Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice (original) (raw)
Abstract
Nitric oxide (NO) can modulate arterial stiffness by regulating both functional and structural changes in the arterial wall. Tissue transglutaminase (TG2) has been shown to contribute to increased central aortic stiffness by catalyzing the cross-linking of matrix proteins. NO _S_-nitrosylates and constrains TG2 to the cytosolic compartment and thereby holds its cross-linking function latent. In the present study, the role of endothelial NO synthase (eNOS)-derived NO in regulating TG2 function was studied using eNOS knockout mice. Matrix-associated TG2 and TG2 cross-linking function were higher, whereas TG2 _S_-nitrosylation was lower in the eNOS−/− compared with wild-type (WT) mice. Pulse-wave velocity (PWV) and blood pressure measured noninvasively were elevated in the eNOS−/− compared with WT mice. Intact aortas and decellularized aortic tissue scaffolds of eNOS−/− mice were significantly stiffer, as determined by tensile testing. The carotid arteries of the eNOS−/− mice were also stiffer, as determined by pressure-dimension analysis. Invasive methods to determine the PWV-mean arterial pressure relationship showed that PWV in eNOS−/− and WT diverge at higher mean arterial pressure. Thus eNOS-derived NO regulates TG2 localization and function and contributes to vascular stiffness.
Keywords: endothelial nitric oxide synthase, nitric oxide, tissue transglutaminase, vascular stiffness, _S_-nitrosylation, pulse-wave velocity, tensile testing
cardiovascular disease is a major contributor of age-related morbidity and mortality, with isolated systolic hypertension being one of the hallmarks of this process (13, 14). It is now well established that vascular stiffness directly leads to systolic hypertension, impaired ventricular-arterial coupling, and an imbalance in myocardial oxygen supply and demand (17, 19). Moreover, multiple studies have conclusively demonstrated that vascular stiffness, as measured by pulse-wave velocity (PWV) or pulse pressure, is an independent predictor of adverse cardiovascular events and can be used to stratify risk patients in the clinical setting (21, 22).
Tissue transglutaminase (TG2) is a multifunctional protein that is ubiquitously expressed in all cell types of the vasculature. TG2 catalyzes the formation of ε-(γ-glutamyl)lysine bonds in a Ca2+-dependent manner and induces protein cross-links to stabilize the extracellular matrix (ECM). TG2 plays an important role in vascular function, including remodeling of resistance vessels (3, 4), increased central aortic stiffness with age (25), and arterial calcification (28). We have previously shown that nitric oxide (NO) regulates TG2 cross-linking activity and trafficking in the vasculature (18, 24). Specifically, decreased NO bioavailability is associated with decreased TG2 _S_-nitrosylation, increased secretion of TG2 to the cell surface and ECM, and increased TG2 cross-linking activity in isolated endothelial, smooth muscle, and fibroblast cells and in the rat aorta. In the previous studies, NO bioavailability was modulated pharmacologically using either NO synthase (NOS) inhibitor _N_G-nitro-l-arginine methyl ester (l-NAME) to decrease NO levels or the NO donor _S_-nitrosoglutathione (GSNO) to increase NO levels. Alternatively, endothelial cells were cocultured with fibroblast and vascular smooth muscle cells in the presence and absence of l-NAME to alter NO bioavailability. In animal models, l-NAME was administered via osmotic infusion pumps to diminish NOS-dependent NO production. In the present study, using genetic approaches, we tested the hypothesis that a decrease in NO produced by endothelial NOS (eNOS) would lead to decreased TG2 _S_-nitrosylation, increased TG2 activity, and increased vascular stiffness. Specifically, TG2 function was examined using eNOS shRNA-mediated knockdown eNOS expression in human aortic endothelial cells (HAEC) and using eNOS−/− mice. We demonstrate that eNOS is critical for inhibiting the activity of TG2 and maintaining central vascular compliance.
MATERIALS AND METHODS
Animals.
Male eNOS knockout mice with their wild-type (WT) background controls (C57BL/6J, 12- to 14-wk-old) obtained from Jackson Laboratories were used in this study. Mice were housed in temperature- and light-controlled conditions and fed and watered ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the Johns Hopkins University School of Medicine.
Cell cultures.
HAEC purchased from Invitrogen were cultured in endothelial cell media (ScienCell Research Labs; ECM media supplemented with 5% FBS, endothelial cell growth supplement, and penicillin/streptomycin) and used between passages 7 and 10.
eNOS knockdown.
Lentivirus-delivered short-hairpin RNA (shRNA; Santa Cruz Biotech) were used to knock down eNOS expression in cells. ECM media was replaced with phenol red-free low serum media containing Polybrene, and samples were treated with eNOS shRNA lentivirus or control lentivirus. After 2 h, media was again exchanged to ECM media, and the cells were cultured for an additional 48 h and used. Knockdown of eNOS was confirmed by Western blotting using eNOS antibody (BD Biosciences).
Protein expression.
The expression levels of different proteins were determined by Western blotting. Cells were lysed in 1× radioimmunoprecipitation assay (RIPA) buffer (Upstate) containing protease inhibitors (Roche) by mechanical scraping, and total protein concentration was determined (Bio-Rad protein assay reagent). Equal amounts of lysate proteins from each sample were resolved by SDS PAGE and electrotransferred to nitrocellulose. Blots were blocked with 3% nonfat dry milk for 1 h at room temperature. The membrane was incubated with primary antibody followed by HRP-conjugated anti-rabbit or anti-mouse antibody (1:1,000) for 1 h at room temperature. GAPDH was used as loading control. Mouse anti-TG2 (1:5,000; Neomarkers) was used to detect TG2 in HAEC, and rabbit anti-TG2 (1:1,000; Neomarkers) was used to detect TG2 in mouse tissue. Rabbit anti-eNOS was from Santa Cruz Biotech (1:1,000), and mouse anti-GAPDH (1:5,000) was from Novus Biologicals. Blots were developed using enhanced chemiluminescence and quantified using ImageJ [National Institutes of Health (NIH)].
Co-immunoprecipitation.
Proteins (50 μg) from homogenates of aorta from eNOS−/− and WT mice were immunoprecipitated using integrin β1-antibody (Armenian hamster anti-integrin β; BD Bioscience). Samples were eluted in Laemmli buffer, resolved by SDS PAGE, and electrotransferred to nitrocellulose membrane. Co-precipitation of TG2 was determined by Western blotting. Integrin β1 was also determined in the sample by Western blotting, using horseradish peroxidase (HRP)-conjugated goat anti-Armenian hamster secondary antibody (Pierce). Proteins from eNOS−/− mouse aorta were immunoprecipitated with isotype IgG as a negative control.
Noninvasive pulse-wave velocity measurement.
Mice were anesthetized in a closed chamber with isoflurane. Anesthesia was maintained by mask ventilation of 1.0–1.5% isoflurane (in 100% O2) with a charcoal scavenging system. Animals were positioned supine on a temperature-controlled printed circuit board (Indus Instruments, Houston, TX) with legs and arms taped to incorporated electrocardiogram electrodes. Body temperature was monitored with a rectal probe (Physitemp, Clifton, NJ) and maintained at 37°C. Doppler spectrograms of aortic outflow were acquired with a 2-mm-diameter, 10-MHz pulsed Doppler probe (Indus Instruments, Webster, TX). Thoracic aortic outflow and abdominal aortic flow profiles were captured. The distance separating the probe locations was also measured. Aortic PWV was calculated as the quotient of the separation distance and the time difference between pulse arrivals, with respect to the R-peak of the electrocardiogram. DSPW software (Indus Instruments) was used for data analysis.
Invasive PWV measurement.
We used a high-fidelity dual-pressure catheter sensor to measure aortic PWV. Mice were anesthetized with an intraperitoneal injection of 1.2% Avertin (2,2,2-tribromoethanol, 240 mg/kg) in 0.9% saline. The animal was positioned supine on the heating pad, with water temperature set to 40°C. Anesthesia was maintained by mask ventilation with 1.0–1.5% isoflurane (in 100% O2), and the reflex response to hind-paw pinching was assessed to monitor depth of anesthesia. After midline neck incision from mandible to sternum, a 1.2-Fr, dual-pressure sensor catheter (Scisense, London, Ontario, Canada) was introduced into the descending thoracic aorta through the left carotid artery without opening the chest cavity (see Fig. 4_A_). The distance between two sensors is fixed at 1 cm. A 30-gauge cannulation needle connected to polyethylene tubing (10) was inserted into left femoral vein for infusion of fluid/drugs. After stabilization of the signal for 10–15 min, baseline blood pressures were recorded. Mean arterial pressure (MAP) was raised and lowered to obtain a full physiological range of blood pressure using intravenous infusion of phenylephrine and sodium nitroprusside, respectively (see Fig. 4_C_). PWV at corresponding MAP was calculated using the foot-to-foot method, the foot being defined by the peak of the second time derivative of two aortic pressures measured simultaneously during each pulse (see Fig. 4_B_). PWV was plotted against MAP to construct phase plots to characterize PWV over a wide range of MAPs from 50 to 150 mmHg in the aorta (see Fig. 4_D_).
Fig. 4.

Vascular stiffness measures in eNOS−/− and WT mice. A: simultaneous pulsed-wave velocity (PWV) and mean arterial pressure (MAP) measurement in mice; a 1.2-Fr catheter was used to measure transit time in vivo. B: example waves/second derivative to determine transit time. C: example of beat-to-beat changes in arterial pressure. D: sample PWV and MAP relationship in a single animal. E: single-point, Doppler-based PWV measurement was higher in eNOS−/− compared with WT. F: MAP was higher in eNOS−/− mice. G: heart rate was similar in both groups. H: PWV-MAP correlation in eNOS−/− mice diverged from WT mice at higher MAPs; dotted lines represent baseline pressure and PWV for eNOS−/− (red) and WT (black) mice. I: intact aorta of eNOS−/− mice were stiffer than WT. Inset: incremental elastic modulus of intact vessels at strain = 0.5. J: decellularized aortic segments of eNOS−/− mice were stiffer than those from WT mice. Inset: incremental elastic modulus of decellularized vessels at strain = 0.8. Values are means ± SE (n = 8). Significant difference by Student's _t_-test for two groups or one-way ANOVA with Bonferroni post hoc analysis for more than two groups: *P < 0.05; **P < 0.01; ***P < 0.001.
In vivo TG inhibition in mice.
Mice were randomized into two groups, and an osmotic pump (Alzet) was implanted and filled with a 4-wk dose of either cystamine (40 mg·kg−1· day−1) or vehicle control.
Pressure-diameter analysis.
Common carotid arteries were isolated from mice and mounted on the glass pipettes of a pressure myograph in a perfusion chamber. The artery was perfused with heated and oxygenated calcium-free Krebs buffer using a peristaltic pump (Cole-Parmer Instrument), which also continuously monitored perfusion pressure. Intraluminal pressure was incrementally increased from 0 to 100 mmHg in steps of 10 mmHg, with each step in 30-s intervals, and corresponding change of vessel outer diameter was simultaneously recorded using microscopic imaging and video dimension analysis (Analog Digital Instruments). Compliance was calculated as previously described (25) using the following equation:
Compliance=ΔVΔP=ΔAΔP=Δd2ΔP
where V is the volume, A is the area, d is the diameter of the vessel, and P is the distending pressure.
TG activity assay.
A dot blot assay was used to determine TG activity (25). The assay is based on the incorporation of TG substrate biotin(amido)pentylamine (BPA; Pierce) into proteins. Intact cells/tissues were incubated with 0.1 mM BPA and 1 mM Ca2+ at 37°C for 4 h in culture media (phenol red-free DMEM supplemented with 2% FBS and penicillin/streptomycin) and then rinsed with PBS three times to remove unreacted BPA. Samples were then lysed/homogenized to recover proteins. Proteins (0.5–1 μg) were loaded onto nitrocellulose membrane (BioDot Dot Blot apparatus; Bio-Rad). The membrane was rinsed and blocked in 3% BSA overnight and probed with HRP-conjugated streptavidin (Amersham Bioscience; 1:10,000 dilution in 3% BSA) to determine BPA incorporation. Blots were then stripped using Restore Plus stripping buffer (Pierce) and reprobed with GAPDH to determine protein loading. BPA incorporation and GAPDH levels were determined by densitometry analysis using ImageJ software (NIH). For each sample, activity was calculated as a ratio of BPA/GAPDH.
Recovery of ECM proteins.
Cells were grown to confluence and treated as indicated. ECM was recovered following established methods (18). In brief, cells were rinsed twice with PBS. Cellular and nuclear materials were extracted by incubation with cell removal solution (0.05% Triton X-100, 50 mM NH4OH, in PBS) until the cells were floating. The matrix was then briefly washed once with rinse buffer (50 mM NH4OH in PBS) and then three times with PBS (Quality Biological). ECM was scraped into 100 μl of lysis buffer, and TG2 expression was determined by Western blotting.
Decellularization of mouse aorta.
The aortas were cleaned of adjacent tissues and fat and decellularized as previously described (18), with minor modifications. Briefly, aortas were incubated in decellularization solution 1 (8 mM CHAPS, 1 M NaCl, and 25 mM EDTA in PBS) for 44 h. Next, samples were incubated in decellularization solution 2 [1.8 mM sodium dodecyl sulfate (SDS), 1 M NaCl, 25 mM EDTA in PBS] for 44 h. Each decellularization solution was changed every 22 h with three 15-min washes in PBS. Samples were washed with and incubated with PBS for 2 days for complete removal of the detergents. All these steps were conducted at room temperature, with continuous shaking under sterile conditions. In the final step, samples were incubated at 37°C for 1 day in endothelial cell media (ScienCell Research Labs) followed by three 15-min washes with PBS to obtain decellularized specimen. Removal of cells was confirmed by the absence of DNA assayed using the Pico Green assay kit (Invitrogen), and absence of GAPDH (cytosolic protein) was confirmed by Western blotting.
S-nitrosylation assay.
TG2 _S_-nitrosylation was determined using the biotin switch assay in cell lysates/tissue homogenates as previously described (18, 25). Because the activity assay also relies on biotinylation, the _S_-nitrosylation assays were performed on a separate set of samples in parallel with the activity assays.
Magnetic twisting cytometry.
Vascular smooth muscle cells were freshly isolated from the aorta using the method of Kobayashi et al. (20) and plated onto fibronectin-coated 96-well strip-well plates. At the level of the single living cell, we measured cell stiffness using forced motions of functionalized beads anchored to the cytoskeleton (CSK) through cell surface integrin receptors using magnetic twisting cytometry (MTC) as described in detail elsewhere (8, 12). For these studies, the stiffness of adherent cells isolated from eNOS−/− and WT mice was measured at a frequency of 0.75 Hz. Cell stiffness is expressed in units of Pascal per nanometer (Pa/nm).
Data analysis and statistics.
Unless otherwise noted, all data are presented as means ± SE, with sample size (n) being indicated for each reported value. All Western blots and dot blots were analyzed by densitometry using the ImageJ software (NIH). Results are expressed as a percentage change relative to the average value measured in the control group. Statistical evaluation was performed by Student's _t_-test for unpaired observations to compare two means. When more than two means were compared, one-way ANOVA with Tukey's post hoc test was used to identify differences. To satisfy the normal distribution assumptions associated with ANOVA, cell stiffness data were converted to log scale before analyses. Vascular response data were analyzed with GraphPad Prism data analysis software. Two-way ANOVA with Bonferroni post hoc tests was employed to compare PWV values at each MAP. A value of P < 0.05 was considered statistically significant.
RESULTS
eNOS regulates TG2 externalization and activity in HAEC.
Decreased expression of eNOS with shRNA treatment was confirmed by Western blotting (Fig. 1_A_). eNOS knockdown resulted in an increase in ECM-associated TG2 (Fig. 1_B_), a decrease in TG2-_S_-nitrosylation (Fig. 1_C_), and an increase in TG activity (Fig. 1_D_) compared with controls (Fig. 1, A–C).
Fig. 1.

Effect of endothelial NO synthase (eNOS) knockdown on transglutaminase (TG2) in human aortic endothelial cells (HAEC). A: lentiviral delivery of eNOS short-hairpin RNA (shRNA) resulted in an ∼50% loss in eNOS protein expression in cultured HAEC. B: TG2 abundance in extracellular matrix (ECM) increased. C: TG2 _S_-nitrosylation decreased. D: TG2 cross-linking function increased with eNOS knockdown. Graphs show means ± SE (n = 6). *Significant difference by Student's _t_-test (P < 0.05).
eNOS−/− mice have altered TG2 activity and localization.
Aortae of eNOS knockout mice were examined to determine the effect of eNOS deficiency on TG2 localization and activity. TG activity was higher in the aortae of eNOS−/− mice compared with WT (C57Bl6) mice (Fig. 2_A_). TG2 abundance in the decellularized tissue scaffold was higher, and _S_-nitrosylation levels were markedly diminished in the aortae of eNOS knockout mice compared with WT controls, whereas total TG2 remained unchanged (Fig. 2, B and C). Immunohistochemical analysis showed that TG2 abundance was similar in eNOS−/− and WT mice, whereas TG2 cross-links were significantly higher in the eNOS−/− mice compared with WT mice (Fig. 2_D_).
Fig. 2.

TG2 localization and function in eNOS−/− mice. A: TG cross-linking activity was higher. B: TG2 abundance in the decellularized aortic matrix was higher (n = 5). C: TG2 _S_-nitrosylation levels were lower in the eNOS−/− mice compared with wild-type (WT) controls (n = 5). D: TG2 abundance was unaltered, whereas TG-specific cross-links were higher in the eNOS−/− mouse (n = 8). *Significant difference by Student's _t_-test (P < 0.05).
Carotid artery compliance is significantly improved in cystamine-treated eNOS−/− mice.
Cystamine is a TG inhibitor that has been widely used in animal models to study the role of TG (2, 7, 9, 10). In the present study, to examine the role of TG2 in mediating vascular properties, cystamine (40 mg·kg−1·day−1) was delivered to WT and eNOS−/− mice for 4 wk using osmotic infusion pumps. eNOS−/− mice exhibited lower carotid artery compliance compared with WT mice. In both eNOS−/− and WT mice, cystamine treatment led to a significant increase in carotid artery compliance compared with their vehicle-treated counterparts (Fig. 3). Although resting blood pressures were higher in the eNOS−/− mice, cystamine treatment did not result in altered blood pressure (WT control: 66.5 ± 5.6 mmHg; WT cystamine: 69.3 ± 6.1 mmHg; eNOS control: 96.8 ± 1.9 mmHg; eNOS cystamine: 98.1 ± 2.8 mmHg) or heart rate (WT control: 503 ± 45 beats/min; WT cystamine: 505 ± 45 beats/min; eNOS control: 513 ± 18 beats/min; eNOS cystamine: 518 ± 26 beats/min).
Fig. 3.
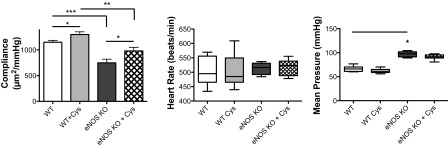
Effect of TG inhibition on vascular compliance. TG inhibitor cystamine (40 mg·kg−1·day−1) was administered for 4 wk to WT and eNOS knockout mice using osmotic infusion pumps. Carotid artery compliance, measured by video-dimension analysis, was lower in eNOS−/− mice compared with WT. Cystamine treatment restored carotid artery compliance toward WT controls. Values are means ± SE (n = 8). Significant difference by one-way ANOVA with Bonferroni post hoc analysis: *P < 0.05; **P < 0.01; ***P < 0.001.
Mechanical properties of the aorta are altered in eNOS−/− mice.
The eNOS−/− mouse is hypertensive and has impaired vasorelaxant responses. Two approaches were used to examine aortic stiffness. First, PWV measurements were obtained using a Doppler-based device. There was a marked increase in PWV in eNOS−/− mice compared with WT controls (Fig. 4_E_) and a concomitant increase in MAP (Fig. 4_F_). Basal heart rates were similar in both groups (Fig. 4_G_). Given the dependence of PWV on wall tension and therefore blood pressure, changes in PWV must be compared at similar MAP to accurately assess changes in arterial stiffness. We therefore measured PWV by varying MAP over a full physiological range of blood pressure by using an invasive dual-pressure sensor, high-fidelity catheter (Fig. 4_H_). At lower pressures (MAP < 125 mmHg), PWVs of eNOS−/− mice were similar to WT controls. At higher MAPs (MAP > 125 mmHg), however, PWVs of eNOS−/− mice were significantly higher than PWVs of WT mice at each probing MAP. Next, the passive mechanical properties of intact and decellularized aorta were examined by tensile testing to determine the stress-strain relationship. Intact aortic rings of eNOS−/− mice were significantly stiffer than age-matched WT controls (Fig. 4_I_); the incremental elastic modulus diverged at 0.5 strain. The decellularized aortic tissue scaffolds of the eNOS−/− mice were also significantly stiffer than those of WT mice; the incremental elastic modulus was significantly altered at 0.8 strain (Fig. 4_J_).
Cell-matrix interactions and CSK stiffness are altered in eNOS−/− mice.
TG2 acts as an integrin-fibronectin binding cofactor at the cell surface, independent of its catalytic function. As determined by co-immunoprecipitation experiments, TG2-integrin β1 interaction was higher in the aorta of eNOS−/− mice compared with WT mice (Fig. 5_A_). Furthermore, using MTC, we found that vascular smooth muscle cells freshly isolated from the aorta of eNOS−/− mice exhibited appreciably higher CSK stiffness than those isolated from WT mice (Fig. 5_B_). Phenylephrine caused 28.97 ± 3.90% and 37.61 ± 7.69% increases in cell stiffness in WT and eNOS knockout cells, respectively. Taken together, these results suggest that TG2 might induce vascular stiffening by increasing both cellular and matrix stiffness.
Fig. 5.

Cell-matrix interactions and CSK stiffness of isolated vascular smooth muscle cells from eNOS−/− and WT mice. A: proteins (50 μg) of aortic homogenates were immunoprecipitated using integrin β1 antibody; higher levels of TG2 co-immunoprecipitated with integrin β1 in eNOS−/− mouse (n = 6); proteins from eNOS−/− mice were immunoprecipitated with isotype IgG as control (right lane). *P < 0.05. B: CSK stiffness of freshly isolated vascular smooth muscle cells from the aorta of eNOS−/− and WT mice was measured using magnetic twisting cytometry. Data are presented as geometric means ± SE (n = 248 cells for eNOS−/−; n = 199 cells for WT).
DISCUSSION
It is well known that the composition and degree of cross-linking of the extracellular matrix influence the mechanical properties of the vessel (29). In the vasculature, TG2 is known to mediate remodeling of the mesenteric artery in response to vasoconstrictive stimuli (3, 4). TG2 secretion, and thus TG2-mediated remodeling, is augmented under reducing conditions and inhibited by NO (18, 24, 25, 27). A major question regarding TG2 function is whether increased cross-linking activity leads to increased vascular stiffness. In this context, it is interesting to note that TG2−/− mice are born with Mendelian frequency and do not exhibit any overt vascular phenotype (16). The PWV and MAP of TG2−/− mice are similar to age-matched WT controls; however, TG2−/− mice are protected from the increase in PWV resulting from NOS inhibition using l-NAME (25). This suggests that, although TG2 may in part mediate vascular stiffness resulting from endothelial dysfunction due to attenuated eNOS function, it may not be significant in normal functioning of the aorta. Indeed, under physiological conditions, with normal levels of NO bioavailability, much of TG2 is _S_-nitrosylated, is confined to the cytosol, and has its cross-linking function held latent, which supports this idea. To further test this hypothesis, we studied the eNOS−/− mouse model, which has been widely used to examine the role of eNOS-derived NO in cardiovascular function. These mice are characterized by the absence of eNOS mRNA and enzymatic activity and show the phenotypes caused by total absence of eNOS. This includes lack of endothelium-derived relaxation factor (EDRF) activity both in isolated arteries and in vivo, as measured by vascular relaxation in response to acetylcholine or other endothelium-dependent vasodilators. These mice are also hypertensive, which underscores the essential role of eNOS-dependent NO in the physiological regulation of blood pressure (15). Again, lack of eNOS-derived NO had a reciprocal correlation with TG2 externalization and activity and coincided with decreased TG2 _S_-nitrosylation. Vascular stiffness in eNOS−/− and WT mice was examined using PWV. This is a well accepted measure of arterial stiffness, which is typically measured as a single-point measurement and reported together with or corrected for MAP. PWV (measured using a Doppler-based device) and MAP are higher in eNOS−/− mice compared with WT controls (26). However, wall stiffness also varies with distending pressures due to the elastic nature of arteries, which in turn depends on the composition and structure of the load-bearing components (i.e., elastin and collagen) in the tissue matrix. Thus, to accurately compare alterations in vascular stiffness between the eNOS−/− and WT mice, PWV changes must be compared at similar arterial pressures or corrected for pressure to accurately determine changes in arterial stiffness, as has been done in previous studies comparing treatments that affect blood pressure (23). Therefore, in this study, we used an invasive approach to determine the pressure dependence of PWV in eNOS−/− and WT mice over a range of physiologically relevant pressures. The eNOS−/− mice had PWV comparable to WT mice at low pressures and significantly higher PWV at higher pressures. This suggests that, although compensatory mechanisms may regulate central aortic stiffness in a normal physiological setting, the eNOS−/− mice will likely have greater remodeling and elevated stiffness when stressed or in pathophysiological states. Tensile testing revealed that the intact aorta and decellularized tissue scaffold of eNOS−/− mice are markedly stiffer than those of WT mice. Increased TG2 abundance and cross-linking function in the aortic matrix of the eNOS−/− mice may contribute to the increased stiffness observed in the decellularized tissue scaffold. Substrates for TG2 in the vascular scaffold could include collagen and fibronectin (27); the biochemical analysis to confirm this remains the focus of ongoing studies. Indeed, increased TG2-mediated cross-linking results in increased matrix stiffness in vitro (6). Interestingly, the tensile properties of intact aorta of eNOS−/− mice diverged from the WT mice earlier than the decellularized scaffolds. This suggests that altered cell-matrix interactions contribute to increased vascular stiffness in the eNOS−/− mouse model. Moreover, TG2-integrin β1 interaction is higher in the eNOS−/− mouse aorta, which can directly facilitate cell-matrix interaction. Smooth muscle cells from eNOS−/− mice are stiffer when placed on a fibronectin matrix, which may in part be mediated by increased TG-integrin β1 interaction, but their response to phenylephrine remains unaltered. Taken together, these results underscore the potential dual role of TG2 in mediating vascular stiffness by altering matrix stiffness (6, 11) and by mediating cell-matrix interaction (1). The specific role of TG2 in this context may be examined by genetic rescue of function experiments wherein catalytically active and C277S catalytically deficient mutant can be delivered to the aorta of TG2−/− mice followed by assessment of mechanical properties. Such experiments are the focus of ongoing studies.
Limitations
The simultaneous measurement of PWV with MAP allows the comparison of two populations with markedly different resting MAPs. However, the use of vasoactive drugs (phenylephrine and sodium nitroprusside) may affect intrinsic vessel tone and thus PWV measurement. However, all groups receive the same drugs, and thus all groups would be prone to the same drug effects. In addition, this protocol in rats has been shown to have no significant effect on large artery stiffness over and above the pressure dependence (5). Although the relatively short distance (1 cm) between the sensors ensures that the PWV measurement is obtained in the upper thoracic trunk, it also imposes limitations on the resolution of transit time detection. The estimated resolution for transit time using the second differential is within 0.5 ms. For the highest PWV values in the pressure range where significance was found, the transit time is of the order of 2 ms. Hence, the resolution is well with the maximum limits. Furthermore, eNOS−/− mice have basal hypertension. This may by itself alter vessel wall stiffness and/or result in TG2 activation mechanisms that were not exhaustively addressed in this study.
In conclusion, this study highlights a critical role for eNOS-dependent NO in regulating TG2 and establishes an inverse relationship between NO bioavailability and TG2 secretion and cross-linking activity. To the best of our knowledge, this is the first study to report simultaneous measurement of PWV and MAP in a mouse model, and contributes a new methodology to address PWV characteristics over a full range of physiological pressures in mice. Divergence of PWV at higher pressures in the eNOS−/− mice compared with WT mice suggests that alterations in vascular stiffness are significant in a hypertensive setting. The response of the eNOS−/− mice to pathophysiological stresses leading to vascular remodeling are likely different than the WT mice. Increase in extracellular TG2 coincides with increased stiffness of the intact aorta, with only a modest increase in the stiffness of the vascular matrix, suggesting a noncatalytic role for TG2 in mediating vascular stiffness. Finally, at resting conditions, the altered vascular phenotype in the eNOS−/− mouse is likely due to endothelial dysfunction and attenuated smooth muscle tone and function occurring due to the absence of NO.
GRANTS
This work was supported by a National Heart, Lung, and Blood Institute Grants 1R01-HL-105296-01 (to D. E. Berkowitz) and HL-107361 (to S. S. An), and an Australian Research Council Grant DP110101134 (to A. Avolio).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.M.J., S.J., J.S., A.B., A.P., E.Y.C., M.B., K.V., and L.S. performed experiments; S.M.J., J.S., A.B., S.S.A., and L.S. analyzed data; S.M.J., J.S., A.B., S.S.A., and L.S. prepared figures; S.M.J. and L.S. drafted manuscript; S.S.A., D.N., A.P.A., D.E.B., and L.S. interpreted results of experiments; S.S.A., D.N., A.P.A., D.E.B., and L.S. edited and revised manuscript; A.P.A., D.E.B., and L.S. conception and design of research; L.S. approved final version of manuscript.
REFERENCES
- 1.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol 148: 825–838, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcock J, Warren AY, Goodson YJ, Hill SJ, Khan RN, Lymn JS. Inhibition of tissue transglutaminase 2 attenuates contractility of pregnant human myometrium. Biol Reprod 84: 646–653, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res 96: 119–126, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Bakker EN, Pistea A, Spaan JA, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow_-_dependent remodeling of small arteries in mice deficient for tissue-type transglutaminase: possible compensation by macrophage-derived factor XIII. Circ Res 99: 86–92, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Butlin M, Hammond A, Lindesay G, Viegas K, Avolio AP. In-vitro and in-vivo use of vasoactive agents in characterising aortic stiffness in rats: testing the assumptions. J Hypertension. In press [Google Scholar]
- 6.Chau DY, Collighan RJ, Verderio EA, Addy VL, Griffin M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials 26: 6518–6529, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Davies JE, Rose C, Sarkar S, Rubinsztein DC. Cystamine suppresses polyalanine toxicity in a mouse model of oculopharyngeal muscular dystrophy. Sci Transl Med 2: 34–40, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS, Sham JS, Liggett SB. Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16: 1299–1304, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elli L, Ciulla MM, Busca G, Roncoroni L, Maioli C, Ferrero S, Bardella MT, Bonura A, Paliotti R, Terrani C, Braidotti P. Beneficial effects of treatment with transglutaminase inhibitor cystamine on the severity of inflammation in a rat model of inflammatory bowel disease. Lab Invest 91: 452–461, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Engholm M, Eftekhari A, Chwatko G, Bald E, Mulvany MJ. Effect of cystamine on blood pressure and vascular characteristics in spontaneously hypertensive rats. J Vasc Res 48: 476–484, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Engler AJ, Griffin MA, Sen S, Bonnemann CG, Sweeney HL, Discher DE. Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J Cell Biol 166: 877–887, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Phys Rev Lett 87: 148102, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Franklin SS. Hypertension in older people: part 1. J Clin Hypertens (Greenwich) 8: 444–449, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franklin SS. Hypertension in older people: part 2. J Clin Hypertens (Greenwich) 8: 521–525, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377: 239–242, 1995 [DOI] [PubMed] [Google Scholar]
- 16.Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89: 991–1023, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Izzo JL., Jr Arterial stiffness and the systolic hypertension syndrome. Curr Opin Cardiol 19: 341–352, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Jandu SK, Webb AK, Pak A, Sevinc B, Nyhan D, Belkin AM, Flavahan NA, Berkowitz DE, Santhanam L. Nitric oxide regulates tissue transglutaminase localization and function in the vasculature. Amino Acids 44: 261–269, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kass DA. Age-related changes in venticular-arterial coupling: pathophysiologic implications. Heart Fail Rev 7: 51–62, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb 12: 138–142, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37: 1236–1241, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation 121: 505–511, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng K, Butlin M, Avolio AP. Persistent effect of early, brief angiotensin-converting enzyme inhibition on segmental pressure dependency of aortic stiffness in spontaneously hypertensive rats. J Hypertens 30: 1782–1790, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Santhanam L, Berkowitz DE, Belkin AM. Nitric oxide regulates non-classical secretion of tissue transglutaminase. Commun Integr Biol 4: 584–586, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM, Berkowitz DE. Decreased _S_-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ Res 107: 117–125, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Soucy KG, Ryoo S, Benjo A, Lim HK, Gupta G, Sohi JS, Elser J, Aon MA, Nyhan D, Shoukas AA, Berkowitz DE. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J Appl Physiol 101: 1751–1759, 2006 [DOI] [PubMed] [Google Scholar]
- 27.van den Akker J, VanBavel E, van Geel R, Matlung HL, Guvenc Tuna B, Janssen GM, van Veelen PA, Boelens WC, De Mey JG, Bakker EN. The redox state of transglutaminase 2 controls arterial remodeling. PLos One 6: e23067, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanbavel E, Bakker EN. A vascular bone collector: arterial calcification requires tissue-type transglutaminase. Circ Res 102: 507–509, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25: 932–943, 2005 [DOI] [PubMed] [Google Scholar]