Systematic Localization of Common Disease-Associated Variation in Regulatory DNA (original) (raw)
. Author manuscript; available in PMC: 2013 Sep 12.
Published in final edited form as: Science. 2012 Sep 5;337(6099):1190–1195. doi: 10.1126/science.1222794
Abstract
Genome-wide association studies (GWAS) have identified many noncoding variants associated with common diseases and traits. We show that these variants are concentrated in regulatory DNA marked by DNase I hypersensitive sites (DHSs). 88% of such DHSs are active during fetal development, and are enriched for gestational exposure-related phenotypes. We identify distant gene targets for hundreds of DHSs that may explain phenotype associations. Disease-associated variants systematically perturb transcription factor recognition sequences, frequently alter allelic chromatin states, and form regulatory networks. We also demonstrate tissue-selective enrichment of more weakly disease-associated variants within DHSs, and the de novo identification of pathogenic cell types for Crohn’s disease, multiple sclerosis, and an electrocardiogram trait, without prior knowledge of physiological mechanisms. Our results suggest pervasive involvement of regulatory DNA variation in common human disease, and provide pathogenic insights into diverse disorders.
Disease- and trait-associated genetic variants are rapidly being identified with genome-wide association studies (GWAS) and related strategies (1). To date, hundreds of GWAS have been conducted, spanning diverse diseases and quantitative phenotypes (2) (fig. S1A). However, the majority (~93%) of disease- and trait-associated variants emerging from these studies lie within noncoding sequence (fig. SIB), complicating their functional evaluation. Several lines of evidence suggest involvement of a proportion of such variants in transcriptional regulatory mechanisms, including modulation of promoter and enhancer elements (3-6), and enrichment within expression quantitative trait loci (eQTL) (3, 7, 8).
Human regulatory DNA encompasses a variety of _cis_-regulatory elements within which the cooperative binding of transcription factors creates focal alterations in chromatin structure. DNase I hypersensitive sites (DHSs) are sensitive and precise markers of this actuated regulatory DNA, and DNase I mapping has been instrumental in the discovery and census of human _cis_-regulatory elements (9). We performed DNase I mapping genome-wide (10) in 349 cell and tissue samples including 85 cell types studied under the ENCODE Project (10) and 264 samples studied under the Roadmap Epigenomics Program (11). These encompass several classes of cell types including cultured primary cells with limited proliferative potential (n=55); cultured immortalized (n=6), malignancy-derived (n=18) or pluripotent (n=2) cell lines; and primary hematopoietic cells (n=4) as well as purified differentiated hematopoietic cells (n=11), and a variety of multipotent progenitor and pluripotent cells (n=19). We also surveyed regulatory DNA by generating DHS maps from 233 diverse fetal tissue samples across post-conception days ~60-160 (late-first to late-second trimester of gestation). We used a uniform processing algorithm to identify DHSs and the surrounding boundaries of DNase I accessibility (i.e., the nucleosome-free region harboring regulatory factors) (12). We defined an average of 198,180 DHSs per cell type (range 89,526-369,920; table SI) spanning on average ~2.1% of the genome. In total, we identified 3,899,693 distinct DHS positions along the genome (collectively spanning 42.2%), each of which was detected in one or more cell/tissue types (median= 5).
Disease- and trait-associated variants are concentrated in regulatory DNA
We examined the distribution of 5,654 noncoding genome-wide significant associations (5,134 unique SNPs; fig. SI, table S2) for 207 diseases and 447 quantitative traits (2) with the deep genome-scale maps of regulatory DNA marked by DHSs. This revealed a collective 40% enrichment of GWAS SNPs in DHSs (fig. SIC, P< 10-55, binomial, compared to the distribution of HapMap SNPs). Fully 76.6% of all noncoding GWAS SNPs either lie within a DHS (57.1%, 2,931 SNPs) or are in complete linkage disequilibrium (LD) with SNPs in a nearby DHS (19.5%, 999 SNPs) (Fig. 1A) (12). To confirm this enrichment, we sampled variants from the 1000 Genomes Project (13) with the same genomic feature localization (intronic vs. intergenic), distance from the nearest transcriptional start site, and allele frequency in individuals of European ancestry. We confirmed significant enrichment both for SNPs within DHSs (P < 10-59, simulation) and also including variants in complete LD (_r_2=1) with SNPs in DHSs (P<10-37, simulation) (fig. S2).
Fig. 1. Disease-associated variation is concentrated in DNase I hypersensitive sites.
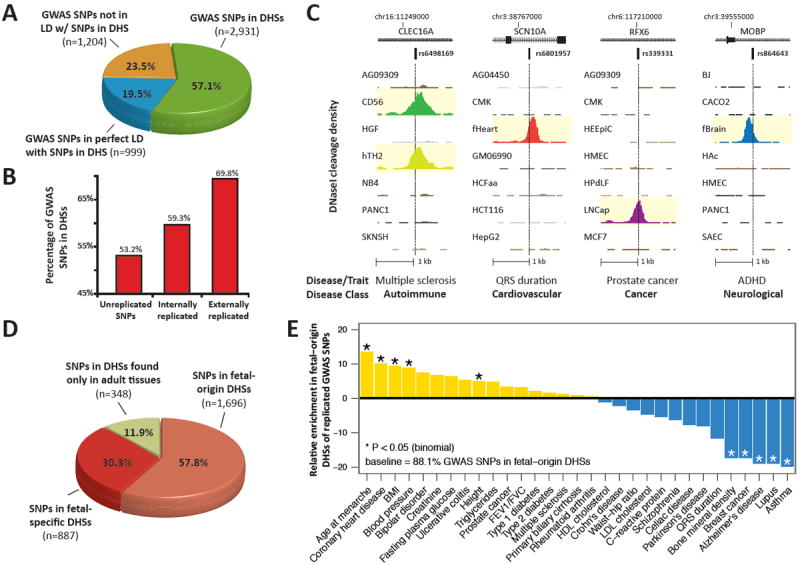
(A) Proportions of noncoding GWAS SNPs localizing within DHSs (green); in complete linkage disequilibrium (_r_2 = 1) with a SNP in a DHS (blue); or neither (yellow). Note that 76.5% of GWAS SNPs are either within or in perfect LD with DHSs. (B) Proportions of GWAS SNPs overlapping DHSs after partitioning by degree of replication. (C) Representative DNase I hypersensitivity (tag density) patterns at diverse disease-associated variants. (D) Proportion of GWAS SNPs localizing in DHSs active in fetal tissues that persist in adult cells (salmon); fetal stage-specific DHSs (red); and adult stage DHSs (green). (E) GWAS SNPs in DHSs show phenotype-specific enrichment for fetal regulatory elements.
In total, 47.5% of GWAS SNPs fall within gene bodies (fig. SIB); however, only 10.9% of intronic GWAS SNPs within DHSs are in strong LD (_r_2≥0.8) with a coding SNP, indicating that the vast majority of noncoding genie variants are not simply tagging coding sequence. Analogously, only 16.3% of GWAS variants within coding sequences are in strong LD with variants in DHSs. We noted that SNPs on widely used genotyping arrays (e.g., Affymetrix) were modestly enriched within DHSs (fig. S2), possibly due to selection of SNPs with robust experimental performance in genotyping assays. However, we found no evidence for sequence composition bias (table S3).
To further examine the enrichment of GWAS SNPs in regulatory DNA, we systematically classified all noncoding GWAS SNPs by the quality of their experimental replication. This disclosed 2,436 unreplicated SNPs; 2,374 ‘internally-replicated’ SNPs (confirmed in a second population in the initial publication); and 324 ‘externally-replicated’ SNPs (confirmed in an independent study) (table S2) (12). We observed a monotonic increase in the proportion of disease/trait variants localizing in DHSs with increasing quality of GWAS SNP experimental replication (Fig. 1B), as well as with increasing strength of association and study sample size (fig. S3). For externally replicated noncoding SNPs, 69.8% lie within a DHS (n=226, P < 10-14, simulation, fig. S2). To exclude the influence of population stratification, we compared the fixation index in African and European populations between GWAS SNPs in DHSs and matched SNPs not in DHSs and found them to be nearly identical (_FST_=0.0843 vs. 0.0847, respectively) (12). The monotonic relationship between evidence for association and SNP concentration in DHSs strongly suggests that many variants are functional and that unreplicated or weaker associations may obscure the true degree of enrichment in DHSs.
GWAS variants localize in cell- and developmental stage-selective regulatory DNA
We observed selective localization within physiologically or pathogenically-relevant specific cell or tissue types, including affected tissues or known or posited effector cell types (Fig. 1C). For a given disorder, cell-selective localization within physiologically or pathogenically-relevant cell types was repeatedly observed for multiple independently-associated SNPs distributed widely around the genome (fig. S4). These results suggest a tissue-specific regulatory role for many common variants, as well as the potential for comprehensive regulatory DNA maps to illuminate associations within disease-relevant cell types.
Many common disorders have been linked with early gestational exposures or environmental insults (14). Because of the known role of the chromatin accessibility landscape in mediating responses to cellular exposures such as hormones (15), we examined if DHSs harboring GWAS variants were active during fetal developmental stages. Of 2,931 noncoding disease- and trait-associated SNPs within DHSs globally, 88.1% (2,583) lie within DHSs active in fetal cells and tissues. 57.8% of DHSs containing disease-associated variation are first detected in fetal cells and tissues and persist in adult cells (‘fetal origin’ DHSs), while 30.3% are fetal stage-specific DHSs (Fig. 1D). GWAS variants in adult stage-specific DHSs localize chiefly in mature hematopoietic cells, connective tissue, endothelial cells, and malignant cells (fig. S6).
We next analyzed the enrichment or depletion of replicated disease-specific GWAS variants in fetal stage DHSs relative to the proportion of total GWAS SNPs in these DHSs. We found the greatest enrichment in phenotypes for which gestational exposures or growth trajectory have been shown to play major roles, including menarche, cardiovascular disease, and body mass index (Fig. 1E) (14, 16). By contrast, we observed relative depletion in fetal DHSs of aging-related diseases, cancer, and inflammatory disorders with presumed (postnatal) environmental triggers. These findings suggest a recurring connection between an exposure-responsive gestational chromatin landscape, regulatory genotype, and risk for specific classes of adult diseases and traits.
DHSs harboring GWAS variants control distant phenotype-relevant genes
Enhancers may lie at great distances from the gene(s) they control (17) and function through long-range regulatory interactions (4, 18), complicating the identification of target genes of regulatory GWAS variants. Most DHSs display quantitative, cell-selective DNase I hypersensitivity patterns which may be systematically correlated with DNase I sensitivity patterns at _cis_-linked promoters. DHSs that are strongly correlated (r > 0.7) with specific promoters function as enhancers that physically interact with their target promoter as detected by chromosome conformation capture methods including 5C and ChlA-PET (10).
To systematically identify the genie targets of DHSs harboring GWAS variants and thereby gain insights into disease mechanisms, we applied the approach described in (10) to the much broader range of cell and tissue types in the present study (12), and intersected the result sets with GWAS data. This analysis revealed 419 DHSs harboring GWAS variants that were strongly correlated (r > 0.7) with the promoter of a specific target gene within ±500 kb of the DHS (table S6, table S7). Among these are numerous examples of target genes that plausibly explain the disease or trait association (Table 1, fig. S7). For example, a SNP (rs385893) associated with platelet count (19) lies in a DHS tightly correlated (r = 0.97) and physically interacting with the 222 kb distant promoter of JAK2, a cytokine-activated signal transducer linked with platelet coagulation and myeloprofilerative disorders (Fig. 2A). Fully 40.8% of correlated DHS-gene pairs span >250 kb (Fig. 2B), and 79% represent pairings with distant promoters vs. those of the nearest gene (table S6, table S7). Notably, these interactions typically extend beyond the range of LD (mean _r_2=0.06; table S6).
Table 1.
Target genes of distal DHSs harboring GWAS variants.
| Disease or trait | r | Target gene | Distance |
|---|---|---|---|
| Amyotrophic lateral sclerosis | 1 | SYNGAP1* - Axon formation; component of NMDA complex | 411 kb |
| Crohn’s disease | 1 | TRIB1* - NFkB regulation | 95 kb |
| Time to first primary tooth | 0.99 | PRDM1* - Craniofacial development | 452 kb |
| C-reactive protein | 0.99 | NLRP3 - Response to bacterial pathogens | 20 kb |
| Multiple sclerosis | 0.98 | AHI1* - White matter abnormalities | 149 kb |
| QRS duration | 0.96 | SCN10A* - Sodium channel involved in cardiac conduction | 181 kb |
| Breast cancer | 0.96 | TACC2* - Tumor suppressor | 411 kb |
| Schizophrenia/brain imaging | 0.95 | KIF1A* - Neuron-specific kinesin involved in axonal transport | 428 kb |
| Brain structure | 0.94 | CXCR6* - Chemokine receptor involved in glial migration | 357 kb |
| Rheumatoid arthritis | 0.94 | CTSB* - Cysteine proteinase linked to articular erosion | 359 kb |
| Ovarian cancer | 0.93 | HSPG2* - Ovarian tumor supressor | 268 kb |
| Multiple sclerosis | 0.93 | ZP1* - Known autoantigen | 153 kb |
| ADHD | 0.93 | PDLIM5* - Neuronal calcium signaling | 328 kb |
| Breast cancer | 0.88 | MAP3K1* - Response to growth factors | 158 kb |
| Amyotrophic lateral sclerosis | 0.88 | CNTN4 - Neuronal cell adhesion | 306 kb |
| Schizophrenia | 0.81 | FXR1* - Cognitive function | 120 kb |
| Type 1 diabetes | 0.75 | ACAD10* - Mitochondrial oxidation of fatty acids | 343 kb |
| Lupus | 0.74 | STAT4 - Mediates IL12 immune response and Th1 differentiation | 113 kb |
Fig. 2. Candidate regulatory roles for GWAS SNPs.
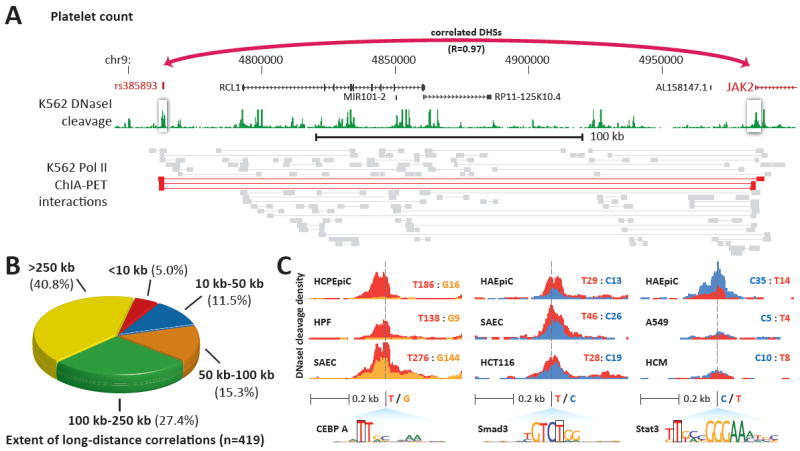
(A) GWAS variant associated with platelet count is connected with the JAK2 gene (myeloproliferative disorders) 222 kb away. Below, ChlA-PET tags (36) validate direct chromatin interactions between this DHS and the JAK2 promoter; red tags demonstrate an interaction between these DHSs. (B) Proportion of DHSs harboring GWAS variants that can be linked to target promoters at the indicated distance. (C) Examples of allele-specific DNase I sensitivity in cell types derived from heterozygous individuals for GWAS variants that alter TF recognition motifs within DHSs (also see table S9). Each cell type track shows DNase I cleavage density scaled by allelic imbalance at the GWAS variant and colored by variant nucleotide (blue = C, green = A, yellow = G, red = T). Total reads from each allele are also shown.
GWAS variants in DHSs frequently alter allelic chromatin state
Next, we examined how GWAS variants in DHSs were distributed with respect to transcription factor recognition sequences, defined using a scan for known motif models at a stringency of P < 10-4 (12). Of GWAS SNPs in DHSs, 93.2% (2,874) overlap a transcription factor recognition sequence. We partitioned GWAS variants into 10 disease/trait classes, and then determined the frequency of GWAS variants associated with a particular disease/trait class that localized within sites for transcription factors independently partitioned into the same classes based on gene ontology annotations (fig. S8) (12). This analysis revealed that common variants associated with specific diseases or trait classes were systematically enriched in the recognition sequences of transcription factors governing physiological processes relevant to the same classes.
Functional variants that alter transcription factor recognition sequences frequently affect local chromatin structure. At heterozygous SNPs altering transcription factor recognition sequences, altered nuclease accessibility of the chromatin template manifests as an imbalance in the fraction of reads obtained from each allele (20, 21). As the concentration of sequence reads and highly overlapping read coverage results in an effective re-sequencing of DHSs, we were able both to detect cell types heterozygous for common SNPs and to quantify the relative proportions of reads from each allele across all cell types (12). This imbalance is indicative of the functional effect of a particular allele on local chromatin state. We detected 584 heterozygous GWAS SNPs with sufficient sequencing coverage, of which 120 showed significant allelic imbalance in chromatin state (at FDR 5%). We identified sites where regulatory variants were associated with allelic chromatin states, with the predicted higher-affinity allele exhibiting higher accessibility (Fig. 2C). In nearly 50% of cases, the magnitude of imbalance was >2:1 (fig. S9). The GWAS SNPs were the sole local sequence difference between haplotypes, indicating that disease-associated variants are responsible for modulating local chromatin accessibility. Further, at sites with very high sequencing depth (>200x), 38.7% (53/137) show significant allelic imbalance (FDR < 5%). As sensitivity to detect allelic imbalance is governed by sequencing depth, this suggests that nearly 40% of GWAS variants in similarly-sequenced DHSs would be expected to show allelic imbalance.
Disease-associated variants cluster in transcriptional regulatory pathways
Transcriptional control of glucose homeostasis and beta cell genesis and function is mediated by a closely-knit transcriptional regulatory pathway defined by specific transcription factors. The Mendelian phenotypes of maturity-onset diabetes of the young (MODY) are caused by separate lesions disrupting the coding sequences of each of these transcription factors (22). Interestingly, we observed clustering of common noncoding variants associated with abnormal glucose homeostasis, insulin and glycohemoglobin levels, and diabetic complications within recognition sites for the same six transcription factors (P < 0.029, binomial; 48% enrichment over random SNPs; Fig. 3A). This suggests that noncoding variants that predispose to dysregulation of glucose homeostasis perturb peripheral nodes of the same regulatory network responsible for Mendelian forms of Type 2 diabetes.
Fig. 3. Common disease-associated variants cluster in regulatory pathways.
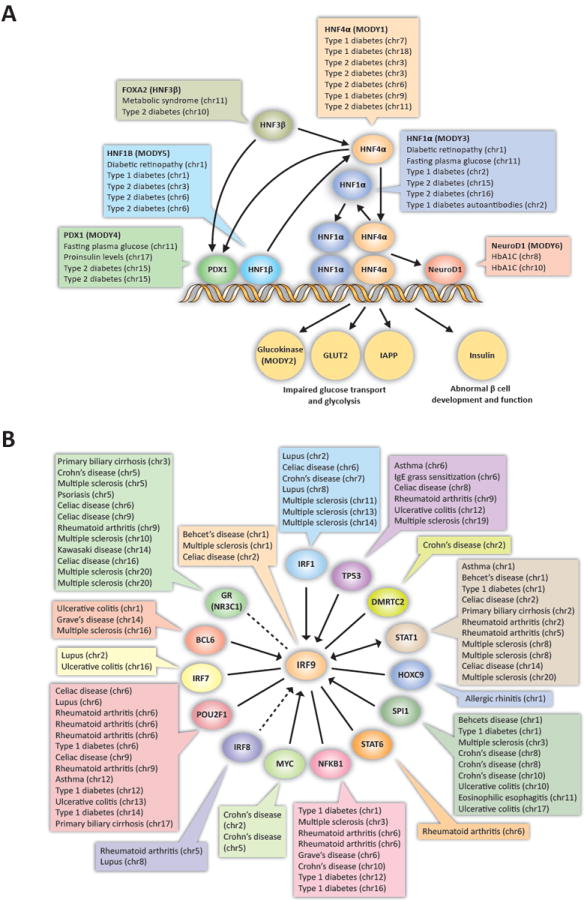
(A) SNPs in DHSs associated with diabetes (Type I and Type II), diabetic complications, and glucose homeostasis localize in recognition sites of transcriptional regulators (labeled ellipses) controlling glucose transport, glycolysis, and beta cell function that are structurally disrupted in the Mendelian phenotypes of maturity-onset diabetes of the young (MODY). Chromosome of each SNP associated with the indicated phenotype is listed (see table S2). (B) 24.4% of SNPs associated with autoimmune disorders that fall within DHSs localize in recognition sequences of TFs that interact with IRF9. Arrows indicate directionality of relationship, dotted lines represent indirect interactions (12). The complete network is shown in fig. S10.
Using known interacting sets of transcription factors, we identified related disease-associated variants in the recognition sequences of a central target factor and its interacting partners (Figs. 3B, S11, S12) for factors involved in autoimmune disease, cancer and neurological development. IRF9 is a transcription factor associated with type I interferon induction (23). Of 26 transcription factors in the IRF9-centered interaction network, 15 represent transcription factors with recognition sequences in multiple distinct DHSs that contain GWAS variants associated with a wide variety of autoimmune disorders (P < 1.6×10-13, binomial; 2.8-fold enrichment vs. random SNPs, Fig. 3B) (12). Notably, 24.4% (64/262) of GWAS SNPs within DHSs of immune cells and associated with autoimmune disease alter one or more of the 15 transcription factor motifs from the IRF9-centered network. This example and those in Figs. S11, S12, illustrate that disease-associated variants from the same or related disorders and traits repeatedly localize within the recognition sequences of transcription factors that form interacting regulatory networks.
Common networks for common diseases
The observation that GWAS variants associated with multiple distinct diseases within the same broader disease class (e.g., inflammation, cancer) repeatedly localize within the recognition sites of interacting transcription factors suggested that cohorts of such transcription factors might form shared regulatory architectures. To explore whether noncoding GWAS SNPs from related diseases perturb different recognition sequences of a common set of transcription factors, we tabulated all transcription factors for which at least 8 recognition sequences in DHSs were perturbed by GWAS SNPs associated with autoimmune diseases (Fig. 4A). Among the 22 factors identified were canonical immune signaling regulators, such as STAT1 and STAT3, NF-κB, and PPARα and PPARγ. These 22 transcription factors comprise a highly significant (P < 9.8 × 10-51, simulation vs. number of factors for random SNPs (12)), shared regulatory architecture that is repeatedly perturbed in a wide range of autoimmune disorders (Fig. 4A).
Fig. 4. Common disease networks.
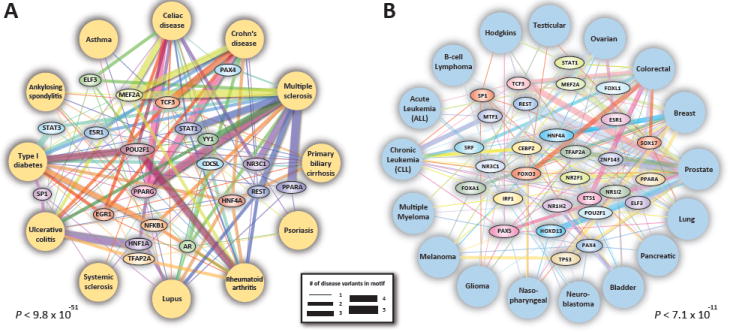
GWAS SNPs from related diseases repeatedly perturb recognition sequences of common transcription factors. Shown are factors whose recognition sequences harbor ≥8 or ≥6 GWAS SNPs in inflammatory/autoimmune diseases (A) and cancer (B), respectively. Edge thickness represents number of associations between TF and disease in DHSs in relevant tissues. Both networks are significantly enriched for overlap with disease-relevant GWAS SNPs, and include many well-studied regulators.
The same analysis in the context of 17 different malignancies exposed a very different network of transcription factors connecting seemingly disparate cancer types (P < 7.1 × 10-11, simulation (12)) including neoplastic regulatory relationships, linking FOXA1 and breast cancer, FOX03 and colorectal cancer, and TP53 and melanoma, breast and prostate cancer (Fig. 4B). We also analyzed six neuropsychiatric disorders, and identified 23 transcription factors whose recognition sequences were perturbed by at least 3 disease-associated variants (fig. S13). Collectively, these results support the hypothesis that shared genetic liability may underlie many related categories of disease (24, 25).
De novo identification of pathogenic cell types
To provide insights into the cellular structure of disease and potentially highlight pathogenic cell types, we explored the selective localization of GWAS SNPs within the regulatory DNA of individual cell types. We further considered the enrichment of all tested variants, not just those with genome-wide significance, and performed serial determination of the cell/tissue-selective enrichment patterns of progressively more strongly associated variants to expose collective localization within specific lineages or cell types. We used all SNPs tested in GWAS meta-analyses of two common autoimmune disorders, Crohn’s disease (26) and multiple sclerosis (MS) (27), and a common continuous physiological trait, cardiac conduction measured by the electrocardiogram QRS duration (28) (n=938,703, 2,465,832, and ~2.5M SNPs, respectively). For SNPs meeting increasingly significant _P_-value cutoffs, we compared the proportion of SNPs in DHSs of each cell type to the proportion of all SNPs in DHSs of the same cell type (Fig. 5). For all three studies, we observed enrichment of more weakly associated variants in regulatory DNA. This enrichment suggests that a large number of functional variants of small quantitative effect act through modulation of regulatory DNA. Additionally, it suggests that conditioning association analyses on regulatory DNA may ameliorate the stringent statistical correction for multiple testing required for genome-wide testing of unselected SNPs.
Fig. 5. Identification of pathogenic cell types.
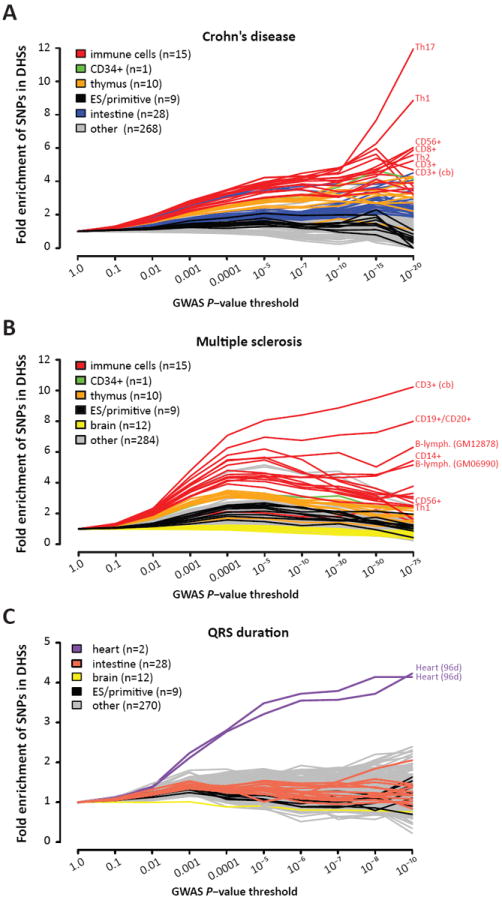
GWAS SNPs are systematically enriched in the regulatory DNA of disease-specific cell types throughout the full range of significance. Shown are SNPs tested for association with the autoimmune disorders Crohn’s disease (A), multiple sclerosis (B) and QRS duration (C).
Furthermore, with progressively stringent _P_-value thresholds, we observed increasingly selective enrichment of disease-associated variants within specific cell types (Fig. 5). Strikingly, in the case of Crohn’s disease, the Th17 (12.0-fold enriched) and Th1 (8.87-fold enriched) T-cell subtypes have a concentration of the most-significant GWAS variants in their DHSs (Fig. 5A). While Crohn’s pathology has classically been associated with Th1 cytokine responses, an emerging consensus points to a defining role for IL17-producing Th17 cells (29). Notably, this analysis was accomplished without any prior knowledge about Crohn’s disease pathology.
In the case of MS, sequential cell-selective enrichment analysis highlighted two cell types: CD3+ T-cells from cord blood, and CD19+/CD20+ B-cells (Fig. SB). While MS has long been thought to be T-cell mediated, a critical role for B-cells has only recently been recognized and has major therapeutic implications (30). It is notable that cord blood CD3+ cells – essentially a naïve population – garner the most highly selective enrichment, particularly in comparison with total adult CD3+ cells or other T-cell subsets, suggesting a role for variants influencing immune education. Also of note, DHSs active in brain tissue were moderately depleted (~10%) for MS-associated variants, suggesting that neural regulatory elements do not play a substantial role in MS pathogenesis, as proposed (31). Analogously, analysis of variants associated with the continuously varying trait of QRS duration revealed similarly specific enrichment within fetal heart DHSs (Fig. 5C). Importantly, in all three cases, the results were obtained without any prior knowledge of physiological mechanisms. These data suggest a generally applicable approach, and highlight the value of extensive maps of regulatory DNA for gaining insights into disease physiology and pathogenesis.
Discussion
Despite a long appreciation of the involvement of regulatory variants in human disease (32-34), difficulty in delineating regulatory DNA regions, particularly in a cell-specific context, has heretofore prevented comprehensive assessment of the relationship between gene regulation and common phenotypes. Our results indicating widespread and systematic localization of variants associated with a wide spectrum of common diseases and traits in regulatory DNA marked by DHSs have many implications for interpreting diverse genotype-phenotype association studies. The connection of numerous DHSs harboring GWAS SNPs with promoters of distant genes expands the genomic horizon of disease and trait associations, and provides a trove of plausible causal genes to explain those associations. The data also unify seemingly unconnected variants associated with related diseases by virtue of their convergent perturbation of common transcription factor networks. Tissue-selective enrichment of phenotype-associated variants raises the possibility of more focused genetic association studies that condition on the regulatory DNA of a known or hypothesized target tissue type. Further, selective enrichment of many more weakly-associated variants within regulatory DNA of pathogenic cell types points to the quantitative contribution of hundreds of variants of small effect size that modulate transcription factor binding characteristics, in contrast to Mendelian variants in transcription factor genes that may perturb entire networks. The results thus highlight a continuous quantitative spectrum of disordered gene regulation between common disease and Mendelian traits, and lend a new perspective on the genetic architecture of common human disease (35).
Supplementary Material
SOM
TableS2
Acknowledgments
We thank many colleagues for their insightful comments and critical readings of the manuscript. We also thank many colleagues who provided individual cell samples for DNase I analysis. This work was supported by National Institutes of Health grants U54HG004592 and U01ES01156 (J.A.S.); P30 DK056465 (S.H.); R01HL088456 (N.S.); and R24HD000836-47 (I.G.). We would like to acknowledge the generous sharing of results from the International MS Genetics, International IBD Genetics, and CHARGE QRS Consortia. DNase I data have been deposited in GEO under accession numbers GSE29692 and GSE18927, and are also available for viewing and download at genome.ucsc.edu, www.uwencode.org/data and www.epigenomebrowser.org.
Footnotes
Supplementary Materials
Materials and Methods
Figs. SI to S13
Tables SI to S12
References (37-49)
References and Notes
- 1.Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindorff L, et al. A Catalog of Published Genome-Wide Association Studies. [Jan 4, 2012]; available at http://www.genome.gov/gwastudies.
- 3.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pomerantz MM, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harismendy O, et al. 9p21 DNA variants associated with coronary artery disease impair interfer-on-γ signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Degner JF, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 10.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012 doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Detailed information on methods and analyses can be found in the supplementary materials available in Science Online.
- 13.1000 Genomes Project Consortium, A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Symonds ME, Sebert SP, Hyatt MA, Budge H. Nutritional programming of the metabolic syndrome. Nat Rev Endocrinol. 2009;5:604–610. doi: 10.1038/nrendo.2009.195. [DOI] [PubMed] [Google Scholar]
- 15.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barker DJ, et al. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–941. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 17.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 18.Lettice LA, et al. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc NatlAcad Sci U S A. 2002;99:7548–7553. doi: 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soranzo N, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight JC, Keating BJ, Rockett KA, Kwiatkowski DP. In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nat Genet. 2003;33:469–475. doi: 10.1038/ng1124. [DOI] [PubMed] [Google Scholar]
- 21.Maurano MT, Wang H, Kutyavin T, Stamatoyannopoulos JA. Widespread site-dependent buffering of human regulatory polymorphism. PLoS Genet. 2012;8:e1002599. doi: 10.1371/journal.pgen.1002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–980. doi: 10.1056/NEJMra002168. [DOI] [PubMed] [Google Scholar]
- 23.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 24.International Schizophrenia Consortium et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotsapas C, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patsopoulos NA, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neural. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sotoodehnia N, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brand S. Crohn“s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn”s disease. Gut. 2009;58:1152–1167. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 30.von Büdingen H-C, Bar-Or A, Zamvil SS. B cells in multiple sclerosis: connecting the dots. Current Opinion in Immunology. 2011;23:713–720. doi: 10.1016/j.coi.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.International Multiple Sclerosis Genetics Consortium (IMSGC) et al. Lack of support for association between the KIF1B rs10492972[C] variant and multiple sclerosis. Nat Genet. 2010;42:469–70. doi: 10.1038/ng0610-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 33.Collins FS, et al. A point mutation in the A gamma-globin gene promoter in Greek hereditary persistence of fetal haemoglobin. Nature. 1985;313:325–326. doi: 10.1038/313325a0. [DOI] [PubMed] [Google Scholar]
- 34.Rockman MV, Wray GA. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol. 2002;19:1991–2004. doi: 10.1093/oxfordjournals.molbev.a004023. [DOI] [PubMed] [Google Scholar]
- 35.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pruitt KD, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neph S, et al. BEDOPS: high-performance genomic feature operations. Bioinformatics. 2012;28:1919–1920. doi: 10.1093/bioinformatics/bts277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: http://www.R-project.org/) [Google Scholar]
- 41.International HapMap 3 Consortium et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aho AV, Kernighan BW, Weinberger PJ. The AWK Programming Language. 1. Addison Wesley; Reading, MA: 1988. [Google Scholar]
- 43.Matys V, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–10. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Portales-Casamar E, et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–10. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37:D77–82. doi: 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SOM
TableS2