E-TALEN: a web tool to design TALENs for genome engineering (original) (raw)
Abstract
Use of transcription activator-like effector nucleases (TALENs) is a promising new technique in the field of targeted genome engineering, editing and reverse genetics. Its applications span from introducing knockout mutations to endogenous tagging of proteins and targeted excision repair. Owing to this wide range of possible applications, there is a need for fast and user-friendly TALEN design tools. We developed E-TALEN (http://www.e-talen.org), a web-based tool to design TALENs for experiments of varying scale. E-TALEN enables the design of TALENs against a single target or a large number of target genes. We significantly extended previously published design concepts to consider genomic context and different applications. E-TALEN guides the user through an end-to-end design process of de novo TALEN pairs, which are specific to a certain sequence or genomic locus. Furthermore, E-TALEN offers a functionality to predict targeting and specificity for existing TALENs. Owing to the computational complexity of many of the steps in the design of TALENs, particular emphasis has been put on the implementation of fast yet accurate algorithms. We implemented a user-friendly interface, from the input parameters to the presentation of results. An additional feature of E-TALEN is the in-built sequence and annotation database available for many organisms, including human, mouse, zebrafish, Drosophila and Arabidopsis, which can be extended in the future.
INTRODUCTION
Transcription activator-like effector nucleases (TALENs) are a promising technique in the field of genome engineering (1,2). A transcription activator-like effector (TALE) contains a series of amino acid repeat domains that each recognizes a certain base. Two hyper variable amino acids within each repeat, the so-called repeat variable di-residues (RVDs), provide binding specificity to a distinct nucleotide. With this approach, RVD repeats (e.g. HD, NG, NH, NI, NK) are assembled sequentially according to the target nucleotide sequence. Thus, every TALE specifically binds its target sequence. Fusion of TALEs to the _Fok_I nuclease results in the generation of TALENs that can be applied for the introduction of DNA double-strand breaks (DSB) at specific target sites (3,4). As the _Fok_I nuclease is only catalytically active as a dimer, TALENs are always designed as pairs that must bind opposing strands of the DNA to allow dimerization of _Fok_I in a spacer region that is bridging the two TALE binding sites (5). In combination with other guidelines, it is possible to design TALEN pairs targeting almost any site in a eukaryotic genome (4,6,7). This technology creates new applications in the fields of genome engineering and functional genomics.
Genome editing processes that follow DSBs are mediated by either non-homologous end joining, randomly leading to short insertion/deletion mutations (indels), or homology-directed repair if a certain template sequence is present as donor matrix (7). In intact cells, the other allele of a gene can serve as a donor matrix (8). By using two TALEN pairs in parallel to target sequences on the same or two different chromosomes, larger mutations like inversions, deletions or translocations can be introduced. Exploiting these properties, TALENs can be used for targeted engineering as well as for reverse genomics approaches.
TALEN-mediated, targeted DNA DSB can for example be used for (i) introduction of endogenous tags into proteins; (ii) targeted introduction of small genomic mutations; (iii) excision and repair of mutations by homologous recombination after DSB; and (iv) introducing mutations resulting in frame-shifts or stop-codons (1,4).
Independent of the specific application, ensuring that the TALEN is specific for its target sequence (showing no off-target effects) and will effectively cut the DNA at the correct locus of the genome is crucial for the success of the intended purpose.
Manual investigation of all factors affecting specificity and effectiveness of the TALENs is a non-trivial and time-consuming task. To date, several specialized software approaches are available for targeted TALEN design or searching for TALENs in pre-calculated libraries (9–11, www.talen-design.de). Although these software/libraries are also available online and fulfil the purpose of finding TALENs targeting for a certain sequence, they mostly do not take genomic context into account, which could have a large effect on the effectiveness of the TALEN construct.
Here, we have developed a comprehensive web-based tool for TALEN design to generate genome editing constructs (12–14). E-TALEN can be used to design specific and efficient TALENs against single genes or large gene sets. E-TALEN is available online at http://www.e-talen.org.
RESULTS
E-TALEN web service
E-TALEN is a straightforward and easy-to-use web service for TALEN design of various scale. The service comprises two different applications: (i) a tool for the design of de novo TALENs and (ii) a tool to re-analyse and re-evaluate existing TALEN designs in their genomic context.
Input parameters have been optimized according to the end user’s intended purpose by providing for example the choice to assess putative designs for a certain experimental purpose, such as knockout or endogenous tagging, thereby restricting the designs to target either the first coding exon or the start or stop codon, respectively. The de novo design tool uses pre-formatted sequence and gene annotation databases to calculate the most suitable TALEN designs for a specific input sequence. Similarly, the re-evaluation tool enables the user to quickly assess whether a particular design is applicable for the intended purpose or not. Both tools are embedded in a user-friendly web environment guiding the scientist through the different steps of a customized design and evaluation of TALENs. The results are provided in HTML output for straightforward interpretation. In addition, TALEN designs can be visually assessed in their genomic context. Moreover, the results can be downloaded in a favoured data format for storage and follow-up analysis.
In short, the E-TALEN web service features: (i) sequence and annotation databases for various organisms; (ii) rules for design optimization towards genome engineering purposes; (iii) scoring schemes for specificity and efficiency assessment; and (iv) a user-friendly system-independent interface.
Implementation
Input
The user must input the sequence (gene or genomic region) to be targeted, which can be either entered directly or uploaded as a file. The input format is flexible, accepting FASTA-formatted nucleotide sequences or RVD-sequences (for re-evaluation), gene symbols or other common database gene IDs (e.g. FBgn numbers for flybase genes, Entrez original gene symbols, Ensembl IDs). A unique feature of E-TALEN is the search for TALENs in up to 50 sequences/genes simultaneously, enabling a parallel design of multiple TALENs for larger experiments.
An additional feature of E-TALEN is that the user, through a series of sections, can customize the search and adjust the task of E-TALEN according to experimental needs. For example, the user can exclude designs that hit putative CpG islands, intron regions or/and intergenic regions. It is important to consider these characteristics of a particular genomic location, as it may affect the efficacy of the TALEN.
Nevertheless, E-TALEN remains flexible enough to use this tool for any nucleotide sequence, as the genomic context is not considered, in case a FASTA input sequence is given but no location stated in the FASTA header.
Sequence index and annotations
If the user enters a gene symbol or ID, the associated sequence is retrieved from a genomic sequence database. Otherwise, the input sequence is read directly from the text entry. The input sequence is directly analysed for CpG islands and scanned for putative TALEN binding sites. To ensure that this search for binding sites is as effective as possible, three techniques are applied (Figure 1):
- A TV [thymidine followed by a non-thymidine (15)] should precede each TALEN binding site. As each TALEN of a TALEN pair binds to opposing DNA strands, a pattern sequence in which an A (adenine) follows a T within a specified number of nucleotides has to be found. For computational efficiency and speed, an index of each T and A in the sequence is calculated. By building a record of positions at which the sequence contains a T or A, the process is converted from a pattern-matching problem to a simple check: For every T is there an A at position T + X (where X is the sum of the left TALE length + the spacer length + the right TALE length).
- Every putative binding site found is annotated with its genomic context, i.e. whether it is contained within an exon, coding sequence, transcript, gene, CpG island and so forth. Annotation of many putative binding sites requires an efficient search of genome annotations. To maximize efficiency and shorten running time, a binary interval tree approach (16) has been used, whereby all genome annotations for the respective organism are stored as a binary interval tree (Supplementary Figure S1). This is an efficient way to store and retrieve interval information and to reduce runtime complexity for the query to O(log(n)), where n is the number of searched intervals. This idea has been successfully applied in annotating short sequencing reads after mapping (16). During the process of target site identification all putative hits are filtered by their nucleotide composition and the acquired annotations according to the user’s requirements (4).
- These criteria ensure that the target binding site is set in the correct genomic context and location and contains the most variable, hence most likely unique nucleotide sequence, similar to that of naturally occurring TALEN binding sites (3,17).
Figure 1.
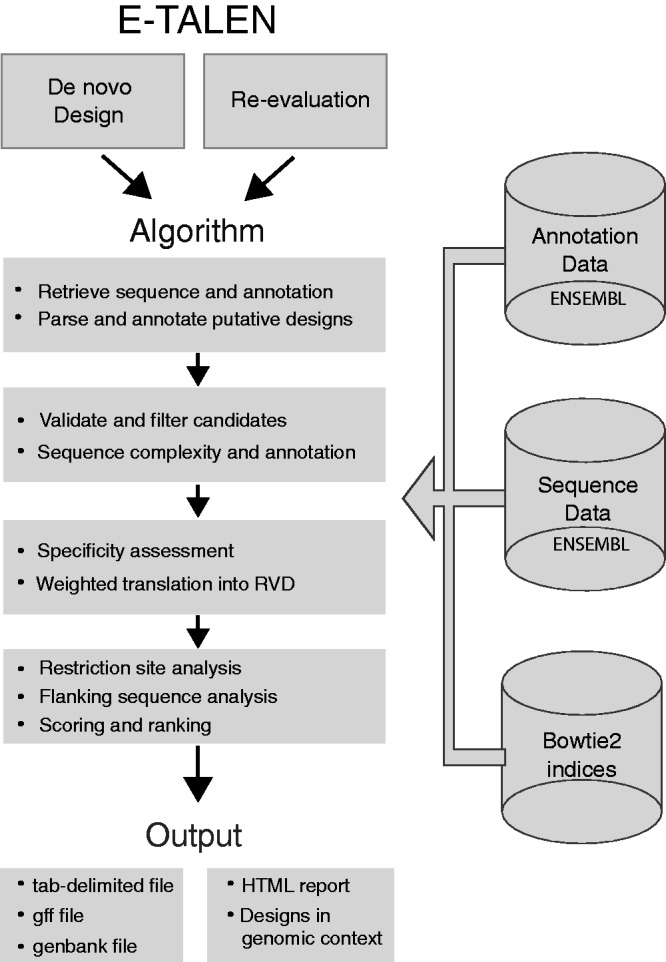
E-TALEN workflow. The E-TALEN workflow can be divided into three parts: the web service/interface, the implemented design algorithm and the output in various file formats and an html report. The web service can be sub-divided into the two different aims of de novo design of TALENs against a specific target and the re-evaluation of existing TALENs to find/re-check their target or genomic context. Depicted on the right are the pre-calculated databases that supply the design algorithm with genomic or sequence information, enabling fast and efficient information management during the design procedure. The main part of E-TALEN consists of design algorithms that find and validate putative TALEN targets, followed by providing comprehensive information on resulting TALENs and their target sites. These two parts of the workflow are hidden from the user. The last part of the E-TALEN workflow is the generation of an output that comprises various computer and human-readable file formats, which are known from high-throughput sequencing, and a visual report shown in the user’s browser.
However, off-target effects can only be excluded on testing whether the binding site is unique in the organism’s genome. To this end, the TALEN binding sites are treated as paired-end sequences, and a Bowtie2 fast alignment (18) is used to map them back to their corresponding genome. Default mapping requirements, which the user can relax, are a perfect un-gapped alignment to opposing strands, including a user-defined spacer. Any putative design that maps to several loci in the genome is considered unspecific and, thus, excluded from follow-up analysis.
Additional features
As a feature of E-TALEN, TALEN designs can additionally be checked against ‘secondary’ off-targets that were artificially introduced into the target genome/cell as for example fluorescent proteins, promoters or any other custom DNA sequences. These efforts all ensure that the resulting TALEN pair specifically cuts at the intended genomic locus/target. A further feature of E-TALEN is the possibility to check for restriction sites in the target sequence or exclusively in the TALEN’s spacer region. In addition, it is possible to retrieve a sequence of a homologous DNA donor matrix of a specified size upstream and downstream of the target sequence, which can be used for homology-directed repair.
Output
TALEN designs passing all filtering steps are translated into RVD sequences by taking the specified assembly kit and certain rules for RVD sequence composition into account as summarized by Streubel et al. (6). As the RVD code is degenerate, different RVDs can bind the same nucleotide with both different specificities and affinities (3). To counter this phenomenon and ensure target specificity, there should be >40% ‘strong’ RVDs (HD, NN). If a sufficient number of ‘strong’ RVDs (>50%) is already present, NK should be used for guanine, otherwise the ‘strong’ RVD NN.
Therefore, the translation of a nucleotide is decided on the basis of the previous translated RVDs in the TALEN to ensure that the overall composition of the resulting TALEN is as balanced as possible and, thus, effective. Accordingly, the resulting TALEN designs are scored by dividing their length by the sum of variances [Equation (1)] of nucleotide and RVD sequences [Equation (2)].
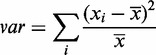
i: Nucleotides (A,C,T,G) or RVDs (NI, HD, NG, NN, NH, NK)
x: Percentage occurrence in the sequence
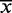 : Mean percentage occurrence in the sequence
: Mean percentage occurrence in the sequence
Equation 1—Sequence variance
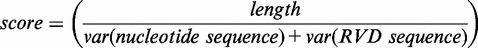
Equation 2—TALEN design score
Consequently, long TALENs with evenly distributed RVD and nucleotide sequences will have a high score and, thus, carry the highest chances to be both specific and effective in binding their targets (4). All results can be saved in multiple data formats as well as in a comprehensive HTML report including a view of the genomic locus.
TALEN targeting PTEN
We applied E-TALEN to design TALENs specifically targeting the tumour suppressor gene Pten in Drosophila melanogaster, Danio rerio and Homo sapiens. Pten codes for the phosphatidylinositol-(3,4,5)-triphosphate phosphatase, a house-keeping gene controlling the intracellular concentration of phosphatidylinositol-(3,4,5)-triphosphate (PI3) and, thus, contributes to the regulation of downstream signal transduction of PI3 kinase (PI3K) and receptor tyrosine kinases (19). Loss of function of Pten plays a role in a broad range of cancers (20).
In Table 1, we summarized the statistics describing the process of TALEN pair design for Pten in three different organisms using E-TALEN.
Table 1.
TALEN designs targeting the second exon of Pten in different organisms
| Pten gene | H. sapiens | D. rerio | D. melanogaster |
|---|---|---|---|
| Gene sequence length (bp) | 108817 | 23926 | 4730 |
| Number of all possible designs | 9052 | 1997 | 370 |
| Number of designs failing by sequence composition | 5059 | 924 | 170 |
| Number of designs failing because not hitting a gene | 0 | 0 | 0 |
| Number of designs failing because not hitting an exon | 3598 | 920 | 51 |
| Number of designs that fails because targeting in a CpG island | 34 | 30 | 0 |
| Number of designs that does not hit the specific exon number 1 | 326 | 94 | 115 |
| Number of designs that does hit multiple target sites | 8 | 0 | 0 |
| Number of designs that result after all filtering | 27 | 29 | 34 |
Concerning the design parameters, the spacer length was restricted to 15 base pairs, and each TALE was set to a length of 18 RVDs. In addition, TALEN constructs were designed to not hit CpG islands, which has been reported to be connected with poor mutagenic outcome of TALENs in vivo (21). As these designs aimed at generating gene knockouts and the first coding exon in Homo sapiens and Danio rerio Pten is covered by CpG islands, the second exon was targeted. Furthermore, the designs had to fulfil the criterion of binding a site in which each of four nucleotides contributes 10–50% of the sequence (4). The user can easily configure all of these parameters on the E-TALEN webpage. In a final step, all designs were tested with the evaluation tool to ensure that they specifically hit the target in the corresponding genetic background.
Table 1 shows the statistical output of E-TALEN depicting every filtering step, the number of successful or failed designs as well as the sequence length. The majority of putative designs failed because the potential binding site had an unfavourable nucleotide composition (e.g. one nucleotide exceeding 50% of the sequence). Likewise, many putative TALEN targets fall into intergenic regions or introns, which are not of interest for generating gene knockouts. In addition, many organisms tend to have the first coding exon and/or promoter region covered by a putatively hyper methylated region (CpG island) (21,22). Consequently, finding designs that target such a region while avoiding hyper methylated regions is more prone to failure as depicted in Supplementary Figure S2 and its corresponding Supplementary Table (S1).
In addition to a statistical output, E-TALEN produces a visualization of the result offering the user the possibility to assess the design’s quality, quantity, location, context and possible obstacles.
Figure 2 depicts a typical output for the Pten gene of three different organisms as explained earlier in the text. Therein, a number of putative TALEN pair designs fulfilling all criteria are depicted for all three organisms.
Figure 2.

TALEN design output with exon 2 of Pten in different organisms as target. TALENs were designed against the tumour suppressor gene Pten with the aim to target the second exon. Shown are independent design runs for the three organisms human, zebrafish and fruit fly. Different transcripts of the gene are coloured orange, different coding sequences are coloured light green and TALENs are shown in yellow. Note that a TALEN is considered valid if any transcript’s second exon is targeted. Targeting the second exon is likely to introduce knockout mutations in the Pten gene.
In contrast, we highlighted important regions that challenge TALEN design in Supplementary Figure S2 with differently coloured boxes. The blue box in the Drosophila melanogaster gene shows that E-TALEN found TALENs targeting the first coding sequence, which will most probably generate a knockout of this gene on introduction into cells. Conversely, E-TALEN did not find any TALENs fulfilling the design criteria applied on the Danio rerio and Homo sapiens gene because of a putative CpG island spanning the first coding exon (SF2 red/green box). As this phenomenon is often occurring in higher organisms (22–24), it can be explained that E-TALEN identified three possible TALEN designs targeting the first coding exon of Pten in Drosophila melanogaster but no single TALEN design for the Homo sapiens and Danio rerio Pten gene.
This example also illustrates that the first exon in the coding sequence is not always the first transcribed exon (SF2 blue box). Thus, targeting the first transcribed exon would result in an inefficient knockout of isoforms in which the first transcribed exon is a non-coding exon.
DISCUSSION
We implemented E-TALEN as an easy-to-use web service that guides the user end-to-end to targeted TALEN design. We successfully combined several aspects of TALEN design such as genomic context, hyper methylation patterns and target site sequence composition. Furthermore, we built E-TALEN into an easily accessible web service, with pre-processed genome annotations and sequences for several common organisms. This database can be easily extended in the future.
An aim of our work was to keep the user’s intention and application of TALENs in the laboratory in mind and to focus on usability. We realized this by offering a set of filtering conditions (tagging and/or knockout) and pre-set parameters optimized for the application of the resulting TALENs. Likewise, we optimized the computational performance of the process to keep the analysis scalable, from a single gene analysis up to library designs targeting genome scale sequences.
E-TALEN, in contrast to other TALEN design tools, does not require any prior knowledge about the target gene. A gene name or any sequence is sufficient, as further information will be retrieved from one of the databases in the background. The analysis of the TALEN design results is simplified by providing tabular data files in common formats (gff, genbank, tab-delimited) or simply an image of the TALENs and their targets in the genomic context.
With E-TALEN, existing designs (custom-made or retrieved from other tools) can be re-evaluated. A TALEN entered for evaluation will be checked for its target and, if that is found, placed in its genomic context. In this way, any TALEN construct (entered in nucleotide or RVD code) can be tested for its targeting specificity and efficiency concerning gene knockouts, tagging approaches or avoidance of hyper methylated regions.
Considering the genomic context is important for choosing an optimal design serving a certain purpose. E-TALEN is the first tool that considers this information and offers the user ways to pre-define the genomic context of the sequence to be targeted with TALENs before the design is performed. We are convinced that this will have a great impact on the specificity and efficiency of TALENs in targeted genome engineering experiments.
Future work includes the expansion of databases and genome annotations to span more organisms. As the application of TALENs is a new technique and design parameters are likely to be refined and altered with further experimental proof, future work will also include the experimental validation of TALEN designs, thereby refining design parameters and validation rules playing a role in TALEN engineering.
REQUIREMENTS
E-TALEN is a web-based tool and, thus, only dependent on an Internet connection and a browser. E-TALEN has been tested to work with Apple’s Safari (≥6.0.4), Mozilla Firefox (≥21.0) and Google Chrome (≥27.0.1453.93). Concerning the server, E-TALEN is installed on an Ubuntu Linux-based operating system with 16 GB RAM and an Intel i7 processor. It requires Perl (≥5.14.2), Bowtie2 (2.0.5) (18) and CpGi (1.3) (25).
The web-service can be accessed at http://www.e-talen.org
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Work in the laboratory of M.B. is supported by an ERC Advanced Grant of the European Commission. Funding for open access charge: Intramural funding.
Conflict of interest statement. None declared.
Supplementary Material
Supplementary Data
ACKNOWLEDGEMENTS
We thank members of the Boutros laboratory for helpful discussions.
REFERENCES
- 1.Bedell V, Wang Y, Campbell J. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark K, Voytas D, Ekker S. A TALE of two nucleases: gene targeting for the masses? Zebrafish. 2011;8:147–149. doi: 10.1089/zeb.2011.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L, Zhou R, Kuo YC, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beurdeley M, Bietz F, Li J. Compact designer TALENs for efficient genome engineering. Nature. 2013;4:1762. doi: 10.1038/ncomms2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Streubel J, Blücher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 2012;30:593–595. doi: 10.1038/nbt.2304. [DOI] [PubMed] [Google Scholar]
- 7.Joung J, Sander J. TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2012;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandt V, Hewitt S, Skok J. It takes two: Communication between homologous alleles preserves genomic stability during V (D) J recombination. Nucleus. 2010;1:23–29. doi: 10.4161/nucl.1.1.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neff K, Argue D, Ma A, Lee H. Mojo hand, a TALEN design tool for genome editing applications. BMC. 2013;14:1. doi: 10.1186/1471-2105-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doyle E, Booher N. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Kweon J, Kim A, Chon J, Yoo J. A library of TAL effector nucleases spanning the human genome. Nature. 2013;31:251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt E, Pelz O, Buhlmann S. GenomeRNAi: a database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 2013;41:D1021–D1026. doi: 10.1093/nar/gks1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arziman Z, Horn T, Boutros M. E-RNAi: a web application to design optimized RNAi constructs. Nucleic Acids Res. 2005;33:W582–W588. doi: 10.1093/nar/gki468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horn T, Sandmann T, Boutros M. Software Design and evaluation of genome-wide libraries for RNA interference screens. Genome Biol. 2010;11:R61. doi: 10.1186/gb-2010-11-6-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cornish-Bowden A. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res. 1985;13:3021–3030. doi: 10.1093/nar/13.9.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renaud G, Neves P, Folador E. Segtor: rapid annotation of genomic coordinates and single nucleotide variations using segment trees. PloS One. 2011;6:e26715. doi: 10.1371/journal.pone.0026715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cermak T, Doyle E, Christian M. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Paluch B, Wang X, Jiang X. PTEN at a glance. J. Cell Sci. 2012;125:4687–4692. doi: 10.1242/jcs.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu P, Cheng H, Roberts T, Zhao J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen S, Oikonomou G, Chiu C. A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res. 2013;41:2769–2778. doi: 10.1093/nar/gks1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J. Research Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010;11:R70. doi: 10.1186/gb-2010-11-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl Acad. Sci. USA. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sati S, Tanwar V, Kumar K, Patowary A. High resolution methylome map of rat indicates role of intragenic DNA methylation in identification of coding region. PloS One. 2012;7:e31621. doi: 10.1371/journal.pone.0031621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takai D, Jones P. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl Acad. Sci. USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data