128.0, 126.3, 125.1, 123.4, 120.3, 115.3, 49.5, 21.7. HRMS (ESI) calcd for C15H15ClNO2 276.0786 (M + H)+, found 276.0790.
4.1.5. 3-(6-Bromopyridin-2-yl)-2-cyano-N-(1,1-dioxo-1H-1λ6-benzo[b]thiophen-6-yl)acrylamide (10)
To a solution of 4 (100 mg, 0.40 mmol) and 6-bromo-pyridine-2-carbaldehyde (112 mg, 0.60 mmol) in EtOH (5 mL) was added piperidine (3 mg, 0.04 mmol) at 0 °C. The mixture was stirred at 90 °C for 0.5 h. A yellow suspension formed during the reaction. The solid was filtered and washed with H2O. The desired product (120 mg, 72%) was obtained as a yellow solid (mp 219–220 °C). HPLC purity 96.0% (tR = 16.57 min). 1H NMR (600 MHz, DMSO-_d_6) δ 10.94 (s, 1H), 8.24 (s, 1H), 8.14 (s, 1H), 7.98 (t, 1H, J = 7.8 Hz), 7.91– 7.93 (m, 2H), 7.84 (d, 1H, J = 7.8 Hz), 7.62 (t, 2H, J = 7.2 Hz), 7.34 (d, 1H, J = 7.2 Hz). 13C NMR (150 MHz, DMSO-_d_6) δ 160.7, 150.8, 147.2, 141.3, 140.8, 140.6, 137.1, 132.7, 130.9, 130.5, 126.7, 126.6, 126.6, 124.8, 114.6, 112.8, 110.8. HRMS (ESI) calcd for C17H11BrN3O3S 415.9699 (M + H)+, found 415.9703.
4.1.6. 5-Chloro-N-(1,1-dioxo-1H-1λ6-benzo[b]thiophen-6-yl)-2-hydroxybenzamide (12)
A solution of 5-chloro-2-hydroxybenzoic acid (2.0 g, 11 mmol) and 4 mL of SOCl2 in 4 mL of toluene was stirred at 110 °C overnight. The mixture was concentrated to give a crude product as a pale yellow oil. To the solution of pyridine (869 mg, 11 mmol) and 1,1-dioxo-1_H_-1λ6-benzo[_b_]thiophen-6-ylamine (200 mg, 1.1 mmol) was added the solution of the acid chloride (500 mg, 2.6 mmol) in DMF (15 mL) dropwise at 0 °C. The mixture was stirred at r.t. for 24 h. The mixture was added to the water solution dropwise. The yellow solid was formed and filtrated. To the mixture of the crude product in THF (8 mL) was added 1 N LiOH (2.2 mL, 2.2 mmol) at 0 °C. The mixture was stirred at 0 °C for 30 min. The mixture was diluted with EtOAc (50 mL) and washed with 2 N HCl (10 mL). The organic layer was separated and dried with anhydrous Na2SO4. The solution was concentrated to afford the crude product, which was washed with CH2Cl2 (20 mL) to give the desired product (150 mg, 39%) as a yellow solid (mp 268–269 °C). HPLC purity 98.3% (tR = 17.24 min). 1H NMR (600 MHz, DMSO-_d_6) δ 11.50 (s, 1H), 10.74 (s, 1H), 8.25 (s, 1H), 7.92 (d, 1H, J = 8.4 Hz), 7.86 (d, 1H, J = 2.4 Hz), 7.59–7.62 (m, 2 H), 7.48 (d, 1H, J = 9.0 Hz), 7.31 (d, 1H, J = 6.6 Hz), 7.04 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, DMSO-_d_6) δ 165.1, 156.2, 140.7, 137.1, 133.1, 132.8, 130.3, 128.6, 126.5, 126.2, 124.6, 122.8, 120.4, 119.0, 112.7. HRMS (ESI) calcd for C15H11ClNO4S 336.0092 (M + H)+, found 336.0098.
4.1.7. 5-Chloro-N-(5-furan-2-yl-[1,3,4]oxadiazol-2-yl)-2-hydroxybenzamide (13)
Compound 13 was prepared in 50% yield by a procedure similar to that used to prepare compound 12. The title compound was obtained as a white solid (mp 196–197 °C). HPLC purity 96.0% (tR = 18.99 min). 1H NMR (600 MHz, CDCl3) δ 10.11 (s, 1H), 8.13 (d, 1H, J = 2.4 Hz), 8.02 (d, 1H, J = 2.4 Hz), 7.64 (dd, 1H, J = 2.4 Hz and 8.4 Hz), 7.49 (dd, 1H, J = 2.4 Hz and 8.4 Hz), 7.21 (d, 1H, J = 9.0 Hz), 6.99 (d, 1H, J = 9.0 Hz). 13C NMR (150 MHz, DMSO-_d_6) δ 164.4, 158.4, 148.3, 135.2, 133.6, 130.8, 130.6, 130.2, 126.2, 125.9, 122.6, 119.7, 115.4. HRMS (ESI) calcd for C13H9ClN3O4 306.0276 (M + H)+, found 306.0274.
4.1.8. N-(1,1-Dioxo-1H-1λ6-benzo[b]thiophen-6-yl)-2,6-diphenyl-isonicotinamide (19)
Compound 19 was prepared in 50% yield by a procedure similar to that used to prepare compound 4. The title compound was obtained as a pale yellow solid (mp 235–236 °C). HPLC purity 97.2% (tR = 22.05 min). 1H NMR (600 MHz, CDCl3) δ 8.76 (s, 1H), 8.34 (d, 1H, J = 6.6 Hz), 8.23 (d, 4H, J = 7.2 Hz), 8.14 (s, 2H) 7.93 (s, 1H), 7.41– 7.55 (m, 6H), 7.40 (d, 1H, J = 8.4 Hz), 7.13–7.15 (m, 1H), 6.26–6.28 (m, 1H). 13C NMR (150 MHz, DMSO-_d_6) δ 164.7, 156.6, 143.9, 141.1, 138.1,137.1, 132.8, 130.4, 129.7,129.0, 126.9,126.6,126.3,124.6,116.7, 112.7. HRMS (ESI) calcd for C26H19N2O3S 439.1111 (M + H)+, found 439.1116.
4.1.9. Acridine-9-carboxylic acid (1,1-dioxo-1H-1λ6-benzo[b] thiophen-6-yl)amide (20)
Compound 20 was prepared in 39% yield by a procedure similar to that used to prepare compound 4. The title compound was obtained as a yellow solid (mp 237–238 °C). HPLC purity 99.4% (tR = 14.48 min). 1H NMR (600 MHz, DMSO-_d_6) δ 8.71 (d, 1H, J = 8.4 Hz), 8.32 (d, 2H, J = 8.4 Hz), 8.08–8.13 (m, 5 H), 7.97 (t, 2H, J = 7.8 Hz), 7.82 (t, 1H, J = 8.4 Hz), 7.68 (t, 2H, J = 7.8 Hz). 13C NMR (150 MHz, DMSO-_d_6) δ 163.4, 147.9, 136.1, 133.9, 132.9, 131.1, 129.5, 128.0, 128.0, 125.2, 121.7, 116.5, 115.5. HRMS (ESI) calcd for C22H15N2O3S 387.0798 (M + H)+, found 387.0802.
4.1.10. N-(1,1-Dioxo-1H-1λ6-benzo[b]thiophen-6-yl) isonicotinamide (21)
Compound 21 was prepared in 56% yield by a procedure similar to that used to prepare compound 4. The title compound was obtained as a pale yellow solid (mp 251–252 °C). HPLC purity 97.0% (tR = 8.81 min). 1H NMR (600 MHz, DMSO-_d_6) δ 10.92 (s, 1H), 8.81– 8.82 (m, 2H), 8.27 (s, 1H), 8.00 (d, 1H, J = 7.8 Hz), 7.88 (t, 2H, J = 2.4 Hz), 7.62 (t, 2H, J = 6.6 Hz), 7.33 (d, 1H, J = 6.6 Hz). 13C NMR (150 MHz, DMSO-_d_6) δ 164.6, 150.4, 141.3, 141.1, 137.1, 132.8, 130.4, 126.5, 126.3, 124.5, 121.6, 112.6. HRMS (ESI) calcd for C14H11N2O3S 287.0485 (M + H)+, found 287.0488.
4.1.11. N-(1,1-Dioxo-1H-1λ6-benzo[b]thiophen-6-yl)-2-phenyl-isonicotinamide (22)
Compound 22 was prepared in 46% yield by a procedure similar to that used to prepare compound 4. The title compound was obtained as a pale yellow solid (mp 285–286 °C). HPLC purity 98.0% (tR = 14.72 min). 1H NMR (600 MHz, DMSO-_d_6) δ 10.97 (s, 1H), 8.89 (d, 1H, J = 4.8 Hz), 8.42 (s, 1H), 8.28 (s, 1H), 8.19 (d, 2H, J = 7.8 Hz), 8.03–8.05 (m, 1H), 7.82 (d, 1H, J = 4.8 Hz), 7.63 (d, 2H, J = 7.8 Hz), 7.56 (t, 1H, J = 7.2 Hz), 7.51 (t, 2H, J = 7.2 Hz), 7.34 (d, 1H, J = 6.6 Hz). 13 C NMR (150 MHz, DMSO-_d_6) δ 164.6, 156.9, 150.4, 142.6, 141.1, 138.1, 137.1, 132.8, 130.4, 129.6, 128.9, 126.8, 126.5, 126.3, 124.6, 120.5, 118.0, 112.7. HRMS (ESI) calcd for C20H15N2O3S 363.0798 (M + H)+, found 363.0791.
4.1.12. Quinoline-3-carboxylic acid (1,1-dioxo-1H-1λ6-benzo[b] thiophen-6-yl)amide (23)
Compound 23 was prepared in 48% yield by a procedure similar to that used to prepare compound 4. The title compound was obtained as a pale yellow solid (mp 209–210 °C). HPLC purity 98.8% (tR = 11.01 min). 1H NMR (600 MHz, acetone-_d_6) δ 10.29 (s, 1H), 9.03 (d, 1H, J = 4.2 Hz), 8.42 (s, 1H), 8.34 (d, 1H, J = 8.4 Hz), 8.14 (d, 1H, J = 8.4 Hz), 8.03–8.05 (m, 1H), 7.83–7.86 (m, 1H), 7.79 (d, 1H, J = 4.8 Hz), 7.68–7.70 (m, 1H), 7.63 (d, 1H, J = 7.8 Hz), 7.57 (d, 1H, J = 7.2 Hz), 7.04 (d, 1H, J = 7.2 Hz). 13C NMR (150 MHz, acetone-_d_6) δ 166.7, 151.0, 149.7, 142.4, 139.1, 133.1, 131.6, 130.7, 128.5, 127.7, 127.2, 126.3, 125.1, 124.8, 124.7, 120.0, 113.3, 113.2. HRMS (ESI) calcd for C18H13N2O3S 337.0641 (M + H)+, found 337.0646.
4.2. Biology
4.2.1. In vitro determination of effects of synthesized compounds on cancer cell proliferation
Cancer cells (breast cancer cell lines MCF-7 and MDA-MB-231, pancreatic cancer cell lines AsPC-1 and Panc-1) were seeded in 96-well plates at a density of 2 × 103 cells/well and treated with DMSO, 0.01, 0.1, 1, 5, 10, and 100 μM of individual STAT3 inhibitors for 72 h. Proliferation was measured by treating cells with the (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2_H_-tetrazolium) (MTS) in a CellTiter 96t Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI, USA). Absorbance of all wells was determined by measuring OD at 550 nm after 1 h incubation at 37 °C on a 96-well iMark™ Microplate Absorbance Reader (BioRad, Hercules, CA). Each individual compound was tested in quadruplicate wells for each concentration.
4.2.2. Molecular docking studies
Compound 5 was docked with the STAT3–SH2 domain using the X-ray structure (PDB code: 1BG1) and AutoDock Vina 1.1.2. Water molecules within the crystal structure were removed and polar hydrogens were added using AutoDockTools. The protein was treated as rigid. Docking runs were carried out using the standard parameters of the program for interactive growing and subsequent scoring, except for the parameters for setting grid box dimensions and center. For all of the docking studies, a grid box size of 30 Å × 30 Å × 30 Å, centered at coordinates 100.452 (x), 75.972 (y), and 68.790 (z) of the PDB structure.
4.2.3. Transient transfection and dual luciferase reporter assays
MDA-MB-231 cells were seeded in 24-well plate at a density of 5 ×104 cells/well in RPMI-1640 medium containing 10% FBS and 1% penicillin–streptomycin. Transient transfections were performed 4 h after plating, using the method described previously [40,51]. Total amount of DNA for transfections was 0.5 μg/well, including pSTAT3-Luc (95%, obtained from Panomics, Cat# LR0077) and internal control vector renilla (5%, from Promega, Madison, WI, USA). 5 h after transfection, the cells were treated with compound 5 for 24 h, then reporter activity was evaluated using dual luciferase reporter assay kit (Promega, Madison, WI, USA) on an Omega™ Microplate Luminometer (BMG LABTECH Inc., NC, USA). Relative luciferase units were the ratio of the absolute activity of firefly luciferase to that of renilla luciferase. Experiments were conducted with triplicates and results are representatives of at least 3 independent experiments.
4.2.4. Western blot analysis
Protein levels were determined by Western blot using the previously described methods [51]. Total cell lysates were prepared from MDA-MB-231 cells. Protein concentrations were measured using the BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Equal amounts of total cellular protein extract (40 μg) were resuspended in denaturing sample loading buffer (0.5 M Tris–HCl, pH 6.8, 10% SDS, 0.1% bromophenol blue, and 20% glycerol), separated by electrophoresis on a 10% polyacrylamide SDS-PAGE gel and then electrophoretically transferred to a nitrocellulose membrane (Thermo Scientific, IL, USA) at 100 V for 1 h at 4 °C. The membrane was then incubated in a blocking solution containing 5% non-fat milk and 1% Tween 20 in TBS for 1 h. The membrane was then incubated with antibodies specific for: phospho-STAT3-pY705 (1:3000, Epitomics, #2236-1), STAT3 (1:2000, Cell Signaling, #4904), Caspase-3-cleaved (1:2000, Epitomics, #1476-1), Cyclin D1 (1:10,000, Epitomics, #2261-1) and β-actin (1:10,000, Sigma, clone AC-15). An anti-rabbit or anti-mouse secondary antibody (Amersham, Piscataway, NJ) was used at 1:4000 dilution. The Western blotted bands were visualized using ECL procedure according to the manufacturer’s instructions (Amersham).
4.2.5. In vivo antitumor activity assays
All procedures including mice and in vivo experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of UT M.D. Anderson Cancer Center (MDACC). Female nude mice were obtained from MDACC and were used for orthotopic tumor studies at 4–6 weeks of age. The mice were maintained in a barrier unit with 12 h light–dark switch. Freshly harvested MDA-MB-231 cells (2.5 × 106 cells per mouse, resuspended in 100 μL PBS) were injected into the 3rd mammary fat pad of the mice, and then randomly assigned into 2 groups (6– 7 mice per group). The mice were given 50 mg/kg compound 5 or vehicle five days per week when the tumor volume reached 200 mm3. All drugs were dissolved in 50% DMSO with 50% polyethylene glycol for in vivo administration. Body weights and tumors volume were measured daily and tumor volume was calculated according to the formula V = 0.5 × L × W2, where L = length (mm) and W = width (mm).
4.2.6. Statistical analysis
Statistical significance was determined using student _t_-test in cell cycle analysis. *Represents a p value less than 0.05.
Supplementary Material
mmc1
Acknowledgments
This work was supported by grants P50 CA097007, P30DA028821, R21MH093844 (J.Z.) from the National Institute of Health, R.A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, John Sealy Memorial Endowment Fund (JZ), the Duncan Family Institute (DFI) Seed Funding Program, and startup fund from MD Anderson Cancer Center (QS). We thank Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance.
Abbreviations
FBDD
fragment-based drug design
STATs
signal transducers and activators of transcription
ER
estrogen receptor
HBTU
2-(1_H_-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
DIPEA
N,_N_-diisopropylethylamine
THF
tetrahydrofuran
SAR
structure–activity relationships
RLU
relative luciferase unit
MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2_H_-tetrazolium)
IC50
half maximal inhibitory concentration
PI
propidium iodide
HRMS
high-resolution mass spectrometry
HPLC
high performance liquid chromatography
TFA
trifluoroacetic acid
DMSO
dimethyl sulfoxide
TLC
thin layer chromatography
NMR
nuclear magnetic resonance
TMS
tetra-methylsilane
EtOAc
ethyl acetate
DMF
dimethylformamide
PBS
phosphate-buffered saline
BCA
bicinchoninic acid
SDS-PAGE
sodium dodecyl sulfate poly-acrylamide gel electrophoresis
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2013.01.023.
Footnotes
Conflict of interest
None.
References
- 1.Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and inter-leukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 2.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 3.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr STAT3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 4.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 5.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 6.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 7.Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–6626. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- 8.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interventions. 2011;11:18–26. doi: 10.1124/mi.11.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]
- 10.Haura EB, Turkson J, Jove R. Mechanisms of disease: insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol. 2005;2:315–324. doi: 10.1038/ncponc0195. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 12.Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 13.Darnell JE., Jr Validating Stat3 in cancer therapy. Nat Med. 2005;11:595–596. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 14.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13:5665–5669. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- 17.Costantino L, Barlocco D. STAT3 as a target for cancer drug discovery. Curr Med Chem. 2008;15:834–843. doi: 10.2174/092986708783955464. [DOI] [PubMed] [Google Scholar]
- 18.Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18:254–267. doi: 10.1038/cr.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schindler CW. Series introduction. JAK-STAT signaling in human disease. J Clin Invest. 2002;109:1133–1137. doi: 10.1172/JCI15644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 21.Matsuno K, Masuda Y, Uehara Y, Sato H, Muroya A, Takahashi O, Yokotagawa T, Furuya T, Okawara T, Otsuka M, Ogo N, Ashizawa T, Oshita C, Tai S, Ishii H, Akiyama Y, Asai A. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med Chem Lett. 2010;1:371–375. doi: 10.1021/ml1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, Sebti S, Yu H, Jove R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent anti-tumor activity. Mol Cancer Ther. 2004;3:1533–1542. [PubMed] [Google Scholar]
- 25.Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280:32979–32988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- 26.Ren X, Duan L, He Q, Zhang Z, Zhou Y, Wu D, Pan J, Pei D, Ding K. Identification of niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. ACS Med Chem Lett. 2010;1:454–459. doi: 10.1021/ml100146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horiguchi A, Asano T, Kuroda K, Sato A, Asakuma J, Ito K, Hayakawa M, Sumitomo M, Asano T. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br J Cancer. 2010;102:1592–1599. doi: 10.1038/sj.bjc.6605691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, Kondo S, Priebe W, Kondo Y. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–2444. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem Lett. 2010;1:85–89. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandal PK, Ren Z, Chen X, Xiong C, McMurray JS. Structure–affinity relationships of glutamine mimics incorporated into phosphopeptides targeted to the SH2 domain of signal transducer and activator of transcription 3. J Med Chem. 2009;52:6126–6141. doi: 10.1021/jm901105k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Jr, Liao WS, McMurray JS. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J Med Chem. 2011;54:3549–3563. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng J, Grande F, Neamati N. Small molecule inhibitors of stat3 signaling pathway. Curr Cancer Drug Targets. 2007;7:91–107. doi: 10.2174/156800907780006922. [DOI] [PubMed] [Google Scholar]
- 34.Zhao M, Jiang B, Gao FH. Small molecule inhibitors of STAT3 for cancer therapy. Curr Med Chem. 2011;18:4012–4018. doi: 10.2174/092986711796957284. [DOI] [PubMed] [Google Scholar]
- 35.Mankan AK, Greten FR. Inhibiting signal transducer and activator of transcription 3: rationality and rationale design of inhibitors. Expert Opin Invest Drugs. 2011;20:1263–1275. doi: 10.1517/13543784.2011.601739. [DOI] [PubMed] [Google Scholar]
- 36.Lavecchia A, Di Giovanni C, Novellino E. STAT-3 inhibitors: state of the art and new horizons for cancer treatment. Curr Med Chem. 2011;18:2359–2375. doi: 10.2174/092986711795843218. [DOI] [PubMed] [Google Scholar]
- 37.Page BD, Ball DP, Gunning PT. Signal transducer and activator of transcription 3 inhibitors: a patent review. Expert Opin Ther Pat. 2011;21:65–83. doi: 10.1517/13543776.2011.539205. [DOI] [PubMed] [Google Scholar]
- 38.Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem. 2012;55:6645–6668. doi: 10.1021/jm300207s. [DOI] [PubMed] [Google Scholar]
- 39.Erlanson DA, McDowell RS, O’Brien T. Fragment-based drug discovery. J Med Chem. 2004;47:3463–3482. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]
- 40.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 41.Hajduk PJ. Fragment-based drug design: how big is too big? J Med Chem. 2006;49:6972–6976. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 42.Babaoglu K, Shoichet BK. Deconstructing fragment-based inhibitor discovery. Nat Chem Biol. 2006;2:720–723. doi: 10.1038/nchembio831. [DOI] [PubMed] [Google Scholar]
- 43.(a) Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]; (b) Barelier S, Krimm I. Ligand specificity, privileged substructures and protein druggability from fragment-based screening. Curr Opin Chem Biol. 2011;15:469–474. doi: 10.1016/j.cbpa.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Liu A, Zhao Z, Xu Y, Lin J, Jou D, Li C. Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3) J Med Chem. 2011;54:5592–5596. doi: 10.1021/jm101330h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3-Beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 47.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernández-Luna JL, Nuñez G, Dalton WS, Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 49.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT, Turkson J. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–9628. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen Q, Uray IP, Li Y, Krisko TI, Strecker TE, Kim HT, Brown PH. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene. 2008;27:366–377. doi: 10.1038/sj.onc.1210643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mmc1