Generation of functional hemangioblasts from human embryonic stem cells (original) (raw)
. Author manuscript; available in PMC: 2013 Sep 7.
Published in final edited form as: Nat Methods. 2007 May 7;4(6):501–509. doi: 10.1038/nmeth1041
Abstract
Recent evidence suggests the existence of progenitor cells in adult tissues that are capable of differentiating into vascular structures as well as into all hematopoietic cell lineages. Here we describe an efficient and reproducible method for generating large numbers of these bipotential progenitors—known as hemangioblasts—from human embryonic stem (hES) cells using an in vitro differentiation system. Blast cells expressed gene signatures characteristic of hemangioblasts, and could be expanded, cryopreserved and differentiated into multiple hematopoietic lineages as well as into endothelial cells. When we injected these cells into rats with diabetes or into mice with ischemia-reperfusion injury of the retina, they localized to the site of injury in the damaged vasculature and appeared to participate in repair. Injection of the cells also reduced the mortality rate after myocardial infarction and restored blood flow in hind limb ischemia in mouse models. Our data suggest that hES-derived blast cells (hES-BCs) could be important in vascular repair.
Although progenitor cells have recently been discovered that can enter the circulation in response to vascular injury and ischemia1–5, defining and isolating these cells has proven problematic. Circulating bone marrow–derived cells have also been shown to be important in normal physiologic maintenance and repair of the body’s vasculature6,7 with approximately 1–3% of endothelial cells at any one time being bone marrow–derived. Furthermore, the entire hematopoietic system has been hypothesized to originate from a transient population of hemangioblasts restricted to embryogenesis8,9. But recent evidence suggests that hemangioblasts or more mature endothelial progenitors may also exist in adult tissues and umbilical cord blood2–4,10,11. More direct proof for their existence was provided when the in vitro equivalent of the hemangioblast was isolated using a mouse embryonic stem cell differentiation system12,13. Recently a human hemangioblast cell population derived from hES cells was also identified using a procedure that consisted of serum-free differentiation in a mixture of cytokines followed by expansion in serum-containing medium14. To date, large-scale generation or functional assessment of hemangioblasts has not been achieved in any of these systems. Here we show that large numbers of what appear to be a distinct population of progenitor cells with both hematopoietic and vascular potential can be efficiently and reproducibly generated from hES cells using a simple two-step procedure with different supplements under fully serum-free conditions.
RESULTS
Generation and characterization of blast cells from hES cells
We developed a simple and efficient method for generating functional blast cells from hES cells with both hematopoietic and endothelial potential in defined media, using a two-step strategy. First, we generated early-stage embryoid bodies (EBs) from hES cells (WA01 (H1)−GFP+) cultured in serum-free medium supplemented with a combination of morphogens and early hematopoietic cytokines, specifically bone morphogenetic protein-4 (BMP-4), vascular endothelial growth factor (VEGF), stem cell factor (SCF), thrombopoietin (Tpo) and fms-related tyrosine kinase 3 ligand (FL), which had been shown to be critical for hematopoietic development of hES cells under serum-free conditions15. We then dissociated EBs and plated individual cells in serum-free semisolid blast-colony growth medium (BGM). Approximately 0.35 ± 0.01% and 0.52 ± 0.06% of individual cells from WA01 hES cells and MA01 hES cells, respectively, developed into grape-like blast colonies. The blast colonies contained <10 cellsat the beginning of day 3, rapidly expanding from days 4 to 6 to >100 cells (Fig. 1a). The colonies were generally less compact and more morphologically homogenous than secondary EBs (Fig. 1a). Cytospin preparation and Wright-Giemsa staining of hES-BCs confirmed morphologic features of immature hES-BCs (Fig. 1a). To extend these results to other hES cell lines (WA07 (H7), WA09 (H9), MA01, MA03, MA09 and MA40), supplements of 50 ng/ml FL and 50 ng/ml Tpo were necessary for sustained growth of the blast colonies. Without FL and Tpo, we obtained smaller clusters of blast colonies (10–20 cells) that died within 4–8 d. The discrepancy observed between different hES cell lines may be due to the intrinsic properties of these cell lines, as previously documented16. Using 3–6 units/ml of human recombinant erythropoietin (Epo) was also essential for blast colony formation and growth in all hES cell lines tested. We examined the time course (from 2 to 6 d) of blast colony formation in EBs. As observed in the mouse system12,13, we also found a short time period during which hES cell-derived EBs generated the greatest numbers of blast colonies. Day 2 EBs generated blast colonies (57.3 ± 7.4 blast colonies per 105 EB cells for WA01 hES cells, mean ± s.e.m., n = 3; 395 ± 10.4 blast colonies per 105 EB cells for MA01 hES cells, n = 3) at a low frequency and, by day 6, only gave rise to hematopoietic (erythroid) progenitors. But day 3.5 EBs generated large numbers of blast colonies (347.4 ± 11.1 blast colonies per 105 EB cells for WA01 hES cells, n = 3; 523.3 ± 60.1 blast colonies per 105 EB cells for MA01 hES cells, n = 3) in all hES cell lines tested, including lines derived from both blastocysts (WA01 and WA09) and single blastomeres (MA01 and MA09)17. We observed variable efficiencies for different hES cell lines and growth conditions (Supplementary Table 1 online). One 6-well plate of WA01-GFP hES cells (12–13 × 106 cells) generated approximately 22.18 ± 3.51 × 106 (expanded for 6–8 d, mean ± s.e.m., n = 17) hES-BCs, whereas roughly the same number of MA01 hES cells generated 49.73 ± 7.23 × 106 (expanded for 6–8 d, n = 9) and 396.4 ± 91.63 × 106 (expanded for 10–13 d, n = 6) hES-BCs. We also examined whether the hES-BCs can be cryopreserved with serum-free medium. After thawing, we plated hES-BCs for both hematopoietic and endothelial lineage development and compared with fresh purified cells. For endothelial cell lineage, we recovered 78 ± 2% (mean ± s.e.m., n = 3) of cells after the freezing process; whereas 61 ± 7% (n = 3) of total hematopoietic colony-forming units (CFU) and 46 ± 4% (n = 3) of cells that form colonies of erythrocytes (CFU-erythrocyte) were retained as compared to fresh hES-BCs. We observed no loss of more primitive multipotential progenitors for cells that form colonies with a mixture of granulocytes, erythrocytes, macrophages and megakaryocytes (CFU-granulocyte-erythrocyte-macrophage-megakaryocyte; CFU-GEMM) after recovering hES-BCs from liquid-nitrogen storage.
Figure 1.

Characterization of hES-BCs. (a) Phase-contrast images of a blast colony (left), secondary EBs (middle) and hES-BCs with Wright-Giemsa staining (right). (b) Phase-contrast images of Wright-Giemsa–stained cells from CFU-erythrocytes (left), cells from CFU-granulocytes (cells that form hematopoietic colonies with granulocytes, middle) and cells from CFU-macrophages (cells that form hematopoietic colonies with macrophages, right). (c) Immunostaining of cells from CFU-erythrocytes with CD235a antibody (left), cells from CFU-granulocytes with CD13 antibody (middle) and cells from CFU-GEMMs with CD45 antibody (right). (d) Analysis of endothelial cells derived from purified hES-BCs: Ac-LDL uptake (red) and immunostaining of vWF (green; left), uptake of Ac-LDL (red) and immunostaining of VE-cadherin (green; middle); immunostaining of vWF (green) and CD31 (red; right). DAPI (blue) staining of cell nuclei. Scale bars, 50 μm.
We next characterized hES-BCs with a battery of antibodies and global gene profiling using Genechip analysis. Immunocytochemical analysis revealed that the hES-BCs expressed several hemangioblast related proteins but no CD31, CD34 and KDR, or other adhesion molecules (Table 1 and Supplementary Fig. 1 online). Molecular profiling using Affymetrix arrays indicated an increase in genes associated with hemangioblast and early primitive erythroblast development as compared to early-stage EBs (Table 1). SCL (SCLY) and LMO2, two genes critical for hemangioblast development18,19, as well as FLT1, a gene encoding a receptor for VEGF, were readily detectable in hES-BCs. Embryonic (ε-globin, 549-fold) and fetal (γ-globin, 817-fold) globin gene expression was dramatically increased in hES-BCs; NF-E2 (12-fold), GATA-1 (sixfold), EKLF (sevenfold), ICAM-4 (fourfold), glycophorins (14-fold) and Epo receptor (fourfold) message levels were also moderately increased.
Table 1.
Characterization of hES-BCs by immunocytochemistry and Affymetrix arrays
| Immunocytochemistry | Affymetrix arrays | |||
|---|---|---|---|---|
| Antibodies | hES-BC staining | Genes | EBs | hES-BCs |
| GATA-2 | + | TAL1/SCL | − | + |
| GATA-1 | + | LMO2 | + | +↑ |
| Tpo-R | + | FLT1 | − | + |
| Epo-R | + | GATA2 | + | + |
| CXCR-4 | + | EPOR | + | +↑ |
| β-catenin | + | GATA1 | − | + |
| CD71 | + | GYPA and GYPB | − | + |
| EGR-1 | + | NFE2 | − | + |
| Integrin α4 | ± | EKLF | − | + |
| Integrin β1 | ± | ICAM4 | − | + |
| VE-cadherin | − | Globins γ and ε | − | + |
| E-cadherin | − | VE-cadherin | − | + |
| KDR | − | MADCAM1 | − | + |
| PECAM-1 (CD31) | − | CD61 (gpIIIa) | − | + |
| CD34 | − | EZFIT | − | + |
| CD41 | − | CXCR4 | + | + |
| CD43 | − | TIE-2 | + | − |
| OCT-4 | − | Integrin β5 | + | − |
| Nanog | – | Integrin β1 | + | +↓ |
| TRA-81 | – | OCT4 | + | − |
| SSEA-4 | − | WNT5α | + | − |
| FGFR | + | − | ||
| AVC-R2B | + | +↓ | ||
| Brachyury | + | − | ||
| BMPR1A | + | − | ||
| PECAM1 | − | − | ||
| KDR | + | − | ||
| NEUROD1 | − | − |
hES-BCs have hemangioblast characteristics
To determine whether hES-BCs have both hematopoietic and endothelial potential, we handpicked blast colonies under a dissection microscope and examined them for the ability to form hematopoietic CFUs as well as the potential to differentiate into functional endothelial cells. When we plated single-cell suspensions in serum-free methylcellulose medium containing a spectrum of cytokines for 10–14 d, we observed colonies of erythroid, myeloid, macrophage and multilineage hematopoietic cells (Supplementary Fig. 2 online). Wright-Giemsa staining (Fig. 1b), immunostaining (Fig. 1c) and FACS analysis (Supplementary Fig. 3 online) using antibodies against CD235a (erythrocytes), CD13 (myeloid) and CD45 (leukocytes) confirmed the identity of their hematopoietic lineages. Most hematopoietic colonies (>50%) were multilineage with an erythroid core and formed within a few days after plating (Supplementary Fig. 2a). Cells in CFU-erythrocytes resembled those of primitive erythroids with brilliant red color; Wright-Giemsa staining showed all erythrocytes were nucleated (Fig. 1b). To distinguish the developmental stage of the erythroids, we analyzed hematopoietic colonies for the expression patterns of the β-cluster globin genes20. All the erythroid and multilineage colonies expressed mainly the embryonic ε-and fetal γ-globin genes; no β-globin gene message was detected (data not shown), suggesting a primitive yolk sac-like status.
To determine their endothelial potential, we grew blast cells derived from all seven hES lines as adherent layers on fibronectin-coated plates. Incubation with AlexaFluor 594-labeled acetylated low-density lipoprotein (Ac-LDL) revealed a punctate staining pattern characteristic of endothelial cells (Fig. 1d); the cells also expressed high levels of von Willebrand factor (vWF; Fig. 1d). Adherent cells in blood vessel–like structures were positive for PECAM-1 (CD31), KDR and VE-cadherin (Fig. 1d and Supplementary Fig. 4 online). After replating on Matrigel-based medium, the adherent cells formed capillary-vascular–like structures (Supplementary Fig. 4 online) that also took up Ac-LDL (data not shown), confirming that these cells were of endothelial lineage.
Blast colonies are clonogenic
We also carried out experiments to determine whether blast colonies were clonal and originated from a common bipotential progenitor cell. To address this, we performed cell-mixing experiments as previously described in mouse studies12,13. We mixed together single-cell suspensions generated from GFP+ WA01 and GFP− MA01 EBs, and examined the resulting blast colonies using both fluorescence and phase-contrast microscopy. In three experiments we found that 100% (77 out of 77) of the blast colonies were either GFP+ or GFP− (we observed no mixed blast colonies; Fig. 2a,b). To rule out the possibility that the GFP− colonies contained cells with an inactive GFP gene, we examined 12 GFP+ and 12 GFP− colonies for the presence of GFP sequence. PCR analysis confirmed the absence of the GFP sequence in all negative colonies (Supplementary Fig. 5 online). We confirmed the clonal origin of blast colonies using limiting-dilution studies. We diluted single EB-cell suspensions at 100 cells/ml in BGM and plated them in 96-well plates. As previously reported13 in mouse embryonic stem cell studies, blast colony development was also cell density– dependent with poor development at low cell number. Only 7 blast colonies developed from fifteen 96-well plates (total of 15,000 plated EB cells). We performed additional limiting dilution studies to determine whether the blast colonies contained cells that have the potential to generate secondary blast colonies. We serially diluted single cells from primary blast colonies and replated them in 96-well plates. A total of 29 secondary blast colonies with identical morphological characteristics as primary blast colonies developed from five 96-well plates. All these experiments confirmed the clonogenicity of both primary and secondary blast colonies (Table 2).
Figure 2.

Clonogenicity of blast colonies. (a,b) Phase-contrast (a) and fluorescence (b) images of two blast colonies developed in a mixture of WA01-GFP and MA01 EBs, which demonstrated the clonal origin of blast colonies. (c) Expansion of a single blast colony in liquid culture; both hematopoietic and endothelial lineages were observed. Scale bars, 100 μm.
Table 2.
Hematopoietic and endothelial lineage development from clonally derived blast colonies
| Number of colonies | Hematopoietic lineage (%) | Endothelial lineage (%) | Hematopoietic and endothelial lineages (%) | |
|---|---|---|---|---|
| Primary blast colonies (condition 1) | 24 | 16 (67) | 23 (96) | 15 (62) |
| Primary blast colonies (condition 2) | 24 | 24 (100) | 22 (92) | 22 (92) |
| Primary blast colonies derived from limited dilution | 7 | 6 (86) | 6 (86) | 5 (71) |
| Secondary blast colonies | 29 | 21 (72) | 20 (69) | 15 (52) |
We used two strategies to demonstrate the bipotential capacity of individual blast colonies. In the first strategy (condition 1), we transferred individual blast colonies to liquid cultures containing growth factors supporting the growth of both hematopoietic and endothelial lineages. Both nonadherent and adherent cells developed in more than 60% (15 of 24) of the colonies (Fig. 2c and Table 2); over 95% (23 of 24) of individual blast colonies generated adherent, stromal-like cells, which formed capillary-vascular-like structures on Matrigel that took up Ac-LDL and expressed vWF (Supplementary Fig. 4b online). Sixty-five percent (16 of 24) of them formed hematopoietic colonies including erythroid, myeloid and multilineage after they were replated in semi-solid medium supplemented with a spectrum of hematopoietic cytokines (Supplementary Fig. 2b online).
In the second strategy (condition 2), we split individual blast colonies from both primary and secondary blast colonies, and tested them for hematopoietic CFU and endothelial progenitor formation. For primary blast colonies, over 90% (22 of 24) gave rise to both hematopoetic and endothelial lineages; 100% and 92% developed hematopoetic and endothelial cells, respectively (Table 2). Similarly, we examined 7 individual primary blast colonies from limiting dilution experiments for their potential to differentiate into hematopoietic and endothelial lineages. Of these, 5 generated both hematopoietic and endothelial progeny (Table 2), showing a lower efficiency when compared with blast colonies from standard experiments, in which EB cells were plated at ~2 × 105 cells/2.5 ml BGM (71% versus 92%), possibly as a result of nonoptimal conditions for development. For the secondary blast colonies, over half (15 of 29) gave rise to both hematopoetic and endothelial lineages, whereas 6 (21%) and 3 (10%) generated only hematopoetic and endothelial cells, respectively (Table 2). Although the primary blast colonies contained heterogeneous populations, including precursors of hematopoietic and endothelial cells, the secondary limiting dilution experiments clearly demonstrated the existence of hemangioblasts in primary blast colonies.
hES-BCs in mouse ischemic retinal vasculature
Although mouse embryonic stem cell–derived blast cells have been discovered almost a decade ago13, it still remains unknown whether these cells are functional in vivo. We used two techniques, intravitreal injection and adoptive transfer, to examine the contribution of the hES-BCs to the in vivo repair process. For intravitreal injection studies, we first purified hES-BCs from day 6 BGM cultures, labeled them with the green dye (PKH-67) and then administered them to mice that had undergone ischemia-reperfusion injury to the retina of one eye. This model of ischemia-reperfusion injury results in damage to the retinal endothelium including vaso-obliteration and generation of acellular capillaries21. We removed the retinas and labeled them with rhodamine-conjugated Ricinus communis to visualize the retinal vasculature. This agglutinin binds to glycoproteins expressed specifically by endothelial cells, and thus is used to fluorescently label the exterior surfaces of blood vessels. Confocal microscopy demonstrated green fluorescent labeling for hES-BC–derived endothelial cells which represented 33.5 ± 10% of the retinal vasculature in the injured eye. We observed no green fluorescence in the control eye (Fig. 3a,b), suggesting that the hES-BCs homed to acellular regions, where they assimilated into injured regions only. We observed this putative repair process in all (n = 17) mice in a uniform manner.
Figure 3.

Repair of ischemic retinal vasculature in a mouse after injection of hES-BCs. (a–e) Mice undergoing ischemia-reperfusion injury were injected cells either intravitreally (a,b) or intravenously (c–e) with fluorescently labeled hES-BCs. Merged images from the ischemic eye of the same mouse 1 d after intravitreal hES-BC cell administration of fluorescently labeled hES-BCs (green channel) (a), or a typical control (uninjured) eye 1 d after saline injection (b). Separate green (hES-BCs) and red channels are shown in the insets. Merged images of the ischemic eyes 2 d (c) and 7 d (d) after systemic hES-BC cell administration and of the uninjured eye of the same mouse (e). (f) hES-BC–derived endothelial cells colocalize to existing injured vasculature in cross-sections of mouse eyes that underwent ischemia-reperfusion injury. High-magnification view of a vascular lumen in the ganglion cell layer adjacent to the inner limiting membrane (lower left) shows lumen surrounded by resident endothelial cells (CD31; green) and hES-BC–derived endothelial cells (human nuclear antigen, red) and the colocalized fluorescence staining of CD31 and human nuclear antigen (yellow). Upper right and left panels are the separate red (human nuclear antigen) and green (CD31) channels used to make composite image. Lower right panel, low magnification of the same region, with the box showing area depicted in all panels. V, vitreous; IPL, inner plexiform layer; RPE, retinal pigment epithelial cell layer; Ch, choroid. Scale bars, 100 μm in a–e (225 μm in insets), and 25 μm in f.
To a second set of mice with ischemia-reperfusion injury in one eye (n = 6) we provided fluorescently labeled hES-BCs by adoptive transfer. Before killing them, we perfusion-fixed the mice with TRITC-dextran to visualize vascular lumen. Red staining depicted patent vessels, green staining demonstrated the hES-BC–derived endothelial cells, and yellow staining showed where hES-BCs have generated a patent vasculature. We observed a large vessel with yellow (red and green) fluorescence 2 d after hES-BC cell injection (Fig. 3c), suggesting that the cells had incorporated into the vascular wall, and that the vessel was perfused. We observed a large number of green microvessels. These green tubes appear to represent new collateral compensatory neovasculature. The vessels are immature and not perfused with TRITC-dextran as they have not yet matured enough to develop patent lumens. The uninjured control eye of the same animal shows only patent TRITC-dextran perfused vessels with no green fluorescence, demonstrating that hES-BCs were not associated with vasculature that has had no injury (Fig. 3d). In retinal flatmounts from mice killed 7 d after hES-BC injection, the number of green tubes (nonlumenized, nonperfused vessels) was less compared to mice killed on day 2. There were more yellow vessels representing hES-BC–derived vessels (green) perfused with TRITC-dextran (red). These should represent fully functional vessels (Fig. 3e). A cross-section of a vessel in the ganglion cell layer in a retina from an injured eye shows colocalization of human nuclear antigen staining with anti-CD31 staining of the vessel and suggests that some endothelial cells in the vasculature were derived from hES-BCs (Fig. 3f).
hES-BCs in the retinal vasculature of diabetic rats
We also studied incorporation of hES-BCs in the retinal vasculature of Bio-Breeding Zucker diabetic rat (BBZDR)/Wor rats with type II diabetes of greater than 3 months. These rats develop many characteristics of preproliferative diabetic retinopathy including acellular capillaries and endothelial dysfunction, pericyte loss and blood retinal barrier breakdown22,23. We prepared retinal flat-mounts in an identical manner as for the mouse studies using rhodamine-conjugated R. communis to visualize vessels. As in the ischemia-reperfusion mouse model, intravitreally administered hES-BCs assimilated with injured resident vasculature. The green cells were easily visible in small as well as large vessels (Fig. 4a,b). We also observed a large vessel that was nearly all green and narrowed very rapidly (Fig. 4b). These ‘pinched-off’ vessels are characteristic of pre-proliferative diabetic retinopathy, where microthrombi result in vessel degeneration and downstream ischemia. In contrast, no green hES-BC colocalized with resident vessels in eyes from the nondiabetic control animal, but rather remained as sheets atop the retina after intravitreal administration of hES-BCs (Fig. 4c).
Figure 4.

Incorporation of hES-BCs into the retinal vasculature of diabetic rats. (a,b) Merged images from the retinal vasculature of two separate diabetic rats 2 d after intravitreal hES-BC administration, showing extensive hES-BC cell incorporation into both large and small vessels. (c) Merged image (green and red channels) from a control (nondiabetic) rat 2 d after hES-BC administration, showing that hES-BCs did not incorporate into vasculature and formed a sheet that lay atop the retina. (d,e) Eye sections from diabetic rats 2 d after intravitreal hES-BC injection, stained with CD31 (green) and human nuclear antigen (red) antibodies, showing colocalized staining of CD31 (green) and human nuclear antigen (red) in endothelial cells lining vessel lumens in the ganglion cell layer of the retina. Arrows, colocalized staining (yellow) of CD31 (green) and human nuclear antigen (red). Insets, separate green and red channels. (f) A section from a nondiabetic control rat 2 d after intravitreal blast cell injection, which is negative for human nuclear antigen staining, but is positive for endothelium staining (CD31, green; arrows). Arrows, positive staining of CD31 (green), but negative for human nuclear antigen (red). Scale bars, 100 μm (a–c), and 25 μm (d–f).
Immunohistochemical analysis showed colocalized staining with CD31 and human nuclear antigen in cells lining vessel lumens in the ganglion cell layer of the diabetic rat retinas (Fig. 4d,e), immediately posterior to the internal limiting membrane that separates the neural retina from the vitreous. This anatomical location is the site of the superficial retinal vascular plexus and is the typical location of vascular pathology in diabetes. This precise localization and incorporation strongly suggests that hES-BCs participate in repair of vessels that are typically involved in diabetic retinopathy. Most sections showed vessels with clearly visible lumens whose peripheries were lined with cells staining for both endothelial (CD31) and human nuclear antigen markers (Fig. 4d,e), whereas there was no evidence of hES-BC cell incorporation in the nondiabetic control rat eyes that were also injected with hES-BCs (Fig. 4f). Although we can not completely rule out the possibility of cell fusion, previous reports using the same model with adult human and mouse cells demonstrated that cell fusion did not have a role in repair of vessels2–4.
hES-BCs in myocardial infarction and hind limb ischemia
We induced myocardial infarction in nonobese diabetic–severe combined immunodeficiency (NOD-SCID) β2 mice by ligation of the left coronary artery, as described previously24. Fifteen minutes after MI induction, we transplanted hES-BCs or cell-free medium in the ischemic and peri-ischemic myocardium. Thirty days post hES-BC cell injection, 71% (12/17) of the animals injected with hES-BCs survived, as compared to 42% (8/19) of mice injected with medium control (Fig. 5a; P < 0.002). Four weeks after injection, the mice were killed and confocal microscopy confirmed the presence of human-specific vWF+ endothelial cells in the lumen of microvessels in the infarcted tissue (Fig. 5b). No human vWF+ cells were detectable in the infarcted heart tissue of control mice (Fig. 5c). It is important to note that cell transplantation has been observed to improve cardiac function in the absence of functional integration25. At this point it is unclear whether, and to what extent, hES-BCs can become and integrate with cardiomyocytes or other cell types.
Figure 5.
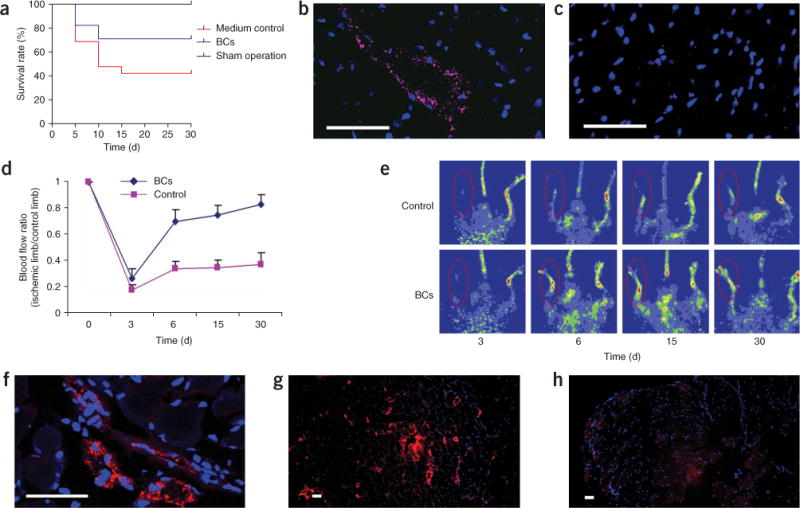
Endothelial differentiation in infarcted heart and in ischemic hind limb muscle after injection of hES-BCs. (a–c) Differentiation of hES-BCs in infarcted hearts: survival curves of mice treated with sham operation, medium control and hES-BCs (a); confocal image of infarcted myocardium section 4 weeks after injection of hES-BCs, immunostained with human-specific vWF antibody (pink) and DAPI (nuclei, blue) (b); confocal image of infarcted myocardium section from control mouse immunostained with human-specific vWF antibody (pink) and DAPI (nuclei, blue), showing no stain of human vWF (c). (d–h) Differentiation of hES-BCs in ischemic hind limb muscles. Restoration of blood flow in surgically induced ischemic limbs by hES-BCs (d). Hind limb blood flow monitored serially for 3–30 d after ligation in mouse receiving 6 × 105 hES-BCs and in mouse receiving medium only. Blood flow is calculated as the ratio of flow in the ischemic limb to that in the non-ischemic limb of the same animal. Laser doppler blood flow images of controls (medium) and ischemic animals injected with blast cells (n = 6 for each group) (e). Confocal microscopy image (f) and regular microscopy image (g) of ischemic hind limb muscle sections 4 weeks after injection of hES-BCs, immunostained with human-specific vWF antibody (red) and DAPI (nuclei, blue); an ischemic hind limb muscle section from a control mouse 4 weeks after PBS injection, immunostained with human specific vWF antibody (red) and DAPI (nuclei, blue), showing no stain of human vWF (h). Scale bars, 40 μm.
We also studied the function of hES-BCs in a mouse hind limb ischemic model. We induced ischemia in the right hind limb by femoral artery ligation surgery and injected hES-BCs or cell free medium into the area of peri-ischemic muscle immediately after surgery. To demonstrate physiological functions of injected hES-BCs, we monitored the right ischemic hind limb blood flow for 4 weeks and compared with blood flow of the left normal hind limb. hES-BCs significantly enhanced the restoration of blood flow rate in ischemic limbs as compared to the medium control (P < 0.0001; Fig. 5d,e). Improvement persisted for four weeks until near-normal flow rates were achieved. Four weeks after injection of hES-BCs into the peri-ischemic muscle, we killed the mice and immunostained them for human-specific vWF. We identified intramuscular areas containing positive staining of human-specific vWF cells showing vascular organization (Fig. 5f,g). Control tissue from infarcted muscle injected with cell-free medium showed no human vWF staining (Fig. 5h).
DISCUSSION
We describe a simple serum-free system that efficiently supports the development of functional hES-BCs. This simplified system can be used to reproducibly generate large numbers of hES-BCs from a range of different hES cell lines. The cells produced using this methodology differentiated into both hematopoietic and endothelial cells in vitro. The results obtained in four different animal models suggest that these cells can repair ischemic vasculature in vivo. The elimination of serum and other animal components from the system, as well as the ability to generate an unlimited supply of hES-BCs, will be important for future preclinical and human studies.
When we transplanted hES-BCs in fetal CD1 mice, low (~0.5%), but clearly detectable levels of human CD45+ cells were observed in the adult mice. Additional studies will be necessary to determine the engraftment potential of hES-BCs in adult SCID mice. Our results suggest that the cells have reparative potential in the animal models studied. hES-BC treatment resulted in functional improvement similar to that previously described using other endothelial precursor populations2–4,26. Adoptive transfer of endothelial precursor cells has been previously shown to restore blood flow and increase capillary density, decreasing limb loss and facilitating recovery from myocardial injury22,23.
Although CD45− FVP cells with hemangioblast-like properties have previously been identified in human EBs27, the rarity of the cells—less than a fraction of a percent of the CD45− FVP cells (0.18%)—precluded preclinical testing. The formation of multipotential colonies from EBs has also been demonstrated28, although it is unclear whether these colonies (which have different markers and morphology than the hES-BCs reported here) can be expanded and/or whether they have any functional activity in vivo. Others have also identified the presence of CD34+ KDR+ bipotential cells in cynomolgus monkey embryonic stem cells29. The cells generated in the present study, however were negative for the presence of these markers and also appear to be a distinctly different cell population. Similarly, several groups have reported the existence of human adult hemangioblasts bearing surface markers for CD34+ KDR+, CD133+, or KDR+ CD31− CD34− (refs. 4,10,11). The blast cells obtained in the present study do not express these markers and thus appear to also be distinct from putative adult hemangioblasts.
The hES-BCs obtained in the present study appear to represent a distinctly different and earlier population of cells than those reported in a recent study14. Unlike the previous study, we used a simple two-step procedure for both the induction of hES-BC progenitors (EB formation) and expansion of hES-BCs. Our system does not require the use of different cytokine combinations at multiple steps, nor does it require serum or conditioned medium at any step, which contain a mixture of cytokines, inducers and inhibitors that differ from batch to batch and could contribute to wide variations in efficiency and reproducibility. We conclusively demonstrated that the hES-BCs in the present study do not express KDR and CD31 by both immunocytochemistry and Genechip analysis; however, when these cells were induced toward endothelial cell differentiation, KDR, VE-Cad and CD31 expression appeared, suggesting that the hES-BCs obtained represent a different, earlier and more expandable population of cells. Replating of the primary blast colonies generated secondary blast colonies that possessed both hematopoietic and endothelial development potential. Our in vivo studies with the hES-BCs suggest that these cells have the ability to migrate to areas of vascular injury and assimilate with the resident vasculature to restore vascular function.
METHODS
hES cell culture and differentiation
The hES cell lines used in this study were previously described H1, H7 and H9 (US National Institutes of Health-registered as WA01, WA07 and WA09, respectively), and four lines (MA01, MA03, MA40 and MA09) were derived at Advanced Cell Technology. We grew hES cells on mitomycin C-treated mouse embryonic fibroblast in complete hES cell medium until they reached 80% confluence, as described previously30. To induce hemangioblast precursor (mesoderm) formation, we plated hES cells (2–5 × 105 cells/ml) on ultra-low dishes (Corning) in serum-free Stemline medium (Sigma) with the addition of BMP-4 and VEGF165 (50 ng/ml each; R&D Systems) and cultured them in 5% CO2. We removed half the medium and added fresh medium 48 h later, with the same final concentrations of BMP-4 and VEGF, plus SCF, Tpo and FLT3 ligand (20 ng/ml each; R&D Systems) to expand the hemangioblast and its precursor. After 3.5 d, we collected EBs and dissociated them by 0.05% trypsin-0.53 mM EDTA (Invitrogen) for 2–5 min, and prepared single-cell suspension by passing through 22G needle 3–5 times and then through a 40-μm strainer. We resuspended cells in 50–200 μl of Stemline medium. To expand hemangioblasts, we mixed the single-cell suspension derived from differentiation of 2–5 × 105 hES cells with 2 ml of BGM containing 1.0% methylcellulose in Isocve’s MDM, 1–2% bovine serum albumin, 0.1 mM 2-mercap-toethanol, 10 μg/ml rh-insulin, 200 μg/ml iron saturated human transferrin, 20 ng/ml rh-GM-CSF, 20 ng/ml rh-IL-3, 20 ng/ml rh-IL-6, 20 ng/ml rh-G-CSF, 3–6 units/ml rh-Epo, 50 ng/ml rh-SCF, 50 ng/ml rh-VEGF and 50 ng/ml rh-BMP-4, with or without 50 ng/ml of Tpo and FL. We plated the cell mixtures (2–5 × 105 cells/2 ml) in one well of six-well ultra-low plates and incubated them at 37°C in 5% CO2 for 4–6 d. After 4–6 d, grape-like blast colonies were visible by microscopy. A complete step-by-step culture protocol is available in the Supplementary Protocol online.
Clonal studies
For the mixing experiments, we mixed cells from EBs of WA01-GFP and MA01 hES cells and plated in BGM. We examined blast colonies 4–6 d later by phase-contrast and fluorescence microscopy. We carried out limiting dilution plating studies using 1.5 × 104 cells from differentiated EBs. After mixing with 150 ml of BGM, we plated these cells in 15 ultra-low 96-well plates (0.1 ml/well) and incubated them at 37°C for hES-BC development. To confirm the clonal origin of blast colonies, we checked these plates without making markers on the plates by two associates 4 h after plating to exclude wells with double or triple cell clumps from further investigation. For secondary blast colonies growth, we hand-picked primary blast colonies, dissociated them into single cells, mixed them with cell densities of 200, 300 or 1,000 cells per 20 ml of BGM, and plated them in two ultra-low 96-well plates for each cell density (0.1 ml/well). Similarly, we excluded wells with double- or triple-cell clumps from the studies.
Single blast colony expansion
For single blast colony expansion (condition 1), we hand-picked individual blast colonies and transferred them to fibronectin-coated 48-well plate containing Stemline with recombinant human SCF (20 ng/ml), Tpo (20 ng/ml), FL (20 ng/ml), IL-3 (20 ng/ml) VEGF (20 ng/ml), G-CSF (20 ng/ml), BMP-4 (15 ng/ml), IL-6 (10 ng/ml), IGF-1 (10 ng/ml), endothelial cell growth supplement (ECGS, 100 μg/ml) and Epo (3 U/ml). After one week in culture, we removed non-adherent hematopoietic cells by gentle pipetting and used them directly for hematopoietic CFU assay. After removal of the nonadherent cells, we cultured the adherent populations for one more week in EGM-2 endothelial cell medium (Cambrex) and then examined them for the expression of vWF. For condition 2, we picked individual blast colonies and divided them into two parts: we plated one half of the cells for hematopoietic CFU assay and another part on fibronectin-coated 48-well plates and cultured them for endothelial cell development. Details are available in the Supplementary Protocol.
Transplantation into mice and rats with diabetes or ischemia-reperfusion injury
All animal studies were conducted with approval from the University of Florida Institutional Animal Care and Use Committee. Detailed methodology is available in Supplementary Methods online. Briefly, we induced the ischemia-reperfusion injury by elevation of intraocular pressure. Seven days after retinal ischemia-reperfusion injury, we injected the mice either systemically via the retro-orbital sinus (n = 13) or intravitreally (n = 4) with fluorescently labeled hES-BCs collected from day 6 blast colonies. For systemic injection, each mouse received 4 × 105 labeled hES-BCs in 100 μl, whereas for intravitreal injection each mouse received 5 × 104 hES-BCs in 2 μl of PBS. The contralateral control eye of the intravitreous injection group was injected with saline. One day later we euthanized the mice. After enucleation, we fixed the eyes, washed and dissected them by a circumferential limbic incision to remove the cornea and lens, followed by the vitreous humor. Next we removed the intact neural retina by gently detaching it from the underlying choroid. We placed the intact retina in permeabilization buffer and then transferred to buffer containing 20 mg/ml rhodamine-conjugated R. communis agglutinin I (Vector Laboratories). After washing, we placed the retinas on glass coverslips and flattened their curvature by 4 or 5 radial incisions extending from the ciliary margin to within 1 mm of the optic disc. We simultaneously captured red and green fluorescence digital images of the mounted retinas with a Zeiss laser scanning confocal microscope using either a 10 × or 20 × objective. We maintained the z-section depth constant at 2 μm, and scanned the entire retinal vasculature through the thickness of the neural retina (which includes the superficial, mid and deep vascular plexi), resulting in typically 25–35 z-section images per fluorescent channel. We captured images from random locations in the mid-periphery of the retinas as these areas are where the most vascular damage is typically observed in these models. The three-dimensional z-stacks were then flattened using ImageJ software (US National Institutes of Health Research Services Branch) so that the three vascular plexi are visible in a single two dimensional plane.
We handled a second set of mice (n = 6) in an identical manner as described above but perfused them with 3–5 ml TRITC-conjugated dextran before killing them to allow visualization of patent vessels.
We used male obese type 2 diabetic BBZDR/Wor rats (Biomedical Research Models) with duration of diabetes of at least 3 months, along with lean age- and sex-matched controls, to examine the ability of hES-BCs to participate in re-endothelialization of damaged capillaries. Rats were immune-suppressed by intramuscular injection of cyclosporine (2 mg/kg per day) beginning one day before administering the cells, and continuing for the duration of the study. For this study, we resuspended GFP-expressing hES-BCs in sterile saline at a concentration of 3 ×104 cells/μl and injected 5 μl of this suspension into each eye of each of six diabetic and control rats. We euthanized the rats 2 d (n = 3 diabetic, n = 3 control) and 7 days (n = 3 diabetic, n = 3 control) after administering the hES-BCs. We processed 4 of the 6 eyes from each treatment group as described for the mouse ischemia-reperfusion study, and detected GFP-expressing hES-BCs using immunohistochemistry. We performed digital image captures by confocal microscopy as described above. We prepared the remaining eyes from each treatment group for cryosections and detection of CD31, human vWF and human nuclear antigen using immunohistochemistry and fluorescence microscopy.
Transplantation into mice with hind-limb ischemia and myocardial infarction
The experimental protocol was approved by the Animal Care Committee of Memorial Sloan-Kettering Cancer Center. We induced myocardial infarction by ligation of the left coronary artery as described previously24 and 15 min after induction of myocardial infarction, we transplanted 3 × 105 hES-BCs or cell-free medium in the ischemic and peri-ischemic myocardium. For the model of hind-limb ischemia, we injected 6 × 105 hES-BCs or cell-free medium into the area of peri-ischemic muscle immediately after the femoral artery ligation surgery, which we performed to induce ischemia of the hind limb26.
Additional methods
Descriptions of methods used in the characterization of the hES-BCs, including in vitro and in vivo experiments, such as the hematopoietic CFU assay, fluorescence-activated cell sorting analysis, endothelial progenitor assay, immunocyto-chemistry, cryopreservation, PCR analysis, gene profiling of individual clones, and analysis of the tissues and blood flow studies are available in Supplementary Methods.
Supplementary Material
Suplementary methods
Supplementaary figure 1
Supplementary figure 2
Supplementary figure 3
Supplementary figure 4
Supplementary figure 5
Supplementary protocol
Supplementary table 1
Acknowledgments
We thank Y. Ivanova, S. Becker, T. Li, C. Luo, I. Klimanskaya (Advanced Cell Technology) and N. Sengupta (University of Florida) for their excellent technical assistance; The Juvenile Diabetes Research Foundation International; NIH grants EY012601; EY007739; EY14818 (to M.B.G.).
Footnotes
Note: Supplementary information is available on the Nature Methods website.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/naturemethods.
References
- 1.Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–712. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- 2.Grant MB, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 3.Bailey AS, et al. Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood. 2004;103:13–19. doi: 10.1182/blood-2003-05-1684. [DOI] [PubMed] [Google Scholar]
- 4.Cogle CR, et al. Adult human hematopoietic cells provide functional hemangioblast activity. Blood. 2004;103:133–135. doi: 10.1182/blood-2003-06-2101. [DOI] [PubMed] [Google Scholar]
- 5.Otani A, et al. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Med. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 6.Crosby JR, et al. Endothelial cells of hematopoietic origin make a significant contribution to adult blood vessel formation. Circ Res. 2000;87:728–730. doi: 10.1161/01.res.87.9.728. [DOI] [PubMed] [Google Scholar]
- 7.Hill JM, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 8.Wagner RC. Endothelial cell embryology and growth. Adv Microcirc. 1980;9:45–75. [Google Scholar]
- 9.Park C, Ma YD, Choi K. Evidence for the hemangioblast. Exp Hematol. 2005;33:965–970. doi: 10.1016/j.exphem.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Loges S, et al. Identification of the adult human hemangioblast. Stem Cells Dev. 2004;13:229–242. doi: 10.1089/154732804323099163. [DOI] [PubMed] [Google Scholar]
- 11.Pelosi E, et al. Identification of the hemangioblast in postnatal life. Blood. 2002;100:3203–3208. doi: 10.1182/blood-2002-05-1511. [DOI] [PubMed] [Google Scholar]
- 12.Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy M, et al. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature. 1997;386:488–493. doi: 10.1038/386488a0. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy M, D’Souza SL, Lynch-Kattman M, Schwantz S, Keller G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian X, Morris JK, Linehan JL, Kaufman DS. Cytokine requirements differ for stroma and embryoid body-mediated hematopoiesis from human embryonic stem cells. Exp Hematol. 2004;32:1000–1009. doi: 10.1016/j.exphem.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Bowles KM, Vallier L, Smith JR, Alexander MR, Pedersen RA. HOXB4 overexpression promotes hematopoietic development by human embryonic stem cells. Stem Cells. 2006;24:1359–1369. doi: 10.1634/stemcells.2005-0210. [DOI] [PubMed] [Google Scholar]
- 17.Klimanskaya I, Chung Y, Becker S, Lu SJ, Lanza R. Human embryonic stem-cell lines derived from single blastomeres. Nature. 2006;444:481–485. doi: 10.1038/nature05142. [DOI] [PubMed] [Google Scholar]
- 18.D’Souza SL, Elefanty AG, Keller G. SCL/Tal-1 is essential for hematopoietic commitment of the hemangioblast but not for its development. Blood. 2005;105:3862–3870. doi: 10.1182/blood-2004-09-3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gering M, Yamada Y, Rabbitts TH, Patient RK. Lmo2 and Scl/Tal1 convert non-axial mesoderm into haemangioblasts which differentiate into endothelial cells in the absence of Gata1. Development. 2003;130:6187–6199. doi: 10.1242/dev.00875. [DOI] [PubMed] [Google Scholar]
- 20.Lu SJ, Li F, Vida L, Honig GR. CD34+CD38− hematopoietic precursors derived from human embryonic stem cells exhibit an embryonic gene expression pattern. Blood. 2004;103:4134–4141. doi: 10.1182/blood-2003-10-3575. [DOI] [PubMed] [Google Scholar]
- 21.Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes 32. Invest Ophthalmol Vis Sci. 2007;48:361–367. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
- 22.Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 23.Murohara T, et al. Transplanted cord blood-derived endothelial precursor cells augment postnatal neovascularization. J Clin Invest. 2000;105:1527–1536. doi: 10.1172/JCI8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, et al. VEGF enhances functional improvement of postinfarcted hearts by transplantation of ESC-differentiated cells. J Appl Physiol. 2002;93:1140–1151. doi: 10.1152/japplphysiol.00307.2002. [DOI] [PubMed] [Google Scholar]
- 25.Dowell JD, Rubart M, Pasumarthi KB, Soonpaa MH, Field LJ. Myocyte and myogenic stem cell transplantation in the heart. Cardiovasc Res. 2003;58:336–350. doi: 10.1016/s0008-6363(03)00254-2. [DOI] [PubMed] [Google Scholar]
- 26.Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest. 2000;106:571–578. doi: 10.1172/JCI9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, et al. Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity. 2004;21:31–41. doi: 10.1016/j.immuni.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood. 2005;106:860–870. doi: 10.1182/blood-2004-11-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umeda K, et al. Identification and characterization of hemoangiogenic progenitors during cynomolgus monkey embryonic stem cell differentiation. Stem Cells. 2006;24:1348–1358. doi: 10.1634/stemcells.2005-0165. [DOI] [PubMed] [Google Scholar]
- 30.Klimanskaya I, McMahon J. Approaches of derivation and maintenance of human ES cells: Detailed procedures and alternatives. In: Lanza R, et al., editors. Handbook of Stem Cells Volume 1: Embryonic Stem Cells. Elsevier/Academic Press; San Diego: 2004. pp. 437–449. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suplementary methods
Supplementaary figure 1
Supplementary figure 2
Supplementary figure 3
Supplementary figure 4
Supplementary figure 5
Supplementary protocol
Supplementary table 1