MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay (original) (raw)
Abstract
Stress granules (SGs) are dynamic cytoplasmic foci at which stalled translation initiation complexes accumulate in cells subjected to environmental stress. SG-associated proteins such as TIA-1, TIAR and HuR bind to AU-rich element (ARE)-containing mRNAs and control their translation and stability. Here we show that tristetraprolin (TTP), an ARE-binding protein that destabilizes ARE-mRNAs, is recruited to SGs that are assembled in response to FCCP-induced energy deprivation, but not arsenite-induced oxidative stress. Exclusion of TTP from arsenite-induced SGs is a consequence of MAPKAP kinase-2 (MK2)-induced phosphorylation at serines 52 and 178, which promotes the assembly of TTP:14-3-3 complexes. 14-3-3 binding excludes TTP from SGs and inhibits TTP-dependent degradation of ARE-containing transcripts. In activated RAW 264.7 macrophages, endogenous TTP:14-3-3 complexes bind to ARE-RNA. Our data reveal the mechanism by which the p38-MAPK/MK2 kinase cascade inhibits TTP-mediated degradation of ARE-containing transcripts and thereby contributes to lipopolysaccharide-induced TNFα expression.
Keywords: AU-rich element, MAPKAP kinase-2, mRNA turnover, TNFα, TTP
Introduction
In response to environmental stress, eucaryotic cells reprogram their translational machinery to allow the preferential synthesis of heat shock proteins (Harding and Ron, 2002; Hinnebusch and Natarajan, 2002). Stress-induced translational reprogramming is triggered by the phosphorylation of eIF2α, a component of the eIF2/GTP/tRNAiMet ternary complex that loads the initiator tRNA onto the small ribosomal subunit to begin protein translation (Clemens, 2001; Anderson and Kedersha, 2002). Phosphorylation of eIF2α reduces the availability of eIF2/GTP/tRNAiMet, resulting in the assembly of a translationally incompetent initiation complex that accumulates, together with a substantial fraction of poly-A mRNA, at discrete cytoplasmic foci known as stress granules (SGs) (Kedersha et al, 1999, 2000, 2002; Kedersha and Anderson, 2001). In cells allowed to recover from stress, eIF2/GTP/tRNAiMet is regenerated, restoring normal protein translation. Because SG-associated mRNAs are not degraded during stress, they are available to restore protein synthesis in cells that recover from stress (Nover et al, 1989).
Whereas transcripts containing AU-rich elements (AREs) are normally labile (Chen and Shyu, 1995), activation of the stress-induced c-Jun N-terminal kinase (JNK) and p38-MAPK pathways leads to their stabilization (Ming et al, 1998; Winzen et al, 1999). We have previously proposed that SGs are sites at which mRNPs are monitored for structure and composition, then sorted for either reinitiation, degradation or packaging into stable nonpolysomal mRNP complexes (Anderson and Kedersha, 2002). This model postulates that RNA-binding proteins that either stabilize or destabilize mRNA will determine the fate of SG-associated transcripts. Thus, factors that stabilize (e.g. HuR; Fan and Steitz, 1998; Peng et al, 1998) or destabilize (e.g. TTP, BRF1; Carballo et al, 1998; Stoecklin et al, 2002) ARE transcripts may be regulated at SGs. To test this hypothesis, we have investigated the contribution of tristetraprolin (TTP), a zinc-finger protein that promotes the degradation of ARE transcripts, to the assembly of SGs and the regulated degradation of ARE transcripts.
TTP binds to AREs located in the 3′ untranslated region (3′UTR) of target mRNAs and directs them to the exosome for rapid degradation (Lai et al, 1999; Chen et al, 2001). Mice lacking TTP develop an inflammatory syndrome characterized by arthritis, dermatitis and cachexia as a consequence of excess tumor necrosis factor α (TNFα) production due to enhanced stability of TNFα mRNA (Taylor et al, 1996; Carballo et al, 1998). p38-MAPK and its downstream substrate MK2 have been reported to phosphorylate TTP, yet the effect of these kinases on TTP activity is controversial (Carballo et al, 2001; Mahtani et al, 2001; Zhu et al, 2001). Phosphorylation promotes the nuclear export of TTP, an effect that is partly dependent upon binding to 14-3-3 proteins (Johnson et al, 2002). 14-3-3 proteins, by interacting with phospho-serine residues, are associated with a large number of partner proteins and thus influence a wide range of cellular processes (Tzivion and Avruch, 2002). Serine 178 of TTP has been identified as a binding site for 14-3-3 (Johnson et al, 2002), but the precise role of TTP:14-3-3 complex formation is not known.
Here we report that TTP is a component of SGs. When bound to 14-3-3, however, TTP is specifically excluded from SGs. We investigated the role of 14-3-3 binding and provide evidence that TTP:14-3-3 complex formation inhibits the decay of ARE-containing transcripts.
Results
Endogenous TTP is associated with SGs upon energy starvation
Several AU-binding proteins including TIA-1, TIAR and HuR are associated with SGs in cells exposed to environmental stress (Kedersha et al, 1999, 2002). To determine whether TTP is also recruited to SGs, three different cell lines were treated with the mitochondrial inhibitor FCCP, a potent inducer of SGs, and subjected to immunofluorescence analysis using an affinity-purified TTP antibody (CARP-3; Brooks et al, 2002). In all three cell lines (DU145, Figure 1A; HeLa, Figure 1B; and COS7, Figure 1C), endogenous TTP is concentrated at discrete cytoplasmic foci (arrows) that colocalize with TIA-1, a robust marker of SGs (left panels show TTP in red, middle panels show TIA-1 in green, right panels show merged view). COS7 cells were also stained for two other markers of SGs, HuR and eIF3η (p116), and perfect colocalization at the cytoplasmic foci indicates that TTP is a component of SGs (Figure 1D and E). TTP is observed in SGs in 41% of FCCP-treated COS7 cells. Surprisingly, TTP is not recruited to SGs assembled in response to arsenite-induced oxidative stress (Figure 1F, arrows).
Figure 1.
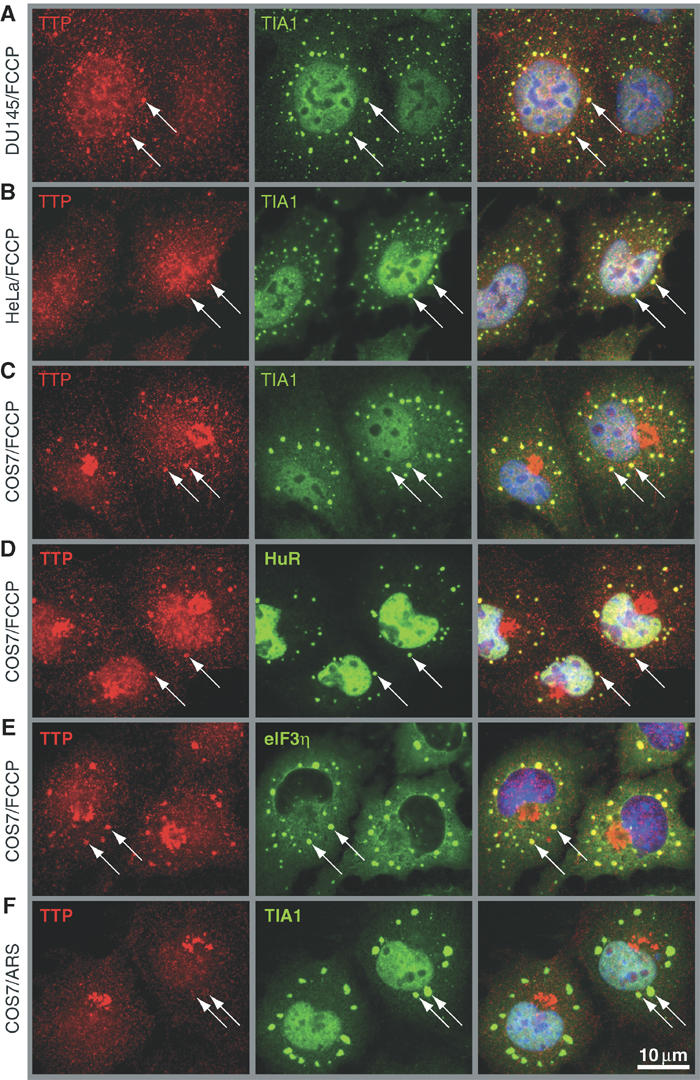
TTP is recruited to SGs upon energy starvation. Subcellular localization of TTP was determined by dual label immunofluorescence using CARP-3, an affinity-purified antibody reactive with TTP (left panels; red), in (A) DU145, (B) HeLa and (C–E) COS7 cells treated for 90 min with FCCP (1 μM). (F) COS7 cells were exposed to oxidative stress by treatment for 60 min with arsenite (ARS, 0.5 mM). SGs were visualized by counterstaining for TIA-1, HuR and eIF3η, as indicated (center panels; green). Merged views are shown in the right panels. Arrows point out SGs; size bar is 10 μm.
Overexpression of TTP promotes the assembly of SGs in the absence of stress
We transiently transfected myc-tagged recombinant TTP (TTP-myc) into COS7 cells to confirm that TTP is a component of SGs. Immunofluorescent staining with an anti-myc antibody revealed that TTP-myc is either diffusely distributed between the nucleus and the cytoplasm, or, in 39% of transfectants (for quantification see Figure 3B), colocalized with TIA-1 at discrete cytoplasmic foci (Figure 2A, left panels show myc-TTP in red, middle panels show TIA-1 in green). Several other markers of SGs are also found at these foci, including HuR, phospho-eIF2α, eIF3η and poly-A RNA (data not shown). Thus, in a subset of cells, overexpression of TTP induces the spontaneous assembly of bona fide SGs.
Figure 3.
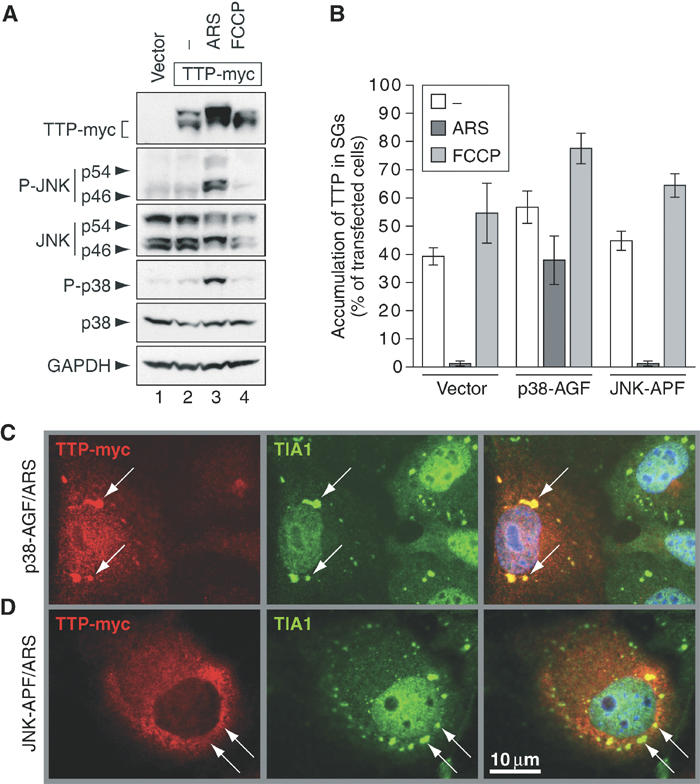
Exclusion of TTP from SGs depends on activation of p38-MAPK. (A) Western blot analysis of cytoplasmic lysates from COS7 cells transfected with vector control (lane 1) or TTP-myc (lanes 2–4). Cells were cultured in media alone (lanes 1 and 2), media containing arsenite (ARS, 0.5 mM, 60 min; lane 3) or FCCP (1 μM, 90 min; lane 4). (B) Quantitative immunofluorescence analysis showing the percentage of transfected cells in which TTP accumulates in SGs (average values±s.e.m. of _n_⩾3 independent experiments). COS7 cells were transfected with TTP-myc together with vector control, p38-AGF or JNK-APF prior to culturing in media alone (open bars) or media containing arsenite (ARS, 0.5 mM, 60 min; dark bars) or FCCP (1 μM, 90 min; shaded bars). (C) Representative immunofluorescence micrographs of arsenite-treated COS7 cells transfected with TTP-myc and p38-AGF or (D) TTP-myc and JNK-APF. Left panels show TTP-myc (red), center panels show TIA-1 as a marker of SGs (green, arrows) and right panels show merged views. Size bar is 10 μm.
Figure 2.
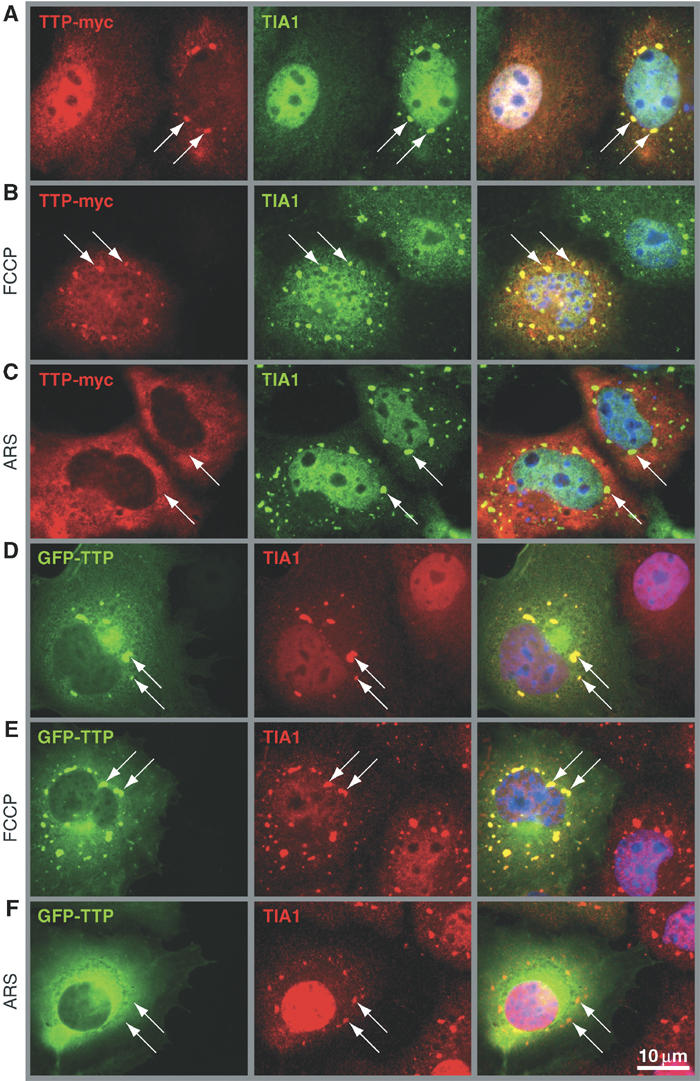
TTP promotes the assembly of spontaneous SGs but is excluded from arsenite-induced SGs. (A) COS7 cells transiently transfected with TTP-myc were cultured in media alone, (B) treated for 90 min with FCCP (1 μM) or (C) treated for 60 min with arsenite (ARS, 0.5 mM). Cells were processed for dual label immunofluorescence using antibodies reactive with myc (left panels; red) and TIA-1 as a maker of SGs (center panels; green). (D) COS7 cells transiently transfected with GFP-TTP were cultured in media alone, (E) treated for 90 min with FCCP (1 μM) or (F) treated for 60 min with arsenite (ARS, 0.5 mM). GFP-TTP is visualized in green (left panels) and TIA-1 in red (center panels). Merged views are shown in the right panels. Arrows point out SGs; size bar is 10 μm.
TTP is excluded from arsenite-induced SGs
In cells treated with FCCP, TTP-myc accumulates at SGs in 55% of transfected cells (Figures 2B and 3B). In contrast, TTP-myc is not recruited to SGs assembled in response to arsenite-induced oxidative stress (Figures 2C and 3B). The differential recruitment of TTP to FCCP- versus arsenite-induced SGs was confirmed using GFP-TTP, which accumulates at SGs in untreated (Figure 2D) and FCCP-treated cells (Figure 2E), but not in arsenite-treated cells (Figure 2F). These results confirm the specificity of SG staining by the TTP antibody and provided evidence that both endogenous and recombinant TTP associate with FCCP-induced, but not arsenite-induced SGs.
Exclusion of TTP from SGs depends on activation of p38-MAPK
TTP is phosphorylated by the stress-activated kinases p38-MAPK and MK2. Because arsenite activates the p38-MAPK/MK2 kinase cascade, phosphorylation of TTP could be responsible for its exclusion from arsenite-induced SGs. We used antibodies specific for phospho-p38 and phospho-JNK to show that arsenite (Figure 3A, lane 3), but not FCCP (lane 4), activates these stress kinases. Arsenite also increases the abundance of a slow-migrating TTP isoform that previously has been shown to arise from phosphorylation (lane 3) (Carballo et al, 2001; Mahtani et al, 2001). Thus, arsenite activates p38-MAPK and JNK, and induces the phosphorylation of TTP.
We used dominant-negative mutants of p38-MAPK (p38-AGF) and JNK (JNK-APF) to determine if either of these kinases is responsible for the exclusion of TTP from arsenite-induced SGs. COS7 cells were cotransfected with TTP-myc and either p38-AGF, JNK-APF or a vector control prior to treatment with arsenite or FCCP. Association of TTP-myc with SGs was assessed by immunofluorescence microscopy and quantified. Figure 3B presents the average percentage (±s.e.m., _n_⩾3) of transfected cells in which TTP-myc accumulates at SGs after incubation in medium alone (open bars), arsenite (dark bars) or FCCP (shaded bars). This analysis clearly shows that p38-AGF, but not JNK-APF, prevents the exclusion of TTP-myc from arsenite-induced SGs. In the representative micrographs prepared from arsenite-treated cells, the association of TTP-myc with SGs is restored in p38-AGF-transfected cells (Figure 3C), but not in JNK-APF-transfected cells (Figure 3D). We conclude that exclusion of TTP from arsenite-induced SGs is dependent on p38-MAPK activation and most likely involves phosphorylation of TTP.
Analysis of TTP truncation and substitution mutants
We next compared the ability of truncation and substitution mutants of TTP to accumulate at SGs. Myc-tagged mutant TTP constructs depicted schematically in Figure 4A were transfected into COS7 cells, and their accumulation at SGs was quantified (Figure 4B). Unlike the full-length protein, TTP mutants lacking the C-terminus (TTP-NZ-myc and TTP-Z-myc) are associated with arsenite-induced SGs. In contrast, TTP mutants lacking the N-terminus (TTP-ZC-myc) are excluded from arsenite-induced SGs. Thus, the C-terminus includes a motif required for exclusion of TTP from arsenite-induced SGs.
Figure 4.
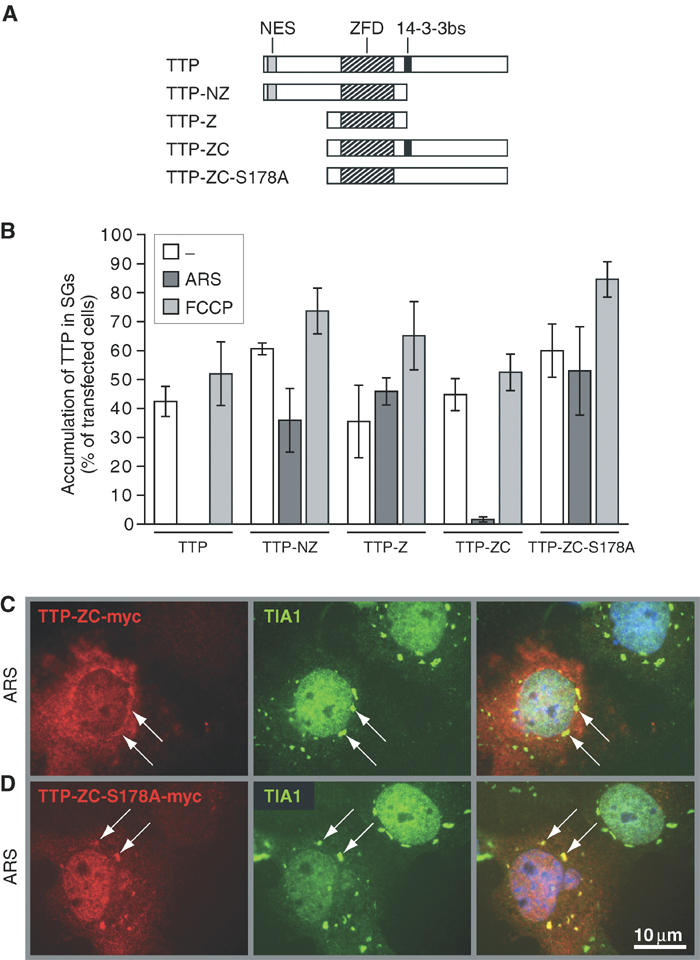
Analysis of TTP truncation and substitution mutants. (A) Schematic representation of wild-type TTP as well as truncation and substitution mutants. NES: nuclear export signal; ZFD: zinc-finger domain; 14-3-3bs: 14-3-3-binding site. (B) Quantitative immunofluorescence analysis showing the percentage of transfected cells in which truncated TTP mutants accumulate in SGs (average values±s.e.m. of _n_⩾3 independent experiments). COS7 cells were transfected with TTP-myc, TTP-NZ-myc, TTP-Z-myc, TTP-ZC-myc and TTP-ZC-S178A-myc, and processed as described for Figure 3B. (C) Representative immunofluorescence micrographs of arsenite-treated COS7 cells transfected with TTP-ZC-myc or (D) TTP-ZC-S178A-myc. Left panels show the myc-tagged TTP mutants (red), center panels show TIA-1 as a marker of SGs (green, arrows) and right panels show merged views. Size bar is 10 μm.
The C-terminus of TTP contains a phosphorylation site at serine 178 that confers binding to 14-3-3 proteins and regulates the nuclear-to-cytoplasmic distribution of TTP (Johnson et al, 2002). We tested whether 14-3-3 binding to phospho-TTP might prevent its recruitment to arsenite-induced SGs. Indeed, TTP-ZC-S178A-myc, a mutant that cannot bind 14-3-3, is efficiently recruited to arsenite-induced SGs, whereas TTP-ZC-myc is not (Figure 4B). Representative immunofluorescence images from arsenite-treated TTP-ZC-myc and TTP-ZC-S178A-myc transfectants are shown in Figure 4C and D, respectively.
MK2 induces TTP:14-3-3 complex formation
Our results suggested that 14-3-3 might sequester TTP from arsenite-induced SGs. Co-precipitation experiments were carried out to assess the assembly of TTP:14-3-3 complexes. COS7 cells were transiently transfected with TTP-myc (also containing a his-tag), and TTP was affinity precipitated from cytoplasmic lysates using nickel beads. After separation on SDS–polyacrylamide gels, co-precipitated 14-3-3 was analyzed by immunoblotting. As shown in Figure 5A, 14-3-3 proteins weakly bind to TTP-myc in untreated (lanes 2 and 6) or FCCP-treated cells (lane 3). In contrast, arsenite is a potent inducer of TTP:14-3-3 binding (lanes 4 and 7). Transfection of TTP-myc together with a dominant-negative mutant of p38-MAPK (p38-AGF; lane 8), but not of JNK (JNK-APF; lane 9), prevents arsenite-induced TTP:14-3-3 complex formation. Thus, there is a precise correlation between the assembly of TTP:14-3-3 complexes and the exclusion of TTP from SGs.
Figure 5.
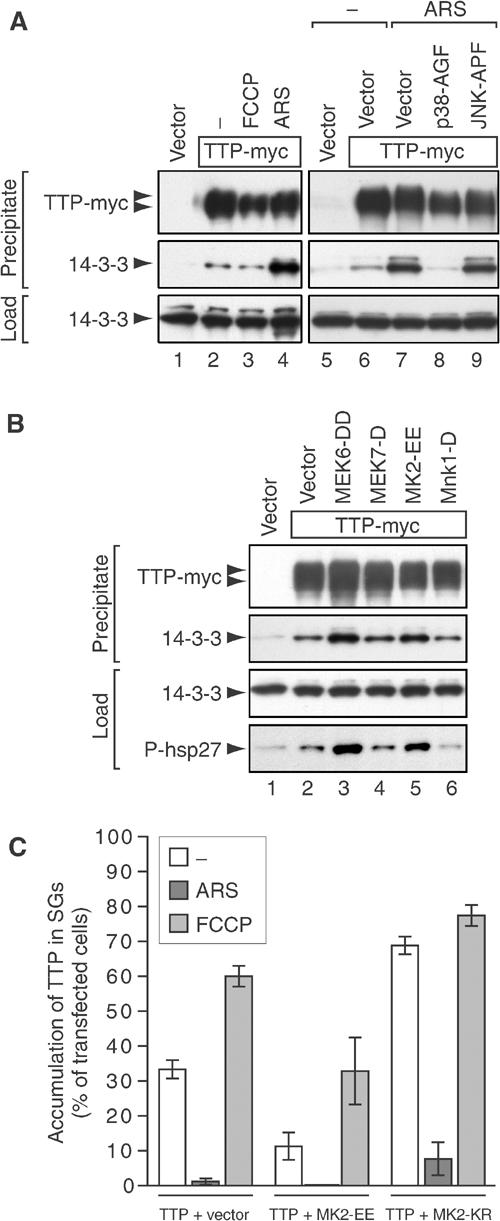
MK2 induces TTP:14-3-3 complex formation. (A) Co-precipitation of TTP and 14-3-3. COS7 cells were transfected with vector control (lanes 1 and 5) or TTP-myc (lanes 2–4 and 6–9), and cotransfected with vector (lanes 6 and 7), p38-AGF (lane 8) or JNK-APF (lane 9). Cells were cultured in media alone (lanes 1, 2, 5 and 6), or media containing FCCP (1 μM, 90 min; lane 3) or arsenite (ARS, 0.5 mM, 60 min; lanes 4 and 7–9). TTP-myc (also containing a his-tag) was affinity precipitated using nickel beads from cytoplasmic lysates and analyzed by Western blot using antibodies reactive with myc and 14-3-3. (B) COS7 cells were transfected with vector control (lane 1) or TTP-myc (lanes 2–6), and cotransfected with vector (lane 2), MEK6-DD (lane 3), MEK7-D (lane 4), MK2-EE (lane 5) or Mnk1-D (lane 6). Cells were cultured in media alone and processed as described in (A). In addition, cytoplasmic lysates (load) were probed using an antibody reactive with phospho(S78)-HSP27. (C) Quantitative immunofluorescence analysis showing the percentage of transfected cells in which TTP accumulates in SGs (average values±s.e.m. of _n_⩾3 independent experiments). COS7 cells were transfected with TTP-myc together with vector control, active MK2-EE or dominant-negative MK2-KR, and processed as described for Figure 3B.
14-3-3 proteins comprise a family of closely related adaptor proteins that bind to a specific sequence motif surrounding phospho-serine or phospho-threonine residues. TTP has been reported to be a substrate for p38-MAPK and its downstream kinase, MK2, as well as other kinases (Taylor et al, 1995; Carballo et al, 2001; Mahtani et al, 2001; Zhu et al, 2001). In order to narrow the candidate kinases responsible for inducing 14-3-3 binding, TTP-myc was cotransfected with several constitutively active recombinant kinases. MEK6-DD (Figure 5B, lane 3), an upstream activator of p38-MAPK, but not MEK7-D (lane 4), an upstream activator of JNK, induces the assembly of TTP:14-3-3 complexes. We next tested MK2 and Mnk1, two kinases directly activated by p38-MAPK. Whereas MK2-EE (lane 5) induces the assembly of TTP:14-3-3 complexes, Mnk1-D (lane 6) does not. The activity of MEK6-DD and MK2-EE was verified using HSP27, a known substrate for MK2 (Gaestel, 2002).
Using immunofluorescence microscopy (quantified in Figure 5C), MK2-EE was found to inhibit the recruitment of TTP-myc to spontaneous SGs (11 versus 33% in control transfectants) or FCCP-induced SGs (33 versus 60%). In contrast, a dominant-negative mutant of MK2 (MK2-KR) increased the recruitment of TTP-myc to spontaneous (79%), FCCP-induced (77%) and, to a lesser extent, arsenite-induced SGs (8 versus 1%). Thus, the MEK6/p38-MAPK/MK2 kinase cascade promotes TTP:14-3-3 complex formation and concomitantly sequesters TTP from SGs.
Identification of a second 14-3-3-binding site at serine 52
14-3-3 proteins generally bind as dimers to their target proteins by interacting with two separate phospho-serine or phospho-threonine motifs. Besides the previously described serine 178 motif at the C-terminus, the N-terminus of TTP contains one sequence motif that conforms to the RXXS/T or RXXXS/T 14-3-3-binding consensus (Figure 6A). Mutation of serine 52 to alanine (TTP-S52A-myc) weakly reduces the assembly of TTP:14-3-3 complexes in arsenite-treated cells (Figure 6B, compare lanes 3 and 5), whereas both the S178A single mutant and the S52A/S178A double mutant (TTP-AA-myc) effectively prevent the assembly of TTP:14-3-3 complexes (lanes 7 and 9).
Figure 6.
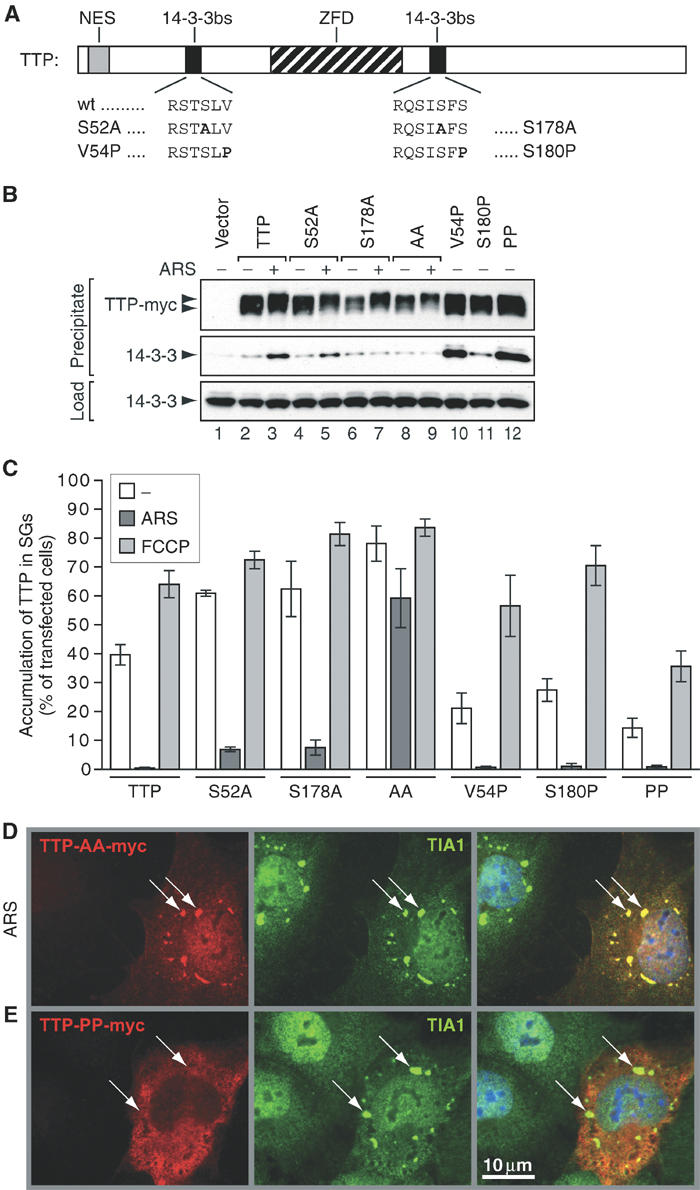
Identification of a second 14-3-3-binding site at serine 52. (A) Schematic depiction of murine TTP showing the wild-type (wt) sequence of 14-3-3-binding sites surrounding serine 52 and 178. Mutations S52A, S178A, V54P and S180P are indicated. NES: nuclear export signal; ZFD: zinc-finger domain; 14-3-3bs: 14-3-3-binding site. (B) Co-precipitation of TTP mutants and 14-3-3. COS7 cells were transfected with vector control (lane 1) or TTP-myc (lanes 2 and 3), TTP-S52A-myc (lanes 4 and 5), TTP-S178A-myc (lanes 6 and 7), TTP-AA-myc (S52A/S178A; lanes 8 and 9), TTP-V54P-myc (lane 10), TTP-S180P-myc (lane 11) and TTP-PP-myc (V54P/S180P; lane 12). Cells were cultured in media alone (lanes 1, 2, 4, 6, 8 and 10–12) or media containing arsenite (ARS, 0.5 mM, 60 min; lane 3, 5, 7 and 9), and processed as described for Figure 5A. (C) Quantitative immunofluorescence analysis showing the percentage of transfected cells in which mutant TTP accumulates in SGs (average values±s.e.m. of _n_⩾3 independent experiments). COS7 cells were transfected with TTP-myc, TTP-S52A-myc, TTP-S178A-myc, TTP-AA-myc, TTP-V54P-myc, TTP-S180P-myc and TTP-PP-myc, and processed as described for Figure 3B. (D) Representative immunofluorescence micrographs of COS7 cells transfected with TTP-AA-myc and treated with arsenite (ARS, 0.5 mM, 60 min) or (E) COS7 cells transfected with TTP-PP-myc and not treated with arsenite. Left panels show the myc-tagged TTP mutants (red), center panels show TIA-1 as a marker of SGs (green, arrows) and right panels show merged views. Size bar is 10 μm.
Immunofluorescence analysis revealed that the single mutants are recruited to arsenite-induced SGs slightly more efficiently than wild-type TTP (7% for S52A and 8% for S178A versus 0.5% for wt; Figure 6C). In contrast, the double-mutant TTP-AA-myc is strongly recruited to arsenite-induced SGs (59%), also exemplified in a representative immunofluorescence micrograph in Figure 6D. We conclude that binding of 14-3-3 to either of the two sites is sufficient for sequestration of TTP from arsenite-induced SGs.
Increased binding of 14-3-3 prevents TTP from associating with SGs
Binding of 14-3-3 to phospho-serine or phospho-threonine residues is greatly enhanced by the presence of a proline residue at position +2 (Yaffe et al, 1997). We altered the 14-3-3-binding sites on TTP to conform to this optimal sequence (V54P and S180P; Figure 6A). The V54P mutant strongly binds to 14-3-3 in the absence of arsenite (Figure 6B, compare lanes 2 and 10). The S180P mutant also binds to 14-3-3 in the absence of arsenite (lane 11), although binding is weaker, presumably because this site belongs to the atypical (mode II: RXXXS/T) category of 14-3-3-binding sites. Mutation of both V54 and S180 to proline (TTP-PP-myc) again confers strong binding to 14-3-3 in the absence of arsenite (lane 12). By immunofluorescence, all the proline mutants associate with SGs at a reduced rate compared to TTP-myc in both untreated and FCCP-treated cells (Figure 6C). The double-mutant TTP-PP-myc shows the strongest exclusion from spontaneous (14 versus 40%) and FCCP-induced SGs (36 versus 64%). In a representative immunofluorescence micrograph shown in Figure 6E, TTP-PP-myc is excluded from SGs of cells not treated with arsenite.
TTP:14-3-3 complex formation prevents decay of ARE-mRNA
To determine whether 14-3-3 binding affects the function of TTP, we compared the ability of wild-type and mutant TTP to degrade an ARE-containing mRNA. COS7 cells were transiently transfected with a β-globin reporter gene that is under the control of a tetracycline-sensitive promoter and has an ARE derived from TNFα inserted into its 3′UTR. Doxycyline was added to inhibit transcription of the reporter gene, and RNA was extracted after the indicated time intervals. Northern blot analysis and quantification by densitometry revealed that decay of the globin-ARE mRNA is relatively slow in cells cotransfected with vector alone (Figure 7A). Cotransfection of TTP-myc, as expected, induces the rapid decay of globin-ARE mRNA (Figure 7B, first panel). The mutant that cannot bind 14-3-3 (TTP-AA-myc) induces rapid decay of globin-ARE mRNA with a similar kinetics (second panel). In contrast, decay induced by the TTP mutant that binds to 14-3-3 constitutively (TTP-PP-myc) is clearly less rapid (third panel). This result suggests that 14-3-3 binding inhibits TTP-mediated degradation of ARE-containing transcripts.
Figure 7.
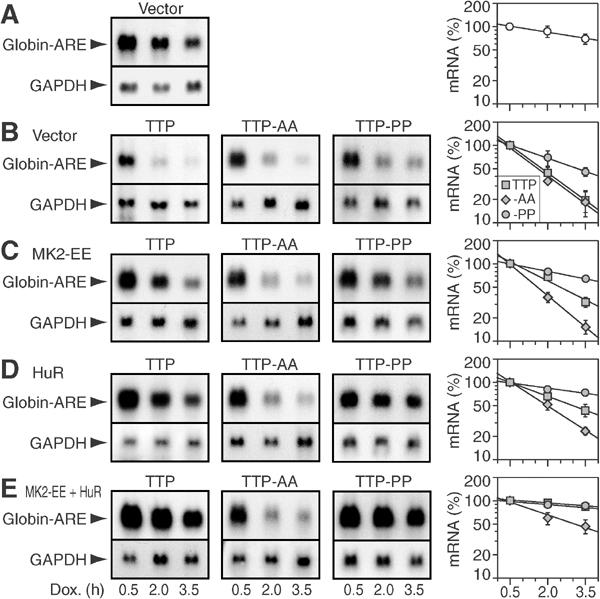
TTP:14-3-3 complex formation inhibits decay of ARE-mRNA. (A) Northern blot analysis was carried out with 10 μg of RNA from COS7 cells transfected with pTet-Off and pTet-7B-ARE, a β-globin reporter construct that contains the ARE of TNFα in the 3'UTR. Cells were treated with doxycycline (1 μg/ml) to block transcription, and total RNA was isolated after 0.5, 2.0 and 3.5 h. Northern blots were hybridized to digoxigenin-labeled probes against globin and GAPDH. The right panel shows quantification of the globin-ARE mRNA. Signal intensities were measured on autoradiograms using a digital camera and chemi-imager software. Globin-ARE signals were normalized to GAPDH signals, and the 0.5 h value was set 100%. Average percentages±s.e.m. of _n_⩾3 independent experiments were plotted against time, and exponential decay curves were calculated by best-fit criteria. (B) In addition to pTet-Off and pTet-7B-ARE, cells were cotransfected with TTP-myc (first panels), TTP-AA-myc (second panels) or TTP-PP-myc (third panels) together with vector, (C) MK2-EE, (D) HuR or (E) both MK2-EE and HuR.
Activation of the p38-MAPK pathway, together with overexpression of HuR, has previously been reported to inhibit TTP and thereby stabilize ARE-mRNA (Ming et al, 2001). Cotransfection of active MK2-EE inhibits TTP-myc-induced ARE-mRNA decay, but not TTP-AA-myc-induced decay (Figure 7C). Similarly, cotransfection of HuR inhibits TTP-myc-induced decay, whereas TTP-AA-myc-induced decay is less affected (Figure 7D). Conversely, decay induced by TTP-PP-myc is more sensitive to inhibition by MK2-EE or HuR. Moreover, cotransfection with both MK2-EE and HuR profoundly inhibits TTP-myc- and TTP-PP-myc-induced decay of ARE-mRNA, whereas TTP-AA-myc-induced decay is less abrogated (Figure 7E). We conclude that stabilization of ARE-mRNA by MK2 and HuR is mediated through inactivation of TTP by the assembly of TTP:14-3-3 complexes.
ARE:TTP:14-3-3 complex formation in RAW 264.7 macrophages
We next confirmed that endogenous TTP:14-3-3 complexes are assembled in lipopolysaccharide (LPS)-activated RAW 264.7 cells. Western blot analysis using affinity-purified TTP antibody reveals that the expression of TTP is strongly induced in response to LPS (Figure 8A, upper panel). Moreover, hyperphosphorylated forms of TTP are observed at the 4 h time point (lane 4). The same antibody efficiently immunoprecipitates TTP and co-precipitates 14-3-3 (bottom panels). Thus, endogenous TTP occurs in a complex with 14-3-3 in LPS-activated RAW 264.7 macrophages.
Figure 8.
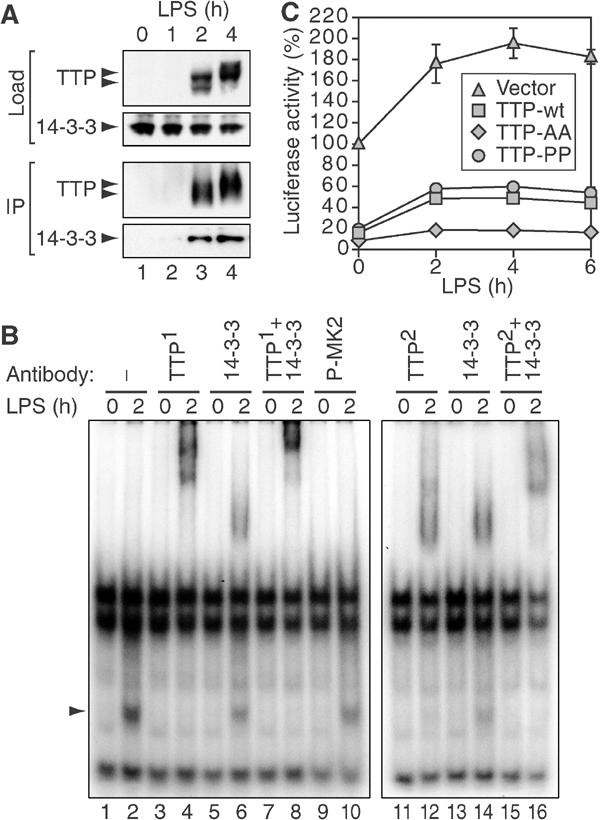
TTP:14-3-3 complex formation in macrophages. (A) TTP was immunoprecipitated with affinity-purified α-TTP (CARP-3) antibody from cytoplasmic lysates of RAW 264.7 cells stimulated with LPS (10 ng/ml) for 0, 1, 2 and 4 h. Load and immunoprecipitated material (IP) were analyzed by Western blotting. Bottom panel shows co-immunoprecipitation of 14-3-3. (B) RNA gel-shift analysis of complexes bound to ARE-RNA. Radiolabeled RNA containing the ARE of TNFα was incubated with cytoplasmic lysates of RAW 264.7 macrophages cultured in medium alone or stimulated with LPS (10 ng/ml) for 2 h. Prior to separation by non-denaturing PAGE, the following antibodies were added to the reaction: no antibody (lanes 1 and 2), CARP-3 rabbit α-TTP (TTP1, lanes 3–4 and 7–8), commercial goat α-TTP (TTP2, lanes 11–12 and 15–16), rabbit α-14-3-3 (lanes 5–8 and 13–16) or rabbit α-phospho-MK2 (lanes 9 and 10). (C) RAW 264.7 macrophages were transiently transfected with luc-UTR, a luciferase reporter containing the 3′UTR of TNFα, and empty vector, TTP-myc, TTP-AA-myc or TTP-PP-myc were cotransfected. After treatment with LPS (10 ng/ml), cytoplasmic extracts were assayed for luciferase activity. Values were normalized to the activity of a luciferase reporter lacking the TNFα 3′UTR to account for transcriptional activation. Mean percentages±s.e.m. of _n_=3 independent experiments were plotted against time.
Further evidence for the existence of an activation-induced complex between endogenous TTP and 14-3-3 was obtained using electrophoretic mobility shift assays. When a radiolabeled RNA containing the TNFα ARE was incubated with lysates from untreated RAW 264.7 cells, three major complexes were resolved by non-denaturing gel electrophoresis (Figure 8B, lane 1). Treatment of the cells with LPS for 2 h induced a fourth complex (lane 2, arrow), which is quantitatively supershifted by the affinity-purified TTP antibody (TTP1, lane 4). The same complex is supershifted by a 14-3-3 antibody (lanes 6 and 14), although not quantitatively. Addition of both antibodies to the reaction further reduces the mobility of the supershifted complexes (lane 8). A similar supershift is observed with a commercial antibody directed against TTP (TTP2, lanes 12–16). As a negative control, the same amount of an unrelated antibody did not shift any of the complexes (lanes 9 and 10). This analysis confirms that TTP is bound to 14-3-3 in LPS-treated RAW 264.7 macrophages, and provides evidence that the TTP:14-3-3 complex can bind to ARE-RNA.
TTP:14-3-3 complex formation contributes to LPS-induced TNFα expression
Finally, we wanted to investigate whether TTP:14-3-3 complex formation plays a role in regulating TNFα expression in macrophages. RAW 264.7 macrophages were transiently transfected with a reporter gene encoding luciferase fused to the 3'UTR of TNFα (luc-UTR) together with either vector control, TTP-myc, TTP-AA-myc or TTP-PP-myc. Cells were treated with LPS over a period of 6 h, and luciferase activity was measured at different time points (Figure 8C). In order to compensate for transcriptional activation, values were normalized to the activity of a luciferase reporter lacking the TNFα 3′UTR. Figure 8C shows that luc-UTR expression increases from 100 to 196% after 4 h of LPS treatment. TTP-myc reduces reporter gene expression to 16%, yet allows for an increase to 49% after 4 h of LPS treatment. TTP-AA-myc, however, is more active by reducing reporter gene expression to 8%, and LPS causes a much weaker increase to 18%. TTP-PP-myc, on the other hand, is slightly less active than TTP-myc, reducing reporter gene expression to 20% and allowing for an increase to 60%. We conclude that phosphorylation of TTP at serines 52 and 178 is responsible, at least in part, for post-transcriptional induction of TNFα expression by LPS.
Discussion
Mammalian cells have evolved post-transcriptional mechanisms that prevent the overexpression of potentially injurious proteins. Pro-inflammatory proteins such as TNFα, interleukin-1β, interleukin-6, cyclooxygenase-2 and matrix metalloproteinase-13 are encoded by short-lived mRNAs that are inefficiently translated (Anderson et al, 2004). This class of mRNA is marked for rapid degradation by AREs found in their 3'UTRs. TTP is one of the ARE-binding proteins that promotes the deadenylation and degradation of these transcripts (Blackshear, 2002). The expression of ARE-containing mRNAs requires the activation of signaling pathways that disable this constitutive decay program. For example, activation of JNK and p38-MAPK/MK2 pathways results in stabilization of ARE-mRNAs (Ming et al, 1998; Winzen et al, 1999). Here we show that p38-MAPK/MK2-induced stabilization of ARE-mRNAs is mediated through the assembly of phospho-TTP:14-3-3 complexes.
Several observations implicate the p38-MAPK/MK2 kinase cascade in the phosphorylation of TTP. Previously, TTP was found to be phosphorylated by both p38-MAPK and MK2 in vitro (Carballo et al, 2001; Mahtani et al, 2001; Zhu et al, 2001). Here we demonstrate that arsenite-induced assembly of phospho-TTP:14-3-3 complexes is prevented by a dominant-negative mutant of p38-MAPK, and that MEK6, the upstream activator of p38-MAPK, as well as MK2 induces the assembly of phospho-TTP:14-3-3 complexes (Figure 5). In addition to the known 14-3-3-binding site at serine 178 (Johnson et al, 2002), we describe a second binding site at serine 52 (Figure 6). In perfect agreement with our findings, Chrestensen et al (2003) have independently identified by mass spectrometry serine 52 and 178 to be the two major sites at which TTP is phosphorylated by MK2. Mutation of either of these sites alone inhibits the assembly of TTP:14-3-3 complexes, but has only a modest effect on exclusion of TTP from arsenite-induced SGs (Figure 6). In contrast, the double mutant is efficiently recruited to arsenite-induced SGs. Thus, the single mutants might permit loose binding of monomeric 14-3-3 that is sufficient to prevent recruitment to SGs, yet the weak interaction appears to be lost during immunoprecipitation.
Activation of the p38-MAPK/MK2 pathway (Winzen et al, 1999; Ming et al, 2001), or overexpression of HuR (Fan and Steitz, 1998; Peng et al, 1998), selectively stabilizes ARE-mRNAs. Our data reveal that MK2-induced stabilization is dependent upon the assembly of TTP:14-3-3 complexes (Figure 7). Mutations that prevent 14-3-3 binding (TTP-AA) render ARE-mRNA decay less sensitive to inhibition by MK2, whereas mutations that enhance 14-3-3 binding (TTP-PP) increase its sensitivity to inhibition. To our surprise, HuR-induced stabilization also appears to be influenced by the ability of TTP to complex with 14-3-3. This result suggests that HuR might affect the assembly or stability of the ARE:TTP:14-3-3 complex.
The p38-MAPK/MK2 kinase pathway and TTP antagonistically regulate the production of TNFα in LPS-activated macrophages. Here we demonstrate that in LPS-activated RAW 264.7 macrophages, TTP occurs in a complex with 14-3-3, but remains bound to ARE-RNA (Figure 8). Moreover, induction of a reporter gene under control of the TNFα 3'UTR is much weaker when mutant TTP-AA is coexpressed as opposed to wild-type TTP. Thus, our results provide an explanation for the paradoxical induction of both TNFα and its antagonist TTP by LPS: MK2-induced phosphorylation of TTP results in the assembly of phospho-TTP:14-3-3 complexes that prevent TTP-induced degradation of TNFα transcripts.
14-3-3 binding causes TTP to move from the nucleus to the cytoplasm (Johnson et al, 2002). Since the degradation of ARE-mRNAs is a cytoplasmic process, this is unlikely to explain the reduced function of TTP. 14-3-3 binding also prevents TTP from associating with SGs (Figures 4, 5 and 6). A simplistic interpretation would be that SGs are the actual site of ARE-mRNA degradation. SGs, however, result from an abrupt translational arrest, whereas ARE-mRNA decay occurs in nonstimulated, nonstressed and translationally competent cells. Therefore, we favor the hypothesis that TTP selects translationally stalled ARE-mRNAs for degradation. A small, microscopically invisible pool of untranslated mRNPs is present in all cells (Civelli et al, 1980). Since complex formation with 14-3-3 does not preclude TTP from binding to ARE-mRNA (Figure 8B), we speculate that 14-3-3 prevents TTP from directing its associated mRNA to the degradative machinery. This implies that TTP and 14-3-3 may be components of polysomal mRNA. This conjecture is supported by the observation that cytoplasmic TTP associates with polysomes in LPS-activated THP-1 macrophages (Brooks et al, 2002). It remains to be determined whether 14-3-3 is also associated with polysomes under these conditions.
The regulated association of TTP with SGs supports our contention that the mRNP composition of the SG-associated transcripts can determine their fate (Anderson and Kedersha, 2002). Another example of a cytoplasmic subdomain that determines the fate of selected mRNAs is the P-body. In yeast, P-bodies have been identified as discrete cytoplasmic foci at which mRNAs accumulate when their degradation is blocked (Sheth and Parker, 2003). Taken together, these independent, but perhaps related findings emphasize the importance of spacially separated mRNA pools within the cytoplasm. Determining the flux of mRNAs between polysomes, SGs, P-bodies, and possibly other sub-domains, will be crucial for a broader understanding of how mRNA metabolism is regulated.
Materials and methods
Plasmid constructs
pcDNA3-TTP-mycHis was cloned by replacing the _Bam_HI fragment in mTTP.tag (Stoecklin et al, 2000) with the _Bam_HI fragment of pCS2-TTP (Johnson et al, 2000). pcDNA3-TTP-S178A-mycHis, pcDNA3-TTP-ZC-mycHis and pcDNA3-TTP-ZC-S178A-mycHis were generated in the same way using the _Bam_HI fragments of pCS2-TTP S178A, pCS2-TTP(Zn-C) and pCS2-TTP(Zn-C) S178A (Johnson et al, 2002). For pcDNA3-TTP-NZ-mycHis and pcDNA3-TTP-Z-mycHis, the _Bam_HI–_Xho_I fragment was excised from pCS2-TTP(N-Zn) and pCS2-TTP(Zn) (Johnson et al, 2002), and ligated into the _Bam_HI and _Xho_I sites of pcDNA3.1/myc-HisA (Invitrogen).
For pcDNA3-TTP-S52A-mycHis and pcDNA3-TTP-AA-mycHis, a portion of the TTP cDNA was amplified by PCR using primer pair G50/G51, digested with _Kpn_I and inserted into the _Kpn_I sites of pcDNA3-TTP-mycHis and pcDNA3-TTP-S178A-mycHis, respectively. pcDNA3-TTP-V54P-mycHis was cloned in a similar fashion using primers G50/G52. For pcDNA3-TTP-S180P-mycHis, two portions of the TTP cDNA were amplified with primer pairs G50/G53 and G54/G55, annealed and reamplified with primers G50/G55. This amplicon was digested with _Bam_HI and inserted into the _Bam_HI sites of pcDNA3-TTP-mycHis. To generate pcDNA3-TTP-PP-mycHis, the _Kpn_I fragment of pcDNA3-TTP-V54P-mycHis was cloned into the _Kpn_I sites of pcDNA3-TTP-S180P-mycHis.
For pcDNA3-Flag-MK2-EE and pcDNA3-Flag-MK2-KR, the cDNAs encoding active and dominant-negative MK2 were amplified with primers G42/G43 from plasmids pcDNA3mycMK2T205E,T317E and pcDNA3mycMK2K76R (Winzen et al, 1999). The amplicons were digested with _Bam_HI and _Xho_I, and inserted into the _Bam_HI and _Xho_I sites of pcDNA3-Flag-BAK. Plasmids encoding GFP-TTP (Johnson et al, 2002) as well as MEK6-DD, MEK-7D, p38-AGF and JNK-APF (Ming et al, 2001) have been published previously. Plasmid pEBG-Mnk1-D was a gift from JA Cooper (Waskiewicz et al, 1999), and pcDNA3-HuR-Flag was kindly provided by JA Steitz (Fan and Steitz, 1998).
For pTet-7B, a T7-tagged rabbit β-globin gene was amplified with primers G18/G19 from puroMXβglobin (Stoecklin et al, 2001). The fragment was digested with _Eco_RV and _Bgl_II, and inserted into the _Eco_RI/blunt–_Bgl_II sites of pTet-BBB-GMCSF (Xu et al, 1998). pTet-7B-ARE was generated by replacing the _Eco_RI–_Bgl_II fragment in pTet-7B with the _Eco_RI–_Bgl_II fragment of puroMXβ-TNFα-ARE53 (Stoecklin et al, 2001).
Stem-ARE was cloned by introducing a 90-nt-long poly-A tail between the _Xho_I and _Kpn_I sites of pBluescript-SK+ (Stratagene), and the following sequence between the blunt-ended _Sac_I and the _Xho_I site: AATTCTAGAAGCCTTTCTAGAACAACAAGTATTTATATTT GCACTTATTATTTATTATTTATTTATTATTTATTTATTT GCTTATGAATGTATTTATTTGGAAGGCCGGGGTGTC.
For pMT2-luc, the luciferase coding region was amplified by PCR using primers 5′luc and 3′luc from pcDNA3.1-Zeo-luc (kindly provided by DA Dixon, Vanderbilt University, Nashville, TN, USA) and inserted into the _Eco_RI–_Xba_I sites of pMT2. An _Xba_I fragment containing the mouse TNFα 3′UTR from pGL3-ARE (kindly provided by V Kruys, Université Libre de Bruxelles, Belgium) was inserted into the _Xba_I site of pMT2-luc to generate pMT2-luc-UTR.
Cell culture and transfection
COS7, DU145, HeLa and RAW 264.7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies) supplemented with 10% fetal calf serum (Sigma) as well as 2 mM L-glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml, all from Mediatech Cellgro). Cells were transfected using Lipofectamine 2000 and Optimem medium (Life Technologies). Sodium arsenite (0.5 mM; Sigma) was added in conditioned medium. For treatment with FCCP (carbonyl cyanide _p_-trifluoromethoxyphenylhydrazone), cells were washed with phosphate-buffered saline (PBS) and cultured in glucose- and pyruvate-free DMEM (Life Technologies) containing 1 μM FCCP. At 24 h prior to addition of doxycycline (1 μg/ml; Sigma), cells were cultured in DMEM containing 5% Tet-system approved fetal bovine serum (BD Biosciences). In COS7 cells, the levels of endogenous TTP are nearly undetectable by Western blotting. Consequently, recombinant TTP is present in large excess over endogenous TTP in the transient transfection experiments.
Fluorescence microscopy
Immunofluorescence was performed as described previously (Kedersha et al, 2000), except that cells were postfixed with PHEM buffer (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2; pH 6.9) containing 0.1% Triton-X instead of 20% methanol when stained for endogenous TTP. Primary antibodies used were as follows: affinity-purified rabbit α-TTP (CARP-3) (Brooks et al, 2002), mouse α-myc (9E10, a kind gift from L Klickstein, Brigham and Women's Hospital, Boston, MA, USA), goat α-TIA-1 (sc-1751, Santa Cruz), mouse α-HuR (3A2, Santa Cruz) and goat α-eIF3η (sc-16377, Santa Cruz). Control antibodies that do not detect SGs have been described previously (Kedersha et al, 1999). To quantify the association of TTP with SGs, 100 transfected cells were analyzed under the microscope and scored positive if TTP showed visible colocalization with TIA-1 in SGs. Three or more (_n_⩾3) independent transfections were analyzed to calculate average percentages±s.e.m.
Western blot analysis
Proteins were separated in 4–20% polyacrylamide gradient Tris-glycine gels as described previously (Kedersha et al, 2000). Primary antibodies used were as follows: mouse α-myc (9E10), mouse α-phospho(T180/Y182)-p38-MAPK (28B10, Cell Signaling), rabbit α-p38-MAPK (sc-7149, Santa Cruz), mouse α-phospho(T183/Y185)-JNK (G-7, Santa Cruz), rabbit α-JNK (#9252, Cell Signaling), rabbit α-phospho(S82) HSP27 (#2401, Cell Signaling), mouse α-GAPDH (6C5, Research Diagnostics Inc.), rabbit α-TTP (CARP-3) and rabbit α-14-3-3β (K-19, Santa Cruz).
Co-(immuno)precipitation
At 24–48 h after transfection, COS7 cells from a 10-cm dish were lysed in 400 μl buffer containing 1% NP-40, 150 mM NaCl, 50 mM Tris–HCl pH 8.0, 1 mM MgCl, 10% glycerol, 20 mM 2-mercaptoethanol, 10 mM imidazole, 1 mM Na-vanadate, 50 mM NaF, 20 nM okadaic acid and EDTA-free complete protease inhibitors (Roche). After incubation of the cytoplasmic fraction for 1 h at 4°C with 40 μl of Ni-NTA magnetic agarose beads (Qiagen), the beads were washed four times in lysis buffer containing 20 mM imidazole and eluted in SDS sample buffer. For co-immunoprecipitation, RAW 264.7 cells from a 10-cm dish were solubilized in 400 μl lysis buffer without 2-mercaptoethanol. Lysates were precleared for 1 h at 4°C with 30 μl Ultralink beads (Pierce) and incubated for 1–2 h with 2 μg of rabbit α-TTP (CARP-3) antibody. Ultralink beads (30 μl) were added for 1 h, washed four times in lysis buffer and eluted at 37°C in SDS sample buffer in the absence of reducing agent.
Northern blot analysis
Total RNA was extracted from subconfluent 10-cm dishes using RNAqueous (Ambion). A measure of 10 μg of RNA was resolved by 1.1% agarose/2% formaldehyde MOPS gel electrophoresis, blotted onto Nytran Supercharge membranes (Schleicher and Schuell) using 8 × SSC and hybridized overnight at 50°C with digoxigenin-labeled DNA probes in DIG Easy Hyb solution (Roche). After washing at 60°C (GAPDH) or 65°C (globin) with 0.5 × SSC/0.1% SDS and 0.2 × SSC/0.1% SDS, 20 min each, the membranes were blocked in Blocking Reagent (Roche) for 30 min at room temperature, probed with alkaline phosphatase-labeled anti-digoxigenin antibody (Roche) for 30 min and washed for 45 min with 130 mM Tris–HCl pH 7.5/100 mM NaCl/0.3% Tween-20. Signals were visualized with CDP-Star (Roche). Probes were generated by PCR using digoxigenin-labeled nucleotides (Roche), β-globin and GAPDH cDNAs and the primer pairs G29/G30 and 5'GAPDH/3'GAPDH, respectively.
RNA gel shift
Conditions for gel-shift analysis were adopted from Mahtani et al (2001) with the following modifications: lysis buffer was complemented with 1 mM Na-vanadate, 40 nM okadaic acid and EDTA-free complete protease inhibitors (Roche). 32P-labeled ARE-RNA was synthesized in vitro using plasmid Stem-ARE linearized with _Xho_I, T3 polymerase and Maxiscript reagents (both Ambion), and purified with Megaclear columns (Ambion). Reactions were carried out at room temperature, and RNase T1 (Ambion) was used at a concentration of 10 U/μl. Prior to non-denaturing 6% polyacrylamide gel electrophoresis, the following antibodies were added for 15 min: 0.4 μg rabbit α-TTP (CARP-3), 0.8 μg goat α-TTP (sc-8458, Santa Cruz), 2 μg rabbit α-14-3-3 (#06-511, Upstate) or 2.5 μg rabbit α-phospho(T222)-MK2 (#07-155, Upstate).
Luciferase assay
RAW 264.7 cells were transiently transfected with pMT2-luc or pMT2-luc-UTR together with vector alone, pcDNA3-TTP-mycHis, pcDNA3-TTP-AA-mycHis or pcDNA3-TTP-PP-mycHis. Luciferase activity was measured using Luciferase Assay System reagents (Promega) and a Monolight 2010 Luminometer (Analytical Luminescence Laboratory).
Primers
Primers used were as follows. G18: 5′-ACATTCGGCCGGGTCGACCACTG-3′; G19: 5′-AATTGATATCCACCATGGCCAGCATGACCGGCGGCCAGCA GATGGGCGTGCATCTGTCCAG-3′; G29: 5′-GGTTGTCTACCCATGGAC-3′; G30: 5′-GTGGTATTTGTGAGCCAG-3′; G42: 5′-ATAAGGATCCCTGTCGGGCTCTCCGGGCC-3′; G43: 5′-TTAACTCGAGTCAGTGGGCGAGAGCCGC-3′; G50: 5′-AGTTAAGCTTGGTACCGAGCTCG-3′; G51: 5′-GGGGTACCCAGCCACAGCTTCGGCCCTCCACCAGGGCAGT GGAG-3′; G52: 5′-GGGGTACCCAGCCACAGCTTCGGCCCTCCGGCAGGCTAGT GGAG-3′; G53: 5′-GGCAAGCCGGGGAAGCTGATGC-3′; G54: 5′-CTTCCCCGGCTTGCCCTCAGG-3′; G55: 5′-GGATCGGATCCCAGAGAGTGG-3′; 5′GAPDH: 5′-TCCTGCACCACCAACTGCTTAGC-3′; 3′GAPDH: 5′-TGATGTCATCATACTTGGCAG-3; 5′luc: 5′-CCGGAATTCATGGAAGACGCCAAAAACAT-3′; 3′luc: 5′-GCTCTAGATTACAATTTGGACTTTCCGC-3′.
Acknowledgments
We thank V Kruys, DA Dixon, JA Cooper, H Holtmann, JA Steitz, AB Shyu and L Klickstein for generously providing plasmids and antibodies. We also thank S Di Marco for helpful advice on gel-shift analysis. PA was supported by National Institute of Health grants AI-33600 and AI-50167 as well as by a Biomedical Science Award from the Arthritis Foundation. TKB was supported by NIH grant CA-84418. GS is the recipient of a stipend from the Swiss National Science Foundation.
References
- Anderson P, Kedersha N (2002) Stressful initiations. J Cell Sci 115: 3227–3234 [DOI] [PubMed] [Google Scholar]
- Anderson P, Phillips K, Stoecklin G, Kedersha N (2004) Post-transcriptional regulation of pro-inflammatory proteins. J Leuk Biol, (in press) [DOI] [PubMed] [Google Scholar]
- Blackshear PJ (2002) Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem Soc Trans 30: 945–952 [DOI] [PubMed] [Google Scholar]
- Brooks SA, Connolly JE, Diegel RJ, Fava RA, Rigby WF (2002) Analysis of the function, expression, and subcellular distribution of human tristetraprolin. Arthritis Rheum 46: 1362–1370 [DOI] [PubMed] [Google Scholar]
- Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ (2001) Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem 276: 42580–42587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Chen C-YA, Shyu A-B (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20: 465–470 [DOI] [PubMed] [Google Scholar]
- Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M (2001) AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107: 451–464 [DOI] [PubMed] [Google Scholar]
- Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW (2004) MK2 phosphorylates tristetraprolin on in vivo sites including S178, a site required for 14-3-3 binding. J Biol Chem, (in press) [DOI] [PubMed] [Google Scholar]
- Civelli O, Vincent A, Maundrell K, Buri J, Scherrer K (1980) The translational repression of globin mRNA in free cytoplasmic ribonucleoprotein complexes. Eur J Biochem 107: 577–585 [DOI] [PubMed] [Google Scholar]
- Clemens MJ (2001) Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol 27: 57–89 [DOI] [PubMed] [Google Scholar]
- Fan XC, Steitz JA (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 17: 3448–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaestel M (2002) sHsp-phosphorylation: enzymes, signaling pathways and functional implications. Prog Mol Subcell Biol 28: 151–169 [DOI] [PubMed] [Google Scholar]
- Harding HP, Ron D (2002) Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51 (Suppl 3): S455–S461 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Natarajan K (2002) Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell 1: 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Geha M, Blackwell TK (2000) Similar but distinct effects of the tristetraprolin/TIS11 immediate-early proteins on cell survival. Oncogene 19: 1657–1664 [DOI] [PubMed] [Google Scholar]
- Johnson BA, Stehn JR, Yaffe MB, Blackwell TK (2002) Cytoplasmic localization of tristetraprolin involves 14-3-3-dependent and -independent mechanisms. J Biol Chem 277: 18029–18036 [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P (2001) Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans 30: 963–969 [DOI] [PubMed] [Google Scholar]
- Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P (2002) Evidence that ternary complex (eIF2–GTP–tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13: 195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P (2000) Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151: 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P (1999) RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol 147: 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol 19: 4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR (2001) Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol 21: 6461–6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Kaiser M, Moroni C (1998) c-jun N-terminal kinase is involved in AUUUA-mediated interleukin-3 mRNA turnover in mast cells. EMBO J 17: 6039–6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Stoecklin G, Lu M, Looser R, Moroni C (2001) Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol Cell Biol 21: 5778–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nover L, Scharf KD, Neumann D (1989) Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol Cell Biol 9: 1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SS, Chen CY, Xu N, Shyu AB (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J 17: 3461–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R (2003) Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300: 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Colombi M, Raineri I, Leuenberger S, Mallaun M, Schmidlin M, Gross B, Lu M, Kitamura T, Moroni C (2002) Functional cloning of BRF1, a regulator of ARE-dependent mRNA turnover. EMBO J 21: 4709–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Ming XF, Looser R, Moroni C (2000) Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol Cell Biol 20: 3753–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Stoeckle P, Lu M, Muehlemann O, Moroni C (2001) Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. RNA 7: 1578–1588 [PMC free article] [PubMed] [Google Scholar]
- Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ (1996) A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4: 445–454 [DOI] [PubMed] [Google Scholar]
- Taylor GA, Thompson MJ, Lai WS, Blackshear PJ (1995) Phosphorylation of tristetraprolin, a potential zinc finger transcription factor, by mitogen stimulation in intact cells and by mitogen-activated protein kinase in vitro. J Biol Chem 270: 13341–13347 [DOI] [PubMed] [Google Scholar]
- Tzivion G, Avruch J (2002) 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J Biol Chem 277: 3061–3064 [DOI] [PubMed] [Google Scholar]
- Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA (1999) Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol 19: 1871–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J 18: 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Loflin P, Chen CY, Shyu AB (1998) A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res 26: 558–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Zhu W, Brauchle MA, Di Padova F, Gram H, New L, Ono K, Downey JS, Han J (2001) Gene suppression by tristetraprolin and release by the p38 pathway. Am J Physiol Lung Cell Mol Physiol 281: L499–L508 [DOI] [PubMed] [Google Scholar]