Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients (original) (raw)
Abstract
Objectives:
To determine the frequency of KCNQ2 mutations in patients with neonatal epileptic encephalopathy (NEE), and to expand the phenotypic spectrum of KCNQ2 epileptic encephalopathy.
Methods:
Eighty-four patients with unexplained NEE were screened for KCNQ2 mutations using classic Sanger sequencing. Clinical data of 6 additional patients with KCNQ2 mutations detected by gene panel were collected. Detailed phenotyping was performed with particular attention to seizure frequency, cognitive outcome, and video-EEG.
Results:
In the cohort, we identified 9 different heterozygous de novo KCNQ2 missense mutations in 11 of 84 patients (13%). Two of 6 missense mutations detected by gene panel were recurrent and present in patients of the cohort. Seizures at onset typically consisted of tonic posturing often associated with focal clonic jerking, and were accompanied by apnea with desaturation. One patient diagnosed by gene panel had seizure onset at the age of 5 months. Based on seizure frequency at onset and cognitive outcome, we delineated 3 clinical subgroups, expanding the spectrum of KCNQ2 encephalopathy to patients with moderate intellectual disability and/or infrequent seizures at onset. Recurrent mutations lead to relatively homogenous phenotypes. One patient responded favorably to retigabine; 5 patients had a good response to carbamazepine. In 6 patients, seizures with bradycardia were recorded. One patient died of probable sudden unexpected death in epilepsy.
Conclusion:
KCNQ2 mutations cause approximately 13% of unexplained NEE. Patients present with a wide spectrum of severity and, although rare, infantile epilepsy onset is possible.
Mutations in KCNQ2 and KCNQ3, encoding the voltage-gated potassium channel Kv7.2 and Kv7.3, are found in 60% to 70% of families with the autosomal dominant self-limiting syndrome of benign familial neonatal seizures (BFNS).1,2 In contrast, we recently identified 8 patients with novel de novo KCNQ2 mutations who presented with a severe neonatal epileptic encephalopathy (NEE). They were identified through screening of KCNQ2 in 80 patients with epileptic encephalopathy (EE) and onset in the first 3 months of life. All individuals carrying a mutation had epilepsy onset in the first week of life. We concluded that KCNQ2 screening should be considered in patients with refractory neonatal seizures of unknown origin.3
In the study presented here, we aimed to determine the frequency of KCNQ2 mutations in a selected cohort of patients with an NEE defined by onset in the first month of life. We furthermore collected detailed clinical information of 6 patients with KCNQ2 encephalopathy diagnosed using an epilepsy gene panel. We describe the clinical and radiologic features of 17 patients, make a first attempt to establish genotype-phenotype correlations, and discuss treatment options.
METHODS
Patients.
We screened a cohort of 84 patients with unexplained NEE. Patients were referred for this study by collaborating child neurologists. All patients had onset of seizures within the first month of life associated with delayed development. Additional neurologic deficits such as hypotonia or spasticity were present in some individuals. Because patients were referred from multiple clinical centers, no uniform etiologic screening protocol was used. However, in all patients, routine diagnostic investigations such as metabolic screening (at least amino acids in blood and urine, organic acids in urine, and blood lactate) and chromosomal analysis were negative. MRI of the brain excluded causal structural abnormalities. An experienced pediatric neuroradiologist reexamined the brain MRI of affected individuals when available. Additionally, we collected clinical data of 6 patients with EE in whom a de novo KCNQ2 mutation was found in a diagnostic setting.
Standard protocol approvals and patient consents.
Parents or the legal guardian of each patient signed an informed consent form for participation. The study was approved by the Commission for Medical Ethics of the University of Antwerp and by the Human Research Ethics Committee of Austin Health.
Mutation analysis.
In the research cohort of 84 patients, mutation analysis of KCNQ2 was performed on genomic DNA extracted from peripheral blood using standard methods as described previously.3 For numbering mutations in KCNQ2, which has several splice variants, we used the longest mRNA transcript (isoform a; NM_172107.2) encoding a protein containing 872 amino acids. Sequenom analysis was performed to check for identified mutations in patients and parents. To confirm paternity, we genotyped 15 short tandem repeat markers located on 10 different chromosomes.
Six patients were referred for diagnostic genetic testing using 1 of 2 different next-generation sequencing epilepsy panels enriched for 50 and 38 genes, respectively, known to be associated with EEs and overlapping phenotypes, as well as respective exon-intron boundaries (as described by Lemke et al.4 in 2012 and at http://www.genedx.com/test-catalog/available-tests/infantile-epilepsy-panel/). Putatively pathogenic variants were validated in patients and parents using direct Sanger sequencing.
RESULTS
Mutation analysis.
We identified 9 novel heterozygous missense mutations in KCNQ2 in 11 of 84 patients (13%) of the screening cohort (table e-1 on the _Neurology_® Web site at www.neurology.org). Six heterozygous missense mutations were detected with the gene panel (table e-2). There were 3 recurrent mutations with 2 mutations present in 2 patients (D, E and J, O) and the other in 3 (G, H, N). Two different mutations in the same codon were also found (F, M). Sixteen mutations arose de novo (parental data not available for patient P). None of the mutations were previously reported in BFNS nor were they identified in the Exome Variant Server of the NHLBI GO Exome Sequencing Project or in the 1000 Genomes Project. All substituted amino acids were highly conserved evolutionary in mammals. PolyPhen-2 and SIFT predicted a very high probability of a damaging effect for all mutations, further supporting their pathogenic nature.
Interestingly, 2 different mutations in the same codon as found in patient C but with a different substitution (p.Ala265Pro and p.Ala265Val) have previously been reported in 2 patients with KCNQ2 encephalopathy.3,5
Clinical features of patients with KCNQ2 mutations.
The clinical features of the 17 patients in whom a mutation was detected are summarized in tables e-1 and e-2. Sixteen patients had seizure onset during the first 3 days of life. The mother of patient H reported rhythmic fetal movements during the last month of pregnancy reminiscent of seizures. One patient (Q) diagnosed using the gene panel had epilepsy onset at the age of 5 months. Fourteen patients presented with daily highly frequent seizures (range: 10 per day—status epilepticus requiring intubation) that were difficult to control for several weeks. Seizures had a prominent tonic component with or without associated clonic jerking of face or limbs and were often associated with apnea and desaturation. Three patients (J, K, O) had infrequent tonic seizures at onset. Patient B presented with daily bursts of myoclonic seizures intermixed with tonic contraction of axial and proximal limb muscles. After the neonatal period, tonic, clonic, tonic-clonic, myoclonic, spasm-like seizures, and focal seizures were seen. Eleven patients eventually became seizure-free between the age of 1 month and early adolescence. Three patients (D, M, N) also had nonepileptic dystonic episodes. Cognitive outcome varied widely with mild to moderate intellectual disability (ID) in 5 patients to severe or profound ID in 10. For 2 patients (N, O) aged, respectively, 4 and 8 months with developmental delay, cognitive outcome could not yet be defined. Only 3 patients were able to walk independently at the time of inclusion in the study. Thirteen patients had severe axial hypotonia, in some associated with limb spasticity.
EEG at onset showed a burst suppression pattern in 12 of 17 patients, which in 2 patients (H, I) was only seen during sleep (figure 1, A and D). In 2 of 17 patients, the initial EEG showed multifocal epileptic activity on an immature background, and in the 3 patients with infrequent seizures at onset, only focal epileptic activity was seen. During follow-up, multifocal epileptic activity was seen, often on an immature or slow background. When available, ictal EEG showed a tonic seizure pattern consisting of a high-voltage slow wave followed by flattening of the background with superimposed low-voltage fast activity, or seizures with a focal rhythmic ictal pattern and onset zone changing between seizures (figure 1, C and E).
Figure 1. Representative early EEG of patients with KCNQ2 encephalopathy.

Each panel contains 20 seconds of EEG, sensitivity 70 μV/mm. (A, B) Interictal EEG of patient D on days 4 and 12, respectively. Burst suppression pattern evolving into a discontinuous and asynchronous background with multifocal epileptic activity. (C) Ictal EEG of patient D on day 4. Slow wave followed by flattening of background (desynchronization) with superimposed low-voltage fast activity. Clinically tonic seizure with bilateral contraction of deltoids as seen in EMG recordings. (D) Interictal EEG of patient G on day 9. Burst suppression pattern. (E) Ictal EEG of patient G on day 9 (seizure duration approximately 60 seconds). Onset of rhythmic epileptic activity in the left hemisphere, with migration to the right hemisphere. Ictal activity in the right hemisphere only during the last 20 seconds of the seizure. Clinically, the seizure starts with opening of the eyes, a minimal tonic contraction, and a subtle vibratory component. During the subsequent 20 seconds, there is clonic eye movement. In the last 30 seconds, tonic asymmetrical posture with clonic eye movement and desaturation occur.
In patients A, D, H, M, N, and O, seizures accompanied by pronounced bradycardia were documented. Figure e-1 shows a seizure with a particularly prolonged bradycardia in patient D. After an initial tachycardia, bradycardia occurred and lasted for more than 3 minutes. It was accompanied by intermittent ventricular escape rhythms and the disappearance of EEG activity for 8 minutes.
Patient B continued to have frequent tonic spasm-like seizures at the age of 2.5 years, mostly on awakening. She was severely impaired and frequently required oxygen supplementation due to respiratory problems. Shortly after she was diagnosed with KCNQ2 encephalopathy, she was found dead in bed. Death due to sudden exacerbation of respiratory distress seemed unlikely and a possible sudden unexpected death in epilepsy (SUDEP) was hypothesized.
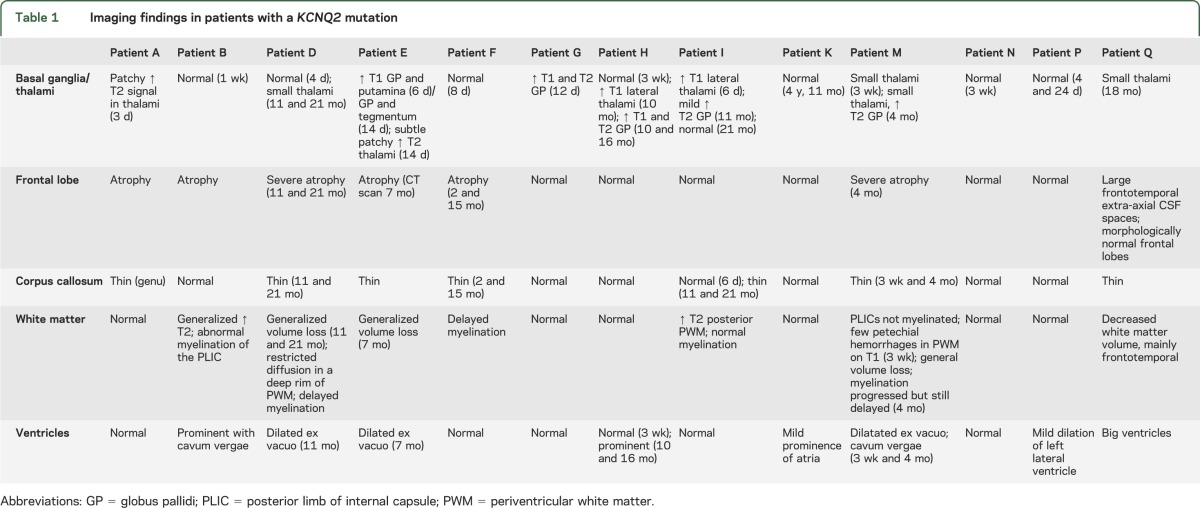
Brain MRI performed shortly after onset of epilepsy was available for review for 11 patients; for patients K and Q, only MRI performed after the first year of life was available. Findings are summarized in table 1.
Table 1.
Imaging findings in patients with a KCNQ2 mutation
DISCUSSION
Since 1998, inherited KCNQ2 mutations and deletions have been known to underlie approximately 60% to 70% of familial cases of BFNS.1,2,6 A minority of the BFNS families has a mutation in KCNQ3.7,8 Although seizures at onset can occur several times a day, they remit in the first months of life and developmental outcome is good. Recently, we described 7 novel missense KCNQ2 mutations in 8 patients with NEE, thereby showing that certain KCNQ2 mutations lead to a more devastating epilepsy disorder.3 Subsequently, 3 novel de novo missense KCNQ2 mutations were detected by whole exome sequencing in 3 of 12 patients with Ohtahara syndrome, confirming its significance in this group of patients.5
In the study presented here, we found KCNQ2 mutations in 11 of 84 patients (13%) with unexplained NEE. Clinical data of 6 additional patients detected with an epilepsy gene panel in a diagnostic setting are also described. Our data confirm that most patients with KCNQ2 encephalopathy present with neonatal seizures with a prominent tonic component and autonomic signs. Seizures are often accompanied by clonic jerking or more complex motor behavior. One patient (F) presented with bursts of myoclonic seizures, but these were often followed by tonic contraction of proximal muscles. The combination of frequent tonic seizures and burst suppression pattern on EEG in 11 of 17 patients might suggest a diagnosis of Ohtahara syndrome at onset. The evolution of the disease differs from the normal course of Ohtahara syndrome, however, as 11 of the patients with KCNQ2 encephalopathy described here became seizure-free, and evolution to West syndrome was rare. Although spasm-like seizures were common later in life, only 2 patients (J, N) had hypsarrhythmia on EEG. Patient J had epileptic spasms at that time but never had Ohtahara syndrome because she never had a burst suppression EEG pattern; patient N on the contrary never had epileptic spasms.
All 28 patients with KCNQ2 encephalopathy described to date had seizure onset in the first week of life except for patient Q, diagnosed using an epilepsy gene panel. This patient presented with epileptic spasms at the age of 5 months. Infantile onset of seizures has also been documented for a few patients with “benign” KCNQ2 mutations.9 Because seizure onset in the first month of life was an inclusion criterion for the current study cohort, patients with KCNQ2 encephalopathy with later onset might be underreported. In our previous study, patients with onset of EE up to 3 months were included however, and all mutation carriers had seizure onset in the first week. Later onset thus seems to be rare, although further studies are needed to elucidate the exact frequency of KCNQ2 mutations in patients with infantile EE.
As seen in BFNS, seizures in KCNQ2 encephalopathy are often accompanied by apnea with desaturation and cyanosis. In 6 patients, at least one seizure with prolonged bradycardia was recorded. Ictal bradycardia normally is a rare phenomenon, reported to occur in only 8% of children with epilepsy and 3% of seizures.10 KCNQ2 subunits are not expressed in the heart so it is unlikely that mutations affect heart function directly.11 Whether bradycardia is an epileptic phenomenon or occurs in response to hypoxia, a reflex often seen in infants, remains to be elucidated with time-locked EEG-ECG-saturation monitoring. In at least 2 patients (M, N), bradycardia occurred only after apnea, so reflex bradycardia seems a plausible mechanism. Whatever the pathophysiologic mechanism is, the simultaneous occurrence of desaturation and bradycardia in an infant with frequent seizures is a hazardous combination and may lead to an increased risk of SUDEP. Indeed, one patient with frequent seizures (B) was found dead in bed shortly after a diagnosis of KCNQ2 encephalopathy was made. Seizure control should therefore be optimized at all times, even though it may be difficult to achieve.
Response to antiepileptic drugs was variable, but 5 patients (D, G, H, I, J) showed at least a temporary good response to carbamazepine. Two of these patients (G, H) also responded well to IV phenytoin during the initial acute phase after failure of several antiepileptic drugs. A trial with a sodium channel blocker should therefore be considered early in the disease. One patient with KCNQ2 encephalopathy described in the literature became seizure-free on adrenocorticotropic hormone (ACTH) therapy.12,13 This could not be confirmed in the 3 individuals (C, M, N) in our study treated with ACTH or steroids because they responded only partially or not at all. Larger studies on the use of ACTH in this disease entity are needed before conclusions on efficacy can be drawn. Finally, one patient was treated with retigabine, an activator of neuronally expressed KCNQ channels.14 Both seizure severity and frequency reduced markedly when high doses were used. He is the only patient with KCNQ2 encephalopathy reported to date in which this designer drug has been trialed. This is probably related to the minimal experience child neurologists have with the drug because it is currently US Food and Drug Administration approved in adults only. Clinical studies on the use of retigabine in infants and children as well as in vitro studies in KCNQ2 mutant animal models will lead to increased confidence in using retigabine in infants with KCNQ2 encephalopathy.
Confirming our previous findings,3 variable T1 and T2 hyperintensities were seen in the basal ganglia and/or thalami in 6 of 11 patients of which brain MRI performed during the first year of life was available for review. Half of the patients (7/13) also had frontotemporal atrophy, a thin corpus callosum, and/or global loss of white matter volume that worsened on sequential imaging in the first 2 years of life.
The current report broadens the KCNQ2 encephalopathy spectrum, as 3 clinical subgroups can be distinguished. The first and largest group (patients A–F, L, M, and Q of the current report and 7 of our 8 original patients) has a stormy onset with frequent therapy-resistant seizures. Some patients become seizure-free after several months to years (10/16 patients; median age of seizure freedom 24 months, range 2 months to adolescence), but all develop a severe to profound ID with axial hypotonia, sometimes accompanied by limb spasticity. Frontotemporal atrophy and/or white matter loss is seen on brain MRI of all patients in this group and in none of the other patients. A second group (patients G, H, I, P, and patient 3 of our first report) presents in a similar way. Nevertheless, seizures are controlled in all patients (median age of seizure freedom 4 months, range 1 to 20 months) and they have only mild to moderate ID. In the last group (patients J, K, and O), seizure frequency is lower at onset and although EEG shows epileptic activity, a typical burst suppression pattern or frequent multifocal epileptic activity is not seen. Despite this apparent less dramatic start similar to BFNS, ID does ensue and outcome ranges from moderate to severe ID.
Our patient groups are too small to define prognostic factors. Seizure severity has a role as seizures are more easily controlled in group 2, and developmental regression occurred at the time of seizure exacerbation in patients J, K, and O. This does not seem to be the sole prognostic factor, however, because patient 8 of the first report also became seizure-free at the age of 2 months and nevertheless profound ID ensued. Remarkably, the 4 patient pairs with the 2 recurrent mutations reported so far (patients 7-8 in the first report, patients D-E, G-H, and J-O in the current report) have a comparable disease evolution. Interestingly, there is also marked similarity in disease severity in the patient pairs F-M and 5-D carrying a mutation in the same codon. This suggests that, perhaps not surprisingly, disease outcome depends to a major extent on the underlying mutation. This is further supported by the fact that all KCNQ2 encephalopathy mutations are novel mutations not reported in BFNS.
The reason KCNQ2 mutations lead to phenotypes ranging from BFNS to KCNQ2 encephalopathy with profound ID remains enigmatic. From the occurrence of KCNQ2 deletions in BFNS, it is known that a total loss of function of one allele leads to the benign phenotype only.1 Haploinsufficiency alone therefore cannot explain the more severe KCNQ2 encephalopathy. Although genetic and environmental background factors might influence the phenotype, it is remarkable that all KCNQ2 encephalopathy mutations published so far are (often recurrent) missense mutations, and never loss-of-function mutations. Most likely, these specific missense mutations exert a dominant negative effect. A conditional mouse model expressing a dominant negative KCNQ2 mutation indeed not only had seizures but also behavioral changes and memory deficits.15
This observation has interesting therapeutic implications. Until now, the focus of treatment of patients with epilepsy in general has always been on treating the symptoms, i.e., seizures. Nevertheless, now that increasing numbers of genetic causes of EE are being discovered, we should start targeting the underlying cause rather than the symptom. In the last few years, gene therapies for neurologic diseases are being intensively studied in neuromuscular disorders and Huntington disease.16–19 Therapies using antisense oligonucleotides are being specifically developed for diseases with known dominant negative mutations. In the field of epilepsy, we are still lagging behind in targeted gene therapy development, and it is time to make the mental shift when we think about developing novel epilepsy therapies. Indeed, specifically inhibiting transcription or translation of the mutated KCNQ2 allele would lead to a loss-of-function situation, mimicking a KCNQ2 deletion, which in turn is known to lead to the milder BFNS phenotype. Such a strategy therefore has the potential to turn a severe EE into a benign neonatal epilepsy syndrome.
In summary, the vast majority of patients with KCNQ2 encephalopathy present in the first week of life with highly frequent therapy-resistant seizures often with a prominent tonic component and autonomous signs. EEG at onset shows a burst suppression pattern or multifocal epileptic activity, and outcome ranges from mild to profound ID. Much fundamental and clinical research remains to be done to fully understand the entity of _KCNQ2_-related epilepsy. We advocate reporting of more patients to improve understanding through genotype-phenotype correlation, and broader screening of patients with infantile epilepsy to identify additional patients with later onset. Given a mutation frequency of 13% in the group of patients with unexplained NEE, we highly recommend screening of KCNQ2 at presentation in infants with frequent neonatal seizures of unknown origin. The physiologic observation of seizure-related bradycardia deserves further study and may offer an avenue to reduce the likelihood of SUDEP. While gaining more experience with the use of retigabine in children with KCNQ2 mutations is an enticing treatment strategy, an early trial of a sodium channel blocker may also prove useful.
Supplementary Material
Data Supplement
Coinvestigators
ACKNOWLEDGMENT
The authors thank the patients and their family members for their cooperation and participation in this study, and the VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be) for the genetic analyses. A.S. is a postdoctoral fellow of the Fund for Scientific Research Flanders (FWO).
GLOSSARY
ACTH
adrenocorticotropic hormone
BFNS
benign familial neonatal seizure
EE
epileptic encephalopathy
ID
intellectual disability
NEE
neonatal epileptic encephalopathy
SUDEP
sudden unexpected death in epilepsy
Footnotes
AUTHOR CONTRIBUTIONS
S.W.: design of the study, interpretation of the data, drafting the manuscript. V.I.: interpretation of the data, drafting the manuscript. R.H.: analysis of the data. R.V.C., H.H., R.S.M., S.G., A.-S.S., B.C., S.B.H., C.E., R.H.: interpretation of the data, revising the manuscript for intellectual content. G.C., T.P., L.G., K.R.: revising the manuscript for intellectual content. E.H., B.A., A.B., I.B., S.S., B.S., R.G., A.P., J.R.L., S.M., I.S., M.A., P.S., C.M.: interpretation of the data, revising the manuscript for intellectual content. A.S.: design of the study, interpretation of the data, drafting the manuscript. P.D.J.: design of the study, interpretation of the data, revising the manuscript for intellectual content.
STUDY FUNDING
Supported by the Fund for Scientific Research Flanders (FWO) (P.D.J.), Methusalem excellence grant of the Flemish Government (P.D.J.), University of Antwerp, the Eurocores program EuroEPINOMICS of the European Science Foundation (P.D.J.), and National Health and Medical Research Council of Australia (I.S.).
DISCLOSURE
S. Weckhuysen, V. Ivanovic, R. Hendrickx, R. Van Coster, H. Hjalgrim, R. Møller, and S. Grønborg report no disclosures. A. Schoonjans is a postdoctoral fellow of the Fund for Scientific Research Flanders (FWO). B. Ceulemans, S. Heavin, C. Eltze, R. Horvath, G. Casara, T. Pisano, L. Giordano, K. Rostasy, E. Haberlandt, B. Albrecht, A. Bevot, I. Benkel, S. Syrbe, B. Sheidley, and R. Guerrini report no disclosures. A. Poduri is supported by the National Institute of Neurological Disorders and Stroke (K23NS069784). J. Lemke and S. Mandelstam report no disclosures. I. Scheffer has served on scientific advisory boards for UCB; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has received speaker honoraria from Athena Diagnostics, UCB, and Janssen-Cilag EMEA; has received payment for development of educational presentations from UCB and Athena Diagnostics; has received funding for travel from Athena Diagnostics, Biocodex, UCB, GlaxoSmithKline, and Janssen-Cilag EMEA; and receives/has received research support from the National Health and Medical Research Council of Australia, Health Research Council of New Zealand, Australian Research Council, The University of Melbourne, American Epilepsy Society, the Jack Brockhoff Foundation, the Shepherd Foundation, and the Perpetual Charitable Trustees. M. Angriman, P. Striano, C. Marini, A. Suls, and P. De Jonghe report no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Heron SE, Cox K, Grinton BE, et al. Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet 2007;44:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 1998;18:25–29 [DOI] [PubMed] [Google Scholar]
- 3.Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25 [DOI] [PubMed] [Google Scholar]
- 4.Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398 [DOI] [PubMed] [Google Scholar]
- 5.Saitsu H, Kato M, Koide A, et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol 2012;72:298–300 [DOI] [PubMed] [Google Scholar]
- 6.Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science 1998;279:403–406 [DOI] [PubMed] [Google Scholar]
- 7.Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet 1998;18:53–55 [DOI] [PubMed] [Google Scholar]
- 8.Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 2003;126:2726–2737 [DOI] [PubMed] [Google Scholar]
- 9.Zara F, Specchio N, Striano P, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia 2013;54:425–436 [DOI] [PubMed] [Google Scholar]
- 10.Moseley BD, Nickels K, Britton J, Wirrell E. How common is ictal hypoxemia and bradycardia in children with partial complex and generalized convulsive seizures? Epilepsia 2010;51:1219–1224 [DOI] [PubMed] [Google Scholar]
- 11.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 2000;1:21–30 [DOI] [PubMed] [Google Scholar]
- 12.Dedek K, Fusco L, Teloy N, Steinlein OK. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res 2003;54:21–27 [DOI] [PubMed] [Google Scholar]
- 13.Serino D, Specchio N, Pontrelli G, Vigevano F, Fusco L. Video/EEG findings in a KCNQ2 epileptic encephalopathy: a case report and revision of literature data. Epileptic Disord 2013;15:158–165 [DOI] [PubMed] [Google Scholar]
- 14.Barrese V, Miceli F, Soldovieri MV, et al. Neuronal potassium channel openers in the management of epilepsy: role and potential of retigabine. Clin Pharmacol 2010;2:225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 2005;8:51–60 [DOI] [PubMed] [Google Scholar]
- 16.Muntoni F, Wood MJA. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov 2011;10:621–637 [DOI] [PubMed] [Google Scholar]
- 17.Magaña JJ, Cisneros B. Perspectives on gene therapy in myotonic dystrophy type 1. J Neurosci Res 2011;89:275–285 [DOI] [PubMed] [Google Scholar]
- 18.Ramaswamy S, Kordower JH. Gene therapy for Huntington's disease. Neurobiol Dis 2012;48:243–254 [DOI] [PubMed] [Google Scholar]
- 19.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 2012;11:125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement
Coinvestigators