Topical Antiangiogenic SRPK1 Inhibitors Reduce Choroidal Neovascularization in Rodent Models of Exudative AMD (original) (raw)
Abstract
Purpose.
Exudative AMD (wet AMD) is treated by monthly injection into the eye of anti-VEGF proteins. VEGF is alternatively spliced to produce numerous isoforms that differ in angiogenic activity. Serine-rich protein kinase-1 (SRPK1) has been identified as a regulator of pro-angiogenic VEGF splicing by phosphorylating serine-rich splicing factor-1 (SRSF1), which binds to VEGF pre-mRNA. We tested the hypothesis that topical (eye drop) SRPK1-selective inhibitors could be generated that reduce pro-angiogenic isoforms, and prevent choroidal neovascularization in vivo.
Methods.
Novel inhibitors were tested for SRPK inhibition in vitro, pro-angiogenic VEGF production in RPE cells by PCR and ELISA, and for inhibition of choroidal neovascularisation in mice and rats.
Results.
A novel disubstituted furan inhibitor was selective for the SRPK family of kinases and reduced expression of pro-angiogenic but not antiangiogenic VEGF isoforms. This inhibitor and previously identified SRPK inhibitors significantly reduced choroidal neovascularisation in vivo. Topical administration of SRPK inhibitors dose-dependently blocked CNV with an EC50 of 9 μM.
Conclusions.
These results indicate that novel SRPK1 selective inhibitors could be a potentially novel topical (eye drop) therapeutic for wet AMD.
Keywords: VEGF, splicing, AMD
Exudative AMD results from excess angiogenic VEGF production. Switching VEGF splicing to the antiangiogenic isoforms by inhibiting serine-rich protein kinase-1 (SRPK1) can be achieved by topical application of novel SRPK1 inhibitors. This could result in novel treatments that do not require injection in wet AMD.
Introduction
AMD, a disease causing vision loss that affects the central area of the macula, is the leading cause of blindness in people over 50 years of age. The most severe form of AMD, wet (exudative) AMD,1 is characterized by choroidal neovascularization (CNV), the leakage of blood and serous fluid from the immature neomicrovasculature beneath the RPE that eventually leads to loss of photoreceptors, retinal detachment, dense macular scarring, and visual loss.2,3 VEGF, a key factor in angiogenesis and vascular leakage,4 is up-regulated during the progression of CNV5–8 and has become the lead therapeutic target for the treatment of exudative AMD.
The vegfa gene is alternatively spliced to form a family of multiple isoforms,9,10 each isoform differing in biologic property, activity, and function.11 Most cells commonly express isoforms VEGF121, VEGF165, and VEGF189, whereas VEGF145 and VEGF206 are comparatively rare. The majority of VEGF isoforms contain exons 1 to 5 (the exception being VEGF11112) but differing portions of exons 6 and 7 that encode heparin sulphate (HS) binding domains. Alterations in the usage of these exons change the biologic properties of alternatively spliced isoforms such as their ability to bind to cell–surface heparin-sulfate proteoglycans and release angiogenic factors.13,14 In 2002, differential splicing of the eighth exon was demonstrated from a proximal splice site (PSS) to a distal splice site (DSS) 66 bases downstream.15,16 Alternative mRNA splicing in this region generated a second family of isoforms (VEGFxxxb) of the same size, but noted for their antiangiogenic properties.17 During pathologic angiogenesis pro-angiogenic isoforms are selectively upregulated,15,18,19 suggesting VEGFxxx and VEGFxxxb may have separate regulatory pathways. These antiangiogenic isoforms, such as VEGF165b and VEGF121b have been shown to be potently antiangiogenic in animal models of retinal and choroidal neovascularisation, when injected intraocularly as recombinant proteins20 and result in both endothelial and retinal epithelial cell cytoprotection.21
The first therapy to be Food and Drug Administration (FDA) approved for the treatment of neovascular AMD in December 2004 was a VEGF165, VEGF189, and VEGF206 specific aptamer, Pegaptanib Sodium (Macugen; OSI Pharmaceuticals, Long Island, NY).22 During clinical trials pegaptanib dose-dependently reduced the risk of severe visual acuity loss and slowed the progression of neovascular AMD, but did not result in significant improvement in vision. In 2006, ranibizumab (Lucentis; Genentech, South San Francisco, CA), a novel humanized anti-VEGF antibody fragment, was FDA approved for the treatment of neovascular AMD. Its approval was based on the results of three clinical trials where, approximately 95% of patients treated monthly with ranibizumab 0.5 mg maintained visual acuity (defined as the loss of <15 letters) and less than or equal to 40% improved vision (defined as the gain of ≥15 letters) at 1 year compared with 11% in the sham control treated group.23–25 Current treatment regimes require ranibizumab administration by intraocular injection as often as monthly.25 Such intraocular injections can result in increased IOP26 and a risk, albeit minor, of endophthalmitis and other severe adverse effects.27 Furthermore, bevacizumab, the anti-VEGF antibody from which ranibizumab was derived, was shown to bind VEGF165b with equal potency to VEGF165, thus, targeting both pro- and antiangiogenic endogenous VEGF isoforms.18
As both the antiangiogenic and angiogenic isoforms of VEGF are derived from the same gene, the control of the isoform family is a result of the control of alternative splicing. We have recently identified some of the pathways that control the splicing of VEGF at the proximal splice site, implicating the RNA binding protein SRSF128,29 and its kinase SRPK130 as key requirements for the decision by cells to use the proximal splice site and, hence, generate pro-angiogenic isoforms of VEGF.29,31 Knockdown of SRPK1 potently reduced VEGF-mediated angiogenesis in vivo in tumors and inhibition of SRPK1 and SRPK2 reduced angiogenesis in vivo.28 The development of new antiangiogenesis agents represents a new era in the treatment of neovascular AMD; however, the search for novel VEGF inhibitors that avoid the need for intravitreal injections, but maintain potency and specificity to pro-angiogenic VEGF isoforms, is imperative. We therefore wished to determine whether new, small molecule inhibitors targeting SRPK1 selectively could be developed and used with therapeutic potential to prevent laser-induced and VEGF-mediated CNV in rodent models. Furthermore, we investigated whether low molecular weight compounds known to inhibit SRPK1 could be used topically to inhibit CNV progression.
Methods
Cell Culture
Primary human RPE isolations were performed on human donor globes obtained within 24 hours postmortem from the Bristol Eye bank (Bristol Eye Hospital [BEH]). Retinas with choroid–RPE sheets were removed to a petri dish, finely chopped, digested in Dulbecco's Modified Eagle Medium (DMEM):F12(1:1)+GlutaMax (Life Technologies, Paisley, UK), and supplemented with 0.3 mg/mL collagenase for 15 minutes at 37°C. Digested choroid–RPE sheets were suspended in media (DMEM:F12+GlutaMax), supplemented with 10% fetal bovine serum (FBS), 0.5% PenStrep (Life Technologies), and spun at 1500 rpm (251_g_) for 10 minutes to pellet cells. Pellets were resuspended in media supplemented with 25% FBS (Life Technologies), grown in cell culture flasks (Greiner, Stonehouse, UK), and split at 80% confluence. ARPE-19 (ATCC, Teddington, UK) cells were cultured in DMEM:F12 plus 10% FBS, split at 80% confluence.
Pharmacological Inhibitor Treatments.
SRPIN340 (N-[2-(1-piperidinyl)-5-(trifluoromethyl)phenyl] iso nicotinamide), MVRL09 (N-[2-(morpholin-4-yl)-5-(trifluoromethyl)phenyl] pyridine-3-carbox amide), and SPHINX (5-methyl-N-[2-(morpholin-4-yl)-5-(tri-fluoromethyl)phenyl] furan-2-carboxamide) were purchased from Ascent Scientific (Bristol, UK), Chembridge (San Diego, CA), and Enamine (Kiev, Ukraine), respectively. Cells at approximately 70% confluence were serum starved for at least 12 hours and treated with 5 or 10 μM compound inhibitors. Twenty-four hours later mRNA was extracted and 48 hours later protein was extracted, for further analyses. Cells pretreated with inhibitors (30 minutes) were activated with epidermal growth factor for 60 minutes before protein extraction.
In Vitro Kinase Assay
Candidate compounds, MVRL09, SPHINX, and SRPIN340 were tested for SRPK1 inhibition using a Kinase-Glo assay (Promega, Southampton, UK32). A reaction buffer containing 9.6 mM MOPS pH of 7 and 0.2 nM EDTA pH of 8 was added to 10 μM SRSF1 Arg-Ser (RS) peptide (NH2-RSPSYGRSRSRSRSRSRSRSRSNSRSRSY-OH) and 0.1 μg of purified SRPK1 kinase. Candidate compounds were serially diluted from 10 μM to 0.5 nM and added to the reaction mixture, wells with omitted SRPK1 kinase and omitted compounds were also added as controls. All wells contained 1% DMSO (Fisher Scientific, Loughborough, UK). One micromolar ATP was added, wells minus ATP were used as background controls. The plate was then incubated at 30°C for 10 minutes. An equal volume of Kinase-Glo (25 μL; Promega) was added to each well and the plate read for luminescence using a Victor X (Perkin Elmer, Waltham, MA).
Semiquantitative: Reverse Transcriptase (RT)-PCR for VEGF
Conventional PCR was used to detect VEGF165 and VEGF165b mRNA. Five percent to 10% of the cDNA was added to a reaction mixture containing: 2× PCR Master Mix (Promega), primers (1 μM each) complementary to exon7b (5′-GGC AGC TTG AGT TAA ACG AAC-3′) and the 3′UTR of exon 8b (5′-ATG GAT CCG TAT CAG TCT TTC CTG G-3′) and DNase/RNase free water. All samples were run in parallel with negative controls (water and cDNA without RT) and positive controls (VEGF165 in a plasmid expression vector [pcDNA] and VEGF165b pcDNA). The reaction mixture was thermo cycled (PCR Express; Thermo Electron Corp., Basingstoke, UK) 30 to 35 times, denaturing at 95°C for 60 seconds, annealing at 55°C for 60 seconds, and extending at 72°C for 60 seconds. PCR products were separated on 2.5% agarose gels containing 0.5 μg/mL ethidium bromide (BioRad, Hemel Hempstead, UK) and visualized under UV transilluminator (BioRad).
Equal cDNA loading was determined by PCR with Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers (Forward: 5′-CAC CCA CTC CTC CAC CTT TGA C-3′; Reverse: 5′-GTC CAC CAC CCT GTT GCT GTA G-3′) or mouse globin primers (F: ACGTGCTAAGCCAGTGACTG, R: CAGCCTTCTCAGCATGAGTC:). Primers result in one amplicon at approximately 112 bp after thermo cycling 30 times, denaturing at 94°C for 45 seconds, annealing at 65°C for 45 seconds, and extending at 72°C for 60 seconds.
PanVEGF and VEGFXxxb ELISA.
One microgram per milliliter pan-VEGF capture antibody (Duoset VEGF ELISA DY-293; R&D Systems, Abingdon, UK) was incubated overnight at room temperature. The plates were blocked (Superblock; Thermo Scientific) and serial dilutions of recombinant human (rh)VEGF165 or rhVEGF165b standards (ranging from 4 ng/mL–16.25 pg/mL) were added, incubated alongside sample lysates, and typically diluted 1:10. The plate was incubated for 1 hour at 37°C with shaking, washed, and incubated with 100 μL/well of either biotinylated goat anti-human VEGF (0.1 μg/ml; R&D Systems) or mouse anti-human VEGF165b (0.25 μg/mL) for 1 further hour at 37°C. After washing, 100 μL/well of horseradish peroxidase (HRP)-conjugated streptavidin (1:200; R&D Systems) was added and plates were left at room temperature for 20 minutes.
The plates were washed and color change induced with substrate A and B (DY-999; R&D Systems) for 20 minutes under light protection. The reaction was stopped by addition of 100 μL/well of 1 M H2SO4 and the absorbance was read immediately in an ELISA plate reader (Opsys MR system plate reader; Dynex Technologies, Worthing, UK) at 450 nm with a control reading at 570 nm. Revelation Quicklink 4.25 software (Dynex Technologies) was also used to calculate a standard curve from mean absorbance values of standards enabling the estimation of VEGF concentration for each sample.
Western Blotting
Protein samples (30–50 μg) were mixed with 1× SDS loading buffer (100 mM Tris-HCl, 4% SDS, 20% glycerol, 0.2% [wt/vol] bromophenol blue and 5% final concentration 2-mercaptoethanol, pH of 6.8). To denature the protein, samples were boiled for 5 minutes at 100°C.
Samples were subjected to PAGE on a 12% SDS-PAGE gel at 90 V in ice cold running buffer (25 mM Tris-HCl, 250 mM glycine, 0.1% SDS, pH of 8.3) for approximately 2.5 hours. The separated proteins were then electrophoretically blotted to a methanol-activated polyvinylidene fluoride (PVDF) membrane (Fisher Scientific) by wet transfer for 2 hours at 90 V in transfer buffer (50 mM Tris-HCl, 38 mM glycine, 20% methanol, pH of 8.3). Membranes were incubated in blocking solution (2.5% nonfat dried milk or 5% BSA in Tris-buffered saline with Tween20 [TBS-T]) with agitation at room temperature for 1 hour and then probed with the primary antibody overnight at 4°C; rabbit polyclonal anti–VEGF-A (A20; sc-152; Santa Cruz, Santa Cruz, CA) diluted 1:1000 to 1:100 in 2.5% nonfat dried milk TBS-T, VEGFxxxb specific mouse monoclonal 56/1 (R&D Systems) diluted 1:1000 to 1:500 in 5% BSA TBS-T, media from mouse hybridoma cell line, mab104 diluted 1:4 in TBS-T, goat polyclonal beta-tubulin (Abcam, Cambridge, UK) diluted 1:1000 in 5% BSA TBS-T, and goat polyclonal beta-actin (Santa Cruz) diluted 1:1000 in 5% BSA TBS-T. Membranes were then washed four times for 10 minutes each with TBS-0.3%T before incubation with secondary HRP-conjugated antibodies: goat anti-mouse IgG, rabbit anti-goat IgG, or goat anti-rabbit IgG (Fisher Scientific) diluted 1:10,000 or fluorescently labeled secondary antibodies (Licor, Cambridge, UK) diluted 1:10,000 in 5% BSA TBS-T or 2.5% nonfat dried milk TBS-T, for 45 minutes at room temperature with agitation. The washes were repeated and the bands were detected by fluorescent visualization (Licor) or using the Enhanced Chemoluminiscence (ECL) SuperSignal West Femto Maximum Sensitivity Substrate kit (Pierce).
Laser Lesion Induction Protocol
Six- to 8- week-old C57/B6 mice (B&K Laboratories, Hull, UK) and adult Norway-Brown rats (Harlan Laboratories, Hillcrest, UK) were anaesthetized with an intraperitoneal injection of a mixture of 50 mg/kg ketamine and 0.5 mg/kg medetomidine. The pupils were dilated with 2.5% phenylephrine hydrochloride and 1% tropicamide. Four photocoagulation lesions were delivered with a krypton red laser (Mice: 250 mW, 100 ms, 75 μm, Rats: 200 mW, 100 ms, 75 μm, 810 nm Oculight Slx laser; IRIS Medical, Iridex, Mountain View, CA) between the retinal vessels in a peripapillary distribution at a distance of 1- to 2-disc diameters from the optic nerve in each eye. Only laser lesions with a subretinal bubble at the time of treatment were included in the study. Immediately following laser photocoagulation the animals either received intravitreal injections in both eyes (day 0 and day 7), or given topical eye drops twice daily (10 μL). Animals were culled on either day 4 or day 14 and eyes were either unfixed for retinal dissection and protein extraction, or fixed and enucleated and choroids stained and examined. Choroidal lesions were imaged and areas traced by a masked observer, lesions area was quantified using Image J (National Institutes of Health, Bethesda, MD).
During topical administration SRPIN340 and SPHINX, were made up into a gel-based drug delivery vehicle to aid duration of drug exposure to the eye,33 0.05% DMSO was used to dissolve the inhibitors and was added to control vehicle.
Mass Spectrometry
A mass spectrometry–based strategy was employed to determine the pharmacokinetics of SRPIN340 in vivo. Initially SRPIN340 and MVRL09 were serially diluted in water (initial stock dissolved in DMSO) from 100 μg/mL to 0 μg/mL and analyzed. The chromatograms produced clear peaks at the expected molecular weights for SRPIN340 (349.1 Da) and MVRL09 (351 Da). The area under the peaks were integrated and plotted against concentration to confirm a linear response was observed. SRPIN340 was investigated in eye tissue following 20 ng intravitreal injection and in eye tissue following a single topical application of 5 μg. After a single administration, mice were killed at time points 1, 4, 8, 24, and 48 hours. Samples, SRPIN340 (the analyte) and control treated, were homogenized and proteins were precipitated out of the samples with an equal volume (100 μL) acetonitrile or acetone. An internal standard of 100 μg/mL MVRL09 was added to samples to account for any loss of samples during preparation. The solutions of 50% sample and 50% acetonitrile and 100 μg/mL MVRL09 were centrifuged for 15 minutes at 4°C to pellet the proteins and the supernatant taken for analysis. Solutions were evaporated at 37°C for 8 hours and resuspended in 30 μL acetonitrile ready for analysis by Liquid chromatography-mass spectrometry using a Waters 2795 HPLC system. Detection was achieved by positive ion electrospray (ESI+) mass spectrometry using a Waters Micromass ZQ spectrometer (Waters, Milford, MA) in single ion monitoring (SIM) mode at 352 m/z units ([M+H]+ SRPIN340) and 350.1 m/z units ([M+H]+ MVRL09). Mass spectrometer settings were as follows: capillary voltage: 3.3 kV; cone voltage: 30 V, desolvation gas flow: 600 L·min−1; cone gas flow: 50 L·min−1. Chromatography (flow rate 1 mL·min−1) was achieved using a Phenomenex Kinetex column (Phenomenex, Macclesfield, UK) (2.6 μ, C18, 100 Å, 4.6 × 50 mm) equipped with a Phenomenex Security Guard precolumn (Luna C5 300 Å). Solvents were: A, HPLC grade H2O containing 0.05% formic acid; B, HPLC grade CH3CN containing 0.05% formic acid. Gradient elution was performed as follows: 0 min, 90% A, 7 minutes, 5% A, 8.5 minutes, 5% A; 8.6 minutes, 90% A; 10 minutes, 90% A. SRPIN340 eluted at 5.9 minutes, and MVRL06 at 4.4 minutes. Peaks occurring at these times in the SIM chromatograms for each compound were integrated using Waters MassLynx software and used to quantify SRPIN340.
Human Ether-a-Go-Go Related Gene (hERG) Assay
Whole cell patch clamp recordings of hERG current (IhERG) were made at 37°C from human embryonic kidney cells (HEK 293) cells stably expressing hERG (generously donated by Craig January34). Cells were maintained in culture and prepared for electrophysiologic recording as described previously.35,36 Cells were superfused with a normal Tyrode's solution containing (in mM) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 Glucose, and 5 HEPES (titrated to pH 7.45 with NaOH). Patch-pipettes were filled with a K+-based dialysate containing (in mM): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, and 10 HEPES (titrated to pH 7.2 with KOH). Pipette resistance typically ranged from 1.5 to 3.5 MΩ and approximately 70% to 80% series resistance could be compensated. Data were acquired using an Axopatch (Axon Instruments [now Molecular Devices], Sunnyvale, CA) 1D amplifier and a CV-4 1/100 headstage and digitized using a Digidata 1200B interface (Axon Instruments). Voltage commands were generated and data acquired using Clampex 8 (Axon Instruments) and data were stored on the hard-disk of a computer (Viglen Ltd, St. Albans, Hertfordshire, UK) for off-line analysis. Data were digitized at 10 kHz with an appropriate bandwidth set on the amplifier. Compounds were applied using a home-built rapid solution exchange device capable of changing superfusate in less than 1 second.
Statistical Analyses
If not indicated otherwise, data are shown as mean ± SEM. All data, graphs, and statistical analyses were calculated with Microsoft Excel (Microsoft Office Software, Seattle, WA), GraphPad Prism (GraphPad Software Inc., La Jolla, CA) and Image J. All results were considered statistically significant at P less than 0.05 (*), P less than 0.01 (**), and P less than 0.001 (***).
Results
Identification of Novel SRPK1 Inhibitors
To identify novel inhibitors for SRPK1 and highly related isoforms SRPK2 and SRPK3, a range of inhibitors were screened in an in vitro kinase assay (Promega32) as well as temperature shift assays.37 The previously identified SRPK inhibitor SRPIN34038 was used as a positive control for the identification of novel candidate compounds, SPHINX (Sr PHosphorylation Inhibitor X) and MVRL09 (N-[2-(morpholin-4-yl)-5-(trifluromethyl) phenyl] pyridine-3-carboxamide), which were tested for SRPK1 activity with the structural derivative of SRPIN340 lacking the trifluoromethyl group, SRPIN349, was used as a negative control (Fig. 1A). SRPIN340, MVRL09, and SPHINX all showed some ability to inhibit the SRPK1 kinase, MVRL09 being the least effective with an IC50 greater than 10 μM. Both SRPIN340 and SPHINX inhibited SRPK1 activity with IC50 values of 0.96 μM and 0.58 μM, respectively (Fig. 1B). The results gained in this assay are consistent with the published IC50 for SRPIN340 (0.89 μM38). Subsequently, the ability of these compounds to inhibit SRPK2 activity was investigated. SRPIN340 inhibited SRPK2 activity with an IC50 of 7.4 μM. MVRL09 showed no inhibition of SRPK2 and SPHINX showed only 10% kinase inhibition at 10 μM (Fig. 1C). All three compounds preferentially target SRPK1 over SRPK2, but SPHINX was the most selective, exhibiting a greater than 10-fold preference for SRPK1. SPHINX was tested against a panel of 58 kinases using temperature shift assays and at 10 μM a temperature shift of greater than 2°C was seen only for SRPK1 (9.82°), SRPK3 (7.93°C), and SRPK2 (3.5°C). All 55 other kinases had less than 2°C shift and 52 of them less than 1°C shift (see Supplementary Table S1).
Figure 1.

(A) Structures of SRPIN340, MVRL09, and SPHINX. (B) Purified SRPK1 protein, the RS peptide of SRSF1 and ATP (1 μM) were used to assess SRPK1 activity. SRPIN340 and SPHINX dose dependently suppressed SRPK1 kinase activity with an IC50 of 0.96 μM and 0.88 μM, respectively. (C) Purified SRPK2 protein, RS peptide, and ATP were used to assess SRPK2 activity. SRPIN340 inhibited SRPK2 activity with an IC50 of 7.4 μM, whereas SPHINX and MVRL09 failed to reach 50% inhibition at 10 μM.
The SRPK inhibitors were tested in a series of in vitro assays to determine the effect of treatment on SR-protein phosphorylation and VEGF isoform expression. Initially, the effect of compound treatment on SR-protein phosphorylation was determined. Previous publications had suggested SRSF1 phosphorylation by SRPK1 leads to the nuclear localization of SRSF1 enabling its binding to VEGF pre-mRNA where it can facilitate splicing.28,29 We observed SR protein phosphorylation could be activated by cell treatment with epidermal growth factor (EGF), as previously described39 (Fig. 2Ai). Pre-incubation of cells with either 10 μM SRPIN340 (P < 0.05) or SPHINX (P < 0.05) blocked EGF-induced phosphorylation of SRSF1 and SRSF2, whereas MVRL09 did not elicit a significant effect. In addition administration of SRPK1 inhibitors without EGF activation significantly reduced SR-protein phosphorylation (Fig. 2Aii).
Figure 2.

Effect of SRPK1 inhibitors on VEGF and SR protein phosphorylation. (A) ARPE-19 cells were treated with 10 μM SRPIN340, MVRL09, SPHINX, or left untreated, and subsequently either treated or not with EGF. Extracted protein was run on Western blots for phospho-SR proteins. (A) Bands were observed at molecular weights corresponding to SRSF1/2, SRSF4, and SRSF6, densitometry analysis was undertaken on SRSF1/2. SRPK inhibition significantly reduced the endogenous phosphorylation of SRSF1/2, moreover pre-incubation of cells with either SRPIN340 or SPHINX significantly blocked an EGF-induced increase in SR protein phosphorylation. (B) Serum-starved primary RPE cells and ARPE-19 cells were treated with inhibitors at 5 μM. Twenty-four hours later, RNA was extracted and cDNA was made. PCR was performed with primers spanning VEGF exon 7b and 8b. Treatment with the inhibitors reduced the expression of VEGF165 relative to GAPDH control either in primary RPE cells (SRPIN340) or both cell lines (SPHINX and MVRL09). Water (W) and sample without RT were used as negative controls. (C) Forty-eight hours after treatment, protein was extracted and assayed for total VEGF by ELISA. Protein was assessed using a VEGFxxxb-specific ELISA and the ratio of VEGFxxxb over total VEGF calculated. The ratio increased in ARPE-19 during treatment with SPHINX and SRPIN340, and in primary RPE during treatment with SPHINX and MVRL09. Total VEGF expression relative to total protein was also calculated. In primary RPE, VEGF expression was reduced following SRPIN340 and MVRL09 treatment, in ARPE-19, VEGF expression was increased following SPHINX and MVRL09 treatment (One-way ANOVA, Bonferroni post hoc. *P < 0.05, **P < 0.01).
Inhibitor treatment for 24 hours at 5 μM reduced the expression of VEGF165 mRNA isoforms relative to GAPDH in primary RPE and ARPE-19 cells (Fig. 2Bi). All three inhibitors reduced the ratio of VEGF165 isoforms relative to VEGF165b isoforms either in primary RPE, ARPE-19, or both cell lines (Figure 2Bii).
We went onto investigate the effect of SRPK1 inhibitors on VEGF protein expression. In primary RPE cells VEGFxxxb expression was increased following SPHINX and MVRL09 treatment, and total VEGF was reduced in the presence of SRPIN340 and MVRL09 mediated by a reduction in VEGFxxx. In ARPE-19 cells VEGFxxxb expression was increased following SRPIN340 and SPHINX treatment, and total VEGF was increased in the presence of SPHINX and MVRL09 suggesting that in this cell type SRPIN340 acts by reducing pro-angiogenic VEGF whereas SPHINX promoted VEGFxxxb expression (Figs. 2Ci–ii).
To determine whether SRPK1 inhibition prevented CNV in animal models, we tested all three compounds in parallel in a laser-induced mouse CNV model. Two intravitreal injections of 10 ng SRPIN340 and SPHINX showed that both inhibitors significantly reduced the neovascular area compared to control injected eyes (P < 0.05, Fig. 3A). MVRL09, the least potent SRPK1 inhibitor, failed to reduce CNV lesion area and had variable effects on VEGF protein expression so was excluded from further investigation.
Figure 3.

SRPK inhibitors are antiangiogenic. (A) Fifteen C57/B6 mice were subjected to laser photocoagulation (IRIDEX Oculight GLX λ, 810 nm, 250 mV, 0.1 second, 75 μm, 4 lesions/eye). On day 0 and day 7 mice received an intraocular (i.o.) injection of SRPIN340, MVRL09, SPHINX (10 ng, 5 ng/μL–2 μL), or saline in the control eye. SRPIN340 and SPHINX both significantly reduced neovascular growth compared with saline-injected controls. Scale bar: 100 μm. (Bi) Norway Brown rats, after induction of CNV, were injected with either 25 ng (10 ng/uL) SRPIN340, 25 ng (10 ng/ul) SPHINX, 1 μg of the anti-mouse VEGF antibody, and saline in the control eye directly after laser procedure (day 0) and on day 7. Staining with isolectin-B4 of the RPE-choroid-sclera complex showed a decrease in lesion size for anti-VEGF and a significant decrease with SRPIN340 and SPHINX. Examples of lesions are shown. Scale bar: 200 μm. (P < 0.05, One-way ANOVA, Bonferronni post hoc). N, number of eyes. (Bi-i-iii) Rats were culled on day 4 (for protein). Protein extracted from retinae four days after laser induction was subjected to Western blot for phospho-SR proteins (Bi-i) or panVEGF (A20) (Bi-ii). SPHINX significantly reduced SRSF1/2 phosphorylation and both SRPIN340 and SPHINX significantly reduced the expression of total VEGF in the retina compared with saline injected eyes, but failed to alter the expression of VEGFxxxb isoforms.
To determine whether SRPIN340 or SPHINX alters VEGF expression in the retina of animals, in which CNV was reduced, a rat model of laser-induced CNV was used, as mouse monoclonal antibodies to VEGF isoforms cannot be easily used in mouse tissues, due to the potential detection of mouse IgG (which runs similarly to VEGF). SRPIN340 and SPHINX were tested alongside a VEGF antibody (Roche, Basel, Switzerland). SRPIN340 or SPHINX injection (25 ng) significantly reduced the CNV area compared with saline injected controls (P < 0.05). The VEGF antibody resulted in a nonsignificant effect on CNV area in this rat model (Fig. 3Bi), although it did result in a reduction in a mouse CNV model.40 Retinal proteins from SRPIN340 and SPHINX-treated eyes 4 days after initial treatment were assessed for SR-protein phosphorylation and VEGF expression by Western blot and compared with vehicle-control injected eyes. A decrease in the detection of SR-protein phosphorylation was observed following SPHINX treatment compared with control, whereas limited n-numbers meant that we were unable to calculate significance for SRPIN340-treated eyes (Fig. 3Bi-i). Decreased expression of panVEGF was observed in both SRPIN340 (P < 0.05) and SPHINX (P < 0.05)-treated eyes but no difference in the expression of VEGFxxxb isoforms was observed compared with vehicle (Figs. 3Bi–ii).
Both previously published SRPIN340 and the novel compound SPHINX used in this study have been equivalent in terms of SRPK1 potency, inhibition of SR proteins phosphorylation, reduction in the expression of pro-angiogenic VEGF, and suppression of CNV formation.
To determine the potency of SRPK1 inhibition in preventing laser-induced CNV, the most extensively characterized SRPK1 inhibitor, SRPIN340, was used in the mouse in a dose-escalation study. SRPIN340 dose-dependently reduced CNV area compared with control-injected eyes with an EC50 of 1.28 ng total injected dose (Fig. 4A). We went on to determine pharmacokinetics of SRPIN340 in the eye following intravitreal injection. Injection of 20 ng resulted in a concentration of greater than 10 ng/mL in the eye over a 2 day period. Curve fitting to the concentrations measured at 1, 4, 8, 24, and 48 hours gave an estimated maximal sustained half-life of 22 hours (Fig. 4B). Although SPHINX was not tested in this trial based on similar structural properties and in vivo effects, we hypothesize that the two compounds (SRPIN340 and SPHINX) will display similar pharmacokinetic profiles.
Figure 4.

SRPK inhibition dose dependently inhibits choroidal angiogenesis. (A) Twenty-two C57/B6 mice were subjected to laser photocoagulation (IRIDEX Oculight GLX λ, 810 nm, 250 mV, 0.1 second, 75 μm, 4 lesions/eye). On day 0 and day 7 mice received i.o. injection of SRPIN340 (5, 3, 1, 0.5, 0.1 ng/μL, 2 μL) or saline in the control eye. SRPIN340 dose-dependently inhibited laser-induced neovascular growth compared with saline controls (P < 0.01, two-way ANOVA) reaching statistical significance at 5 ng/μL (P < 0.01) with an EC50 of 0.32 ng/μL, total injection of 1.28 ng. Scale bar: 50 μm. (B) Twenty nanograms of SRPIN340 was injected intravitreally into the left and right eyes of 10 CD-1 mice. Two mice were culled at each of the following time points; 1, 4, 8, 24, and 48 hours, eyes were removed, tissue homogenized, proteins precipitated out, and sample lysate analyzed by mass spectrometry. SRPIN340 expression in the eye tissue decreased over time with a sustained half-life of 22 hours (one-phase exponential decay, prism).
Based on their low molecular weight and favorable drug qualities, we hypothesized that SRPIN340 (MW349, LogP 3.68, and satisfies Lipinski's rule of 5) or SPHINX (MW351, LogP 3.09, and satisfies Lipinski's rule of 5) may be capable of preventing VEGF-mediated CNV when administered topically as an eye drop. We performed a single dose test of 10 μg/mL SRPIN340 or SPHINX and compared them with a vehicle of saline + 0.05% DMSO. Twice daily topical SRPIN340 (P < 0.05) and SPHINX (P < 0.05) significantly reduced CNV lesion area compared with vehicle-treated eyes (Fig. 5).
Figure 5.
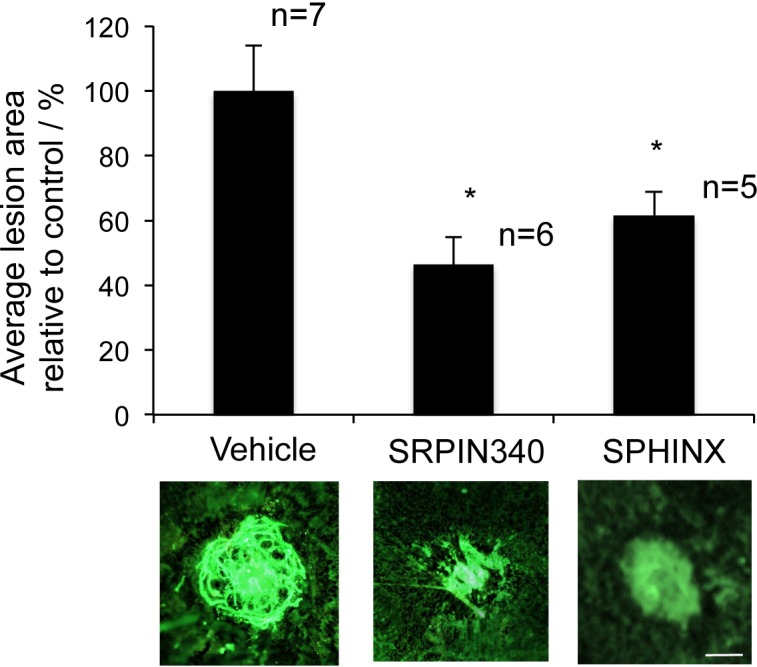
SRPK inhibition as eye drops can inhibit choroidal neovascularization. Ten C57/B6 mice were subjected to four laser-lesions (250 mW; 75 μm; 01 second; 810 nm) to the back of the eye on day 0. From day 1 to 14 mice received twice daily topical treatments of 10 μg/mL SRPIN340 or SPHINX (10 μL volume per drop) in the contralateral eye and vehicle control in the ipsilateral eye. On day 14 mice were culled, eyes enucleated, and the sclerochoroidal complexes stained for isolectin-B4. Flatmounted choroids were imaged and analyzed (ImageJ). SRPIN340 and SPHINX significantly inhibited CNV formation. *P < 0.05 compared with vehicle. Scale bar: 100 μm.
As previously, SRPIN340 was selected to determine the potency of topical eye drops in a dose escalation study. SRPIN340 was dissolved in a previously described drug delivery vehicle (Doukas et al.33) and administered twice daily. Fourteen days following laser insult, animals were killed by cardiac puncture, and blood collected for mass spectrometry analysis. Choroids were fixed and flatmounted for isolectin staining and retinae were extracted for RNA analysis. Topical SRPIN340 drops dose-dependently reduced CNV lesion area with an EC50 of 640 ng total dose, 500 times the concentration required to achieve the same effect following SRPIN340 intravitreal injection (Fig. 6A). SRPIN340 was not detected systemically by mass spectrometry after 14 days of twice daily eye drops (data not shown). Furthermore, VEGF165 mRNA expression in the retinae was reduced following SRPIN340 treatment at 100 μg/mL, but no significant effect was observed following 10 and 1 μg/mL SRPIN340 relative to mouse globin internal control (Fig. 6B).
Figure 6.

SRPIN340 as eye drops inhibited choroidal neovascularization dose dependently. Twelve C57/B6 mice were subjected to four laser-lesions (250 mW; 75 μm; 01 second; 810 nm) to the back of the eye on day 0. From day 1 to 14 mice received 20 topical treatments of 10 μL SRPIN340 in a drug delivery vehicle at varying doses (100–0.1 μg/mL) in the contralateral eye and vehicle control in the ipsilateral eye. On day 14 mice were culled, eyes enucleated, and the sclerochoroidal complexes stained for isolectin-B4. Retinal protein was taken for RNA analysis. (A) Flatmounted choroids were imaged and analyzed (ImageJ). SRPIN340 demonstrated dose-dependent inhibition of neovascularisation with an EC50 of 3.187 ug/ml. Images show a vehicle-treated lesion and a 100 μg/mL SRPIN340-treated lesion. (B) VEGF165 mRNA expression relative to mouse globin was significantly reduced in 100 μg/mL SRPIN340-treated eyes compared with saline but not at 10 μg/mL or 1 μg/mL (P < 0.05; one-way ANOVA, Bonferroni post hoc). (C) Five micrograms of SRPIN340 in 10 μL was dropped on the left and right eyes of 10 CD-1 mice. Two mice were culled at each of the following time points; 1, 4, 8, 24, and 48 hours, eyes were removed, washed, and dissected into anterior and posterior compartments. Tissue was homogenized, proteins precipitated out, and sample lysate analyzed by mass spectrometry. SRPIN340 was detected in the eye after 1 hour at 175 ng/mL, 3.5% of the total applied dose. Expression in the eye tissue decreased over time with an initial half-life of 1 hour in the whole eye (one-phase exponential decay, prism). (D) The half-life in the posterior chamber was 35 minutes, and at 12 hours 0.15% of the applied dose was detected.
To confirm SRPIN340 was detectable in the eye following topical administration mass spectrometry was performed. A single drop of 5 μg in 10 μL vehicle SRPIN340 was administered to one eye of CD-1 mice and a control vehicle was administered to the other. After 1, 4, 8, 24, and 48 hours mice were culled and eye tissue from the anterior and posterior chamber of the eye was separately analyzed for SRPIN340 expression. SRPIN340 was detected both in the whole eye and specifically in the posterior chamber. After 1 hour 3.5% of the total applied dose was detected in the eye (Fig. 6C). In the posterior chamber, composed of retina, sclerochoroidal complex and residual vitreous, 0.15% of the applied dose was detected after 12 hours (Fig. 6D). Exponential decay analysis using prism software generated an initial half-life in the posterior chamber of 35 minutes.
The development of small molecule inhibitors for use in humans requires that they have minimal effects on the activity of the hERG cardiac potassium channel, as pharmacologic inhibition of this channel can lead to QT interval prolongation on the electrocardiogram, with its associated risk of potentially fatal torsades de pointes arrhythmia.41 To determine whether these compounds had any activity on hERG, the current passing through the hERG channel (IhERG) was measured. The protocol used for IhERG pharmacology (shown in the lower panels of Figs. 7A, 7B) is identical to that used in prior studies36,42,43. The amplitude of IhERG ‘tails' was measured relative to the instantaneous current elicited by a brief (50 ms) prepulse to −40 from −80 mV. During 2 minutes of exposure to vehicle control there was little decline in IhERG amplitude (Figs. 7A, 7C, 7D). At the end of this period, cells were exposed to 3 μM of the Class Ia antiarrhythmic quinidine, which is well established to inhibit IhERG with a submicromolar IC50 value.42,43 This led to a rapid reduction in IhERG, blocking the tail current by approximately 75% (Figs. 7A, 7C, 7D). In separate experiments, cells were exposed to 10 μM of either SRPIN340 or SPHINX. Figure 7B shows representative traces from an experiment in which SPHINX was applied, whilst Figure 7C shows mean time-course data for both SPHINX and SRPIN340, superimposed with vehicle control data. Two minutes of perfusion of 10 μM SRPIN340 reduced the hERG tail current (Itail) by 19.87 ± 1.50% (n = 5 cells). Ten μM SPHINX reduced Itail by 10.43 + 1.42% (n = 5 cells). 0.02% DMSO blocked Itail by 2.58 ± 0.56% (n = 4 cells). Three micrometres of quinidine blocked Itail by 74.49 ± 2.92%. Thus, SPHINX at 10 μM (17-fold greater than the IC50 for SRPK1), produced only a small (∼10%) reduction in IhERG amplitude, whilst SRPIN340 produced a slightly larger (∼20%) reduction in current. Whilst these effects were significantly different from the vehicle control (P < 0.05; one-way ANOVA with Bonferroni post-test), they represent only modest effects of the compounds on IhERG at concentrations far exceeding those effective against their intended target and were much smaller than the positive control value for quinidine (P < 0.001 versus both SRPIN340 and SPHINX).
Figure 7.

(A) Example hERG current (IhERG) traces in vehicle control (0.02% DMSO, black line), after 2 minutes perfusion of vehicle control solution (grey line) and in the presence of 3 μM quinidine (light grey line). The lower panel shows the voltage protocol. (B) Representative IhERG records in vehicle control (black line) and in the presence of 10 μM SPHINX (grey line) elicited by the protocol shown in the lower panel. (C) Mean data (± SEM) showing the continuous time-plot of effect of 10 μM SPHINX (solid circle, n = 5 cells) and 10 μM SRPIN340 (open circle, n = 5 cells) on Itail. The solid square plot shows the effect of 0.02% DMSO on Itail as vehicle control and 3 μM quinidine as positive control (n = 4 cells). For each cell, the hERG tail current amplitude was normalized to the maximal Itail observed in control. (D) Bar chart showing the mean fractional IhERG tail inhibitory levels produced by 10 μM SRPIN340, 10 μM SPHINX, vehicle control, and 3 μM quinidine.
Discussion
We have used three distinct small molecule inhibitors of SRPK1, all containing a trifluoromethyl phenyl group, but with differing primary groups (disubstituted furan-morphonyl [SPHINX], piperidinyl-isonicotinamide [SRPIN340], and morphonyl-nicotinamide [MVRL09]) attached to the phenyl ring to reduce the expression of pro-angiogenic VEGF165 and inhibit CNV in vivo. The trifluoromethyl carboxamide group was necessary, but not sufficient alone, to inhibit SRPK1, as SRPIN349, which lacks the trifluoromethyl group, did not inhibit SRPK1.
The kinase catalytic domain of SRPK family members are characterized by unique domain insert in the kinase hinge region. Structures of SRPK kinases revealed that a helix of this SRPK insertion domain packs parallel to the kinase hinge region offering the possibility to anchor inhibitors to this SRPK-specific structural feature. The developed inhibitors used this structural feature explaining the high specificity of these compounds for SRPK1 and SRPK2.
The interaction of SRSF1 with SRPK1 is well described in the literature.44–46 SRSF1 is a proto-oncogene that regulates the alternative splicing of numerous genes, including VEGF.30 Here, we show administration of both SRPIN340 and SPHINX to ARPE-19 cells, reduced phosphorylation of SR-proteins, and blocked phosphorylation induced by EGF treatment. EGF initiates a signaling pathway acting through Akt whereby SR-proteins are phosphorylated by SRPK1.38 The third, less potent inhibitor, MVRL09 failed to significantly alter SR-protein phosphorylation. Furthermore, in both primary RPE and ARPE-19 cells all three SRPK1 inhibitors reduced pro-angiogenic VEGF expression at mRNA level. At a protein level, the results were more variable showing SRPIN340 reduced pro-angiogenic VEGF in primary RPE and increased VEGFxxxb in ARPE-19, SPHINX increased VEGFxxxb expression in both primary RPE and ARPE-19, and MVRL09 reduced pro-angiogenic VEGF, and promoted VEGFxxxb in primary RPEs but increased pro-angiogenic VEGF in ARPE-19 cells. We observed a consistent reduction in the pro-antiangiogenic VEGF ratio and decrease in SR-protein phosphorylation following SRPIN340 and SPHINX treatment.
When tested in vivo in a mouse laser-induced CNV model SRPIN340 and SPHINX but not MVRL09, significantly reduced the area of neovascular lesions. MVRL09 was the least potent SRPK inhibitor, unable to significantly alter SR-protein phosphorylation, had variable effects on VEGF expression and failed to reduce CNV in vivo. Based on these results MVRL09 was excluded from further investigation.
Although a change in the balance of VEGF isoforms was observed during SRPK1 inhibition in human primary RPE and ARPE-19 cell lines, expression of VEGFxxxb isoforms remains to be definitively shown in mouse tissues. A recent study has questioned the existence of VEGFxxxb isoforms,50 suggesting that they are rarely expressed in mice, and difficult to detect. We, and others, have demonstrated clearly and repeatedly that expression of the VEGF isoforms in humans and rats is relatively straightforward, using protocols in which positive and negative controls are employed to ensure that the methodologies used are effective. In mouse tissues, VEGF165b mRNA (detected by qPCR47–49) and protein (identified by Western blot48) expression have been described, although, in mice it is critical to include stringent controls (to exclude IgG contamination with a mouse antibody on mouse tissue, or cross amplification by using insufficiently stringent annealing conditions50). It has been suggested that the alternative splicing of VEGF transcripts increases during evolution with VEGF165b expression being very low in healthy mice increasing through rabbits and pigs to highest expression in primates47 and interestingly, anti-VEGF165b antibodies are pro-angiogenic in rat and mouse development,49,51 indicating a role for these isoforms in rodents. Thus, in the mouse model, SRPK1 inhibitors may prevent CNV by reducing the expression of pro-angiogenic VEGF isoforms either by preventing the use of the proximal splice site, resulting in reduction of mature RNA, or through indirect alterations in transcriptional or translational regulation. Currently, no investigations have identified the regulators of VEGF alternative splicing in mice.
To confirm if the results achieved in mice are reproducible in other models a laser-induced rat CNV model was performed. SRPK inhibition by SRPIN340- and SPHINX-reduced CNV area, SRSF1/2 phosphorylation and total VEGF protein expression in rat ocular tissues compared with vehicle-injected controls. VEGFxxxb expression was investigated in the rat retinae, although VEGFxxxb was detected, no change in expression was observed during SRPIN340 and SPHINX treatment compared with saline controls at that time point, suggesting in rodent models SRPK1 inhibition may be antiangiogenic by reducing pro-angiogenic VEGF expression.
We went on to determine the EC50 of one of our investigated compounds, SRPIN340, on CNV area in the mouse model. SRPIN340 was selected over SPHINX for this dose-escalation study as both compounds were equivalent in our studies, but SRPIN340 had previously been more characterized as an SRPK1 inhibitor. SRPIN340 produced an EC50 of 1.28 ng and the dose range of SRPIN340 used, 0.2 to 10 ng/eye, was similar to the dose of rhVEGF165b previously used to inhibit CNV.20 Hua and colleagues determined an effective rhVEGF165b dose in humans of 3 μg/eye, 1000- to 10-fold lower than ranibizumab or bevacizumab.24 It is difficult to directly compare human-specific ranibizumab to the SRPIN340 efficacy data in mouse, but compared with rhVEGF-A165b, SRPIN340 was 160 times less potent,20 but required a dose 125 times less than pegaptanib in rodents.52 The data suggests SRPK inhibition by either SRPIN340 or SPHINX has the potential to achieve similar reductions in CNV to current treatment of ranibizumab, but at lower doses following intravitreal injection. One of the drawbacks of ranibizumab treatment is the need for monthly intraocular injections, thus, we investigated whether SRPK inhibition could potently inhibit CNV following topical administration. Twice daily topical administration of either SRPIN340 or SPHINX significantly suppressed CNV in mice at 10 μg/ml. Once again SRPIN340 was selected as an example compound for a topical dose-response trial in mice. SRPIN340 showed an EC50 of 3.19 μg/mL, 500 times greater than injected SRPIN340. Pharmacokinetic analysis suggested 3.5% of the topically applied dose was detected in the eye after 1 hour reflecting previous estimations that due to poor ocular bioavailability only 5% of a topically applied drug will get into the eye.53 Furthermore, this SRPK inhibitor was still detectable in the eye 48 hours after a single topical administration, but was not detected systemically. Despite only approximately 5% of applied SRPIN340 being likely to penetrate the eye the dose required to achieve 50% CNV inhibition using topical SRPIN340 compares favorably with other topical therapies. In 2008, Doukas and colleagues33 identified TG100801 an inactive prodrug that generates TG100572 by de-esterification. TG100801 targets Src kinases and selected tyrosine receptor kinases including VEGFR-1 and VEGFR-2, twice daily topical application of 1%, but not 0.6% TG100801 significantly reduced CNV lesion area,33 however, 0.01% SRPIN340 solution in the same drug delivery vehicle produced a significant inhibition of CNV area. To our knowledge SRPIN340 is the first topical compound to alter the expression of pro-angiogenic VEGF isoforms. Following the dose-response trial of topical SRPIN340, mouse retinae were assessed for VEGF165 expression and showed a significant reduction in VEGF165 at 100 μg/mL but not at 10 μg/mL or 1 μg/mL relative to mouse globin expression. VEGF165, the more potently mitogenic VEGF isoform54 is sufficient for physiologic neovascularization during retinal development.55 This study has focused on investigating VEGF165 expression following SRPK1 inhibition; however, we predict the expression other pro-angiogenic VEGF isoforms such as VEGF121 will also be reduced following SRPK1 inhibition. Indeed, it has been shown that SRSF1 binds to human VEGF pre-mRNA in a region 35 nucleotides prior to the VEGF exon eight PSS, a splice site conserved both across species and by all pro-angiogenic VEGF isoforms.28 Although we have demonstrated a reduction in pro-angiogenic VEGF during SRPK1 inhibition it is possible that other genes involved in angiogenesis are also affected by SRPK1 inhibition. In particular, SRSF1 regulates the alternative splicing of numerous genes, several of which are involved in angiogenesis. For example, SRSF1 mediated alternative splicing of both Ron and TEAD-1, have been linked to angiogenic growth factor expression and VEGF expression, respectively.56,57 SRSF1 may, thus, impact VEGF expression through both direct regulation and indirect regulation through other as yet undetermined pathways.
The data presented in this study suggest that SRPIN340 and a novel furan molecule are potent and selective SRPK1 inhibitors and are effective in reducing pro-angiogenic VEGF-mediated CNV associated with AMD. Furthermore, we have shown that SRPK inhibition is effective at reducing CNV following topical administration in mice, suggesting that topical administration of SRPK inhibitors may be a novel therapeutic strategy for exudative AMD.
Supplementary Material
Supplemental Data
Acknowledgments
Supported by grants from Fight for Sight, Skin Cancer Research Fund, the Richard Bright VEGF Research, the British Heart Foundation (PG11/20/28792), MRC grant numbers G10002073, MR/K020366/1, MR/K013157/1, and BBSRC BB/J007293/1, and the Japanese Society for the Promotion of Science. Heart Research UK (HRUK RG2594/11/13 [CD and JCH]). The SGC, a registered charity (number 1097737 [Sk and OF]) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, Takeda, AbbVie, Boehringer Ingelheim, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation and the Wellcome Trust (092809/Z/10/Z).
Disclosure: M.V. Gammons, P; O. Fedorov, None; D. Ivison, None; C. Du, None; T. Clark, None; C. Hopkins, None; M. Hagiwara, None; A.D. Dick, None; R. Cox, None; S.J. Harper, P; J.C. Hancox, None; S. Knapp, None; D.O. Bates, P
References
- 1.Ferris FL III, Fine SL, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy.Arch Ophthalmol. 1984; 102: 1640–1642 [DOI] [PubMed] [Google Scholar]
- 2.Campochiaro PA, Nguyen QD, Shah SM, et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: results of a phase I clinical trial.Hum Gene Ther. 2006; 17: 167–176 [DOI] [PubMed] [Google Scholar]
- 3.Fine SL, Berger JW, Maguire MG, Ho AC. Age-related macular degeneration.N Engl J Med. 2000; 342: 483–492 [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis.Am J Pathol. 1995; 146: 1029–1039 [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye.Am J Ophthalmol. 2002; 134: 411–431 [DOI] [PubMed] [Google Scholar]
- 6.Das A, Fanslow W, Cerretti D, Warren E, Talarico N, McGuire P. Angiopoietin/Tek interactions regulate mmp-9 expression and retinal neovascularization.Lab Invest. 2003; 83: 1637–1645 [DOI] [PubMed] [Google Scholar]
- 7.Shima DT, Adamis AP, Ferrara N, et al. Hypoxic induction of endothelial cell growth factors in retinal cells: identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen.Mol Med. 1995; 1: 182–193 [PMC free article] [PubMed] [Google Scholar]
- 8.Spilsbury K, Garrett KL, Shen WY, Constable IJ, Rakoczy PE. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization.Am J Pathol. 2000; 157: 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen.Science. 1989; 246: 1306–1309 [DOI] [PubMed] [Google Scholar]
- 10.Jingjing L, Xue Y, Agarwal N, Roque RS. Human Müller cells express VEGF183, a novel spliced variant of vascular endothelial growth factor.Invest Ophthalmol Vis Sci. 1999; 40: 752–759 [PubMed] [Google Scholar]
- 11.Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA.Mol Endocrinol. 1991; 5: 1806–1814 [DOI] [PubMed] [Google Scholar]
- 12.Mineur P, Colige AC, Deroanne CF, et al. Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents.J Cell Biol. 2007; 179: 1261–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tischer E, Mitchell R, Hartman T, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing.J Biol Chem. 1991; 266: 11947–11954 [PubMed] [Google Scholar]
- 14.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors.FASEB J. 1999; 13: 9–22 [PubMed] [Google Scholar]
- 15.Bates DO, Cui TG, Doughty JM, et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma.Cancer Res. 2002; 62: 4123–4131 [PubMed] [Google Scholar]
- 16.Woolard J, Wang WY, Bevan HS, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression.Cancer Res. 2004; 64: 7822–7835 [DOI] [PubMed] [Google Scholar]
- 17.Perrin RM, Konopatskaya O, Qiu Y, Harper S, Bates DO, Churchill AJ. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor.Diabetologia. 2005; 48: 2422–2427 [DOI] [PubMed] [Google Scholar]
- 18.Varey AH, Rennel ES, Qiu Y, et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy.Br J Cancer. 2008; 98: 1366–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pritchard-Jones RO, Dunn DB, Qiu Y, et al. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma.Br J Cancer. 2007; 97: 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hua J, Spee C, Kase S, et al. Recombinant human VEGF165b inhibits experimental choroidal neovascularization.Invest Ophthalmol Vis Sci. 2010; 51: 4282–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magnussen AL, Rennel ES, Hua J, et al. VEGF-A165b is cytoprotective and antiangiogenic in the retina.Invest Ophthalmol Vis Sci. 2010; 51: 4273–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gragoudas ES, Adamis AP, Cunningham ET Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration.N Engl J Med. 2004; 351: 2805–2816 [DOI] [PubMed] [Google Scholar]
- 23.Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration.N Engl J Med. 2006; 355: 1419–1431 [DOI] [PubMed] [Google Scholar]
- 24.Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration.N Engl J Med. 2006; 355: 1432–1444 [DOI] [PubMed] [Google Scholar]
- 25.Brown DM, Michels M, Kaiser PK, Heier JS, Sy JP, Ianchulev T. Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study.Ophthalmology. 2009; 116: 57–65 e55 [DOI] [PubMed] [Google Scholar]
- 26.Good TJ, Kimura AE, Mandava N, Kahook MY. Sustained elevation of intraocular pressure after intravitreal injections of anti-VEGF agents.Br J Ophthalmol. 2010; 95: 1111–1114 [DOI] [PubMed] [Google Scholar]
- 27.Jager RD, Aiello LP, Patel SC, Cunningham ET Jr. Risks of intravitreous injection: a comprehensive review.Retina. 2004; 24: 676–698 [DOI] [PubMed] [Google Scholar]
- 28.Amin EM, Oltean S, Hua J, et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing.Cancer Cell. 2011; 20: 768–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowak DG, Amin EM, Rennel ES, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis.J Biol Chem. 2010; 285: 5532–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanford JR, Ellis JD, Cazalla D, Caceres JF. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor.Proc Natl Acad Sci U S A. 2005; 102: 15042–15047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nowak DG, Woolard J, Amin EM, et al. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by known splicing and growth factors.J Cell Sci. 2008; 121: 3487–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koresawa M, Okabe T. High-throughput screening with quantitation of ATP consumption: a universal non-radioisotope, homogeneous assay for protein kinase.Assay Drug Dev Technol. 2004; 2: 153–160 [DOI] [PubMed] [Google Scholar]
- 33.Doukas J, Mahesh S, Umeda N, et al. Topical administration of a multi-targeted kinase inhibitor suppresses choroidal neovascularization and retinal edema.J Cell Physiol. 2008; 216: 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, Gong Q, Ye B, et al. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature.Biophys J. 1998; 74: 230–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng H, Zhang Y, Du C, Dempsey CE, Hancox JC. High potency inhibition of hERG potassium channels by the sodium-calcium exchange inhibitor KB-R7943.Br J Pharmacol. 2012; 165: 2260–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du CY, El Harchi A, Zhang YH, Orchard CH, Hancox JC. Pharmacological inhibition of the hERG potassium channel is modulated by extracellular but not intracellular acidosis.J Cardiovasc Electrophysiol. 2011; 22: 1163–1170 [DOI] [PubMed] [Google Scholar]
- 37.Federov O, Niesen FH, Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. In: Kuster B. ed_Kinase Inhibitors: Methods and Protocols_. New York: Humana Press; 2011: 109–118 [DOI] [PubMed] [Google Scholar]
- 38.Fukuhara T, Hosoya T, Shimizu S, et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication.Proc Natl Acad Sci U S A. 2006; 103: 11329–11333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Qiu J, Liu W, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus.Mol Cell. 2012; 47: 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rennel ES, Regula JT, Harper SJ, Thomas M, Klein C, Bates DO. A human neutralizing antibody specific to Ang-2 inhibits ocular angiogenesis.Microcirculation. 2011; 18: 598–607 [DOI] [PubMed] [Google Scholar]
- 41.Hancox JC, McPate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes.Pharmacol Ther. 2008; 119: 118–132 [DOI] [PubMed] [Google Scholar]
- 42.El Harchi A, Melgari D, Zhang YH, Zhang H, Hancox JC. Action potential clamp and pharmacology of the variant 1 Short QT Syndrome T618I hERG K(+) channel.PLoS One. 2012; 7: e52451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McPate MJ, Duncan RS, Hancox JC, Witchel HJ. Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs.Br J Pharmacol. 2008; 155: 957–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aubol BE, Chakrabarti S, Ngo J, et al. Processive phosphorylation of alternative splicing factor/splicing factor 2.Proc Natl Acad Sci U S A. 2003; 100: 12601–12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngo JC, Chakrabarti S, Ding JH, et al. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2.Mol Cell. 2005; 20: 77–89 [DOI] [PubMed] [Google Scholar]
- 46.Velazquez-Dones A, Hagopian JC, Ma CT, et al. Mass spectrometric and kinetic analysis of ASF/SF2 phosphorylation by SRPK1 and Clk/Sty.J Biol Chem. 2005; 280: 41761–41768 [DOI] [PubMed] [Google Scholar]
- 47.Xu J, Dou T, Liu C, et al. The evolution of alternative splicing exons in vascular endothelial growth factor A.Gene. 2011; 487: 143–150 [DOI] [PubMed] [Google Scholar]
- 48.Zhao M, Shi X, Liang J, et al. Expression of pro- and anti-angiogenic isoforms of VEGF in the mouse model of oxygen-induced retinopathy.Exp Eye Res. 2011; 93: 921–926 [DOI] [PubMed] [Google Scholar]
- 49.Caires KC, de Avila JM, Cupp AS, McLean DJ. VEGFA family isoforms regulate spermatogonial stem cell homeostasis in vivo.Endocrinology. 2012; 153: 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris S, Craze M, Newton J, et al. Do anti-angiogenic VEGF (VEGFxxxb) isoforms exist? A cautionary tale.PLoS One. 2012; 7: e35231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McFee RM, Rozell TG, Cupp AS. The balance of proangiogenic and antiangiogenic VEGFA isoforms regulate follicle development.Cell Tissue Res. 2012; 349: 635–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization.J Exp Med. 2003; 198: 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geroski DH, Edelhauser HF. Drug delivery for posterior segment eye disease.Invest Ophthalmol Vis Sci. 2000; 41: 961–964 [PubMed] [Google Scholar]
- 54.Keyt BA, Berleau LT, Nguyen HV, et al. The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency.J Biol Chem. 1996; 271: 7788–7795 [DOI] [PubMed] [Google Scholar]
- 55.Stalmans I, Ng YS, Rohan R, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms.J Clin Invest. 2002; 109: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thobe MN, Gurusamy D, Pathrose P, Waltz SE. The Ron receptor tyrosine kinase positively regulates angiogenic chemokine production in prostate cancer cells.Oncogene. 2010; 29: 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das S, Anczukow O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC.Cell Rep. 2012; 1: 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data