Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation (original) (raw)
Abstract
Vascular dysfunction is emerging as a key pathological hallmark in Alzheimer’s disease (AD). A leaky blood–brain barrier (BBB) has been described in AD patient tissue and in vivo AD mouse models. Brain endothelial cells (BECs) are linked together by tight junctional (TJ) proteins, which are a key determinant in restricting the permeability of the BBB. The amyloid β (Aβ) peptides of 1–40 and 1–42 amino acids are believed to be pivotal in AD pathogenesis. We therefore decided to investigate the effect of Aβ 1–40, the Aβ variant found at the highest concentration in human plasma, on the permeability of an immortalized human BEC line, hCMEC/D3. Aβ 1–40 induced a marked increase in hCMEC/D3 cell permeability to the paracellular tracer 70 kD FITC-dextran when compared with cells incubated with the scrambled Aβ 1–40 peptide. Increased permeability was associated with a specific decrease, both at the protein and mRNA level, in the TJ protein occludin, whereas claudin-5 and ZO-1 were unaffected. JNK and p38MAPK inhibition prevented both Aβ 1–40-mediated down-regulation of occludin and the increase in paracellular permeability in hCMEC/D3 cells. Our findings suggest that the JNK and p38MAPK pathways might represent attractive therapeutic targets for preventing BBB dysfunction in AD.
Keywords: Alzheimer’s disease, amyloid β, blood–brain barrier, brain endothelial cells, occludin, JNK, p38MAPK
Introduction
The blood–brain barrier (BBB) restricts the movement of hydrophilic molecules across brain capillaries [1]. Cerebral capillaries are lined with brain endothelial cells (BECs), which are sparingly covered by pericytes on the abluminal membrane. BECs and pericytes are anchored within a basal lamina, which is surrounded by astrocytic end feet. The intimate association between astrocytes, pericytes and BECs is often referred to as the neurovascular unit as each cell type is important for the physiological function of the BBB [2]. BECs are interconnected by adherens and tight junctions (TJs), which confer the low permeability of the BBB. Although vascular adherens junctions are ubiquitous, continuous TJ are not found in endothelial cells in most other tissues. TJs consist of the transmembrane proteins junctional adhesion molecule 1 (JAM-1), occludin and claudin (cldn) family members and of intracellular proteins, namely, zona occludens family members (ZO-1, 2), cingulin, AF-6 and 7H6 [2], which serve to anchor the TJ to the actin cytoskeleton. Increased BBB permeability is correlated with disruption of TJ protein organization in many diseases, including multiple sclerosis and cerebral ischaemia [2]. More recently, BBB dysfunction is emerging as a key pathological hallmark in Alzheimer’s disease (AD).
AD is characterized by the accumulation of amyloid β (Aβ) plaques [3] and oligomers [4], intraneuronal neurofibrillary tangles of tau [5], neuronal atrophy/dysfunction [6] and activated astrocytes and microglia [7]. Increased Aβ levels, in particular Aβ 1–40 and 1–42, are believed to play a major role in AD pathologies, inducing neuronal apoptosis and inflammation [8]. Recently, several features of vascular dysfunction in AD patients have been demonstrated, including brain hypoperfusion [9], decreased Aβ clearance [10], reduced glucose transporter 1 expression [11], BEC morphological alterations [12] and increased BBB permeability [13, 14].
An increase in the permeability of the BBB could have potentially disastrous consequences for the neuronal homeostatic environment, and has been demonstrated in AD patients by the abnormal presence of plasma proteins in the CSF or brain parenchyma, although some contrasting results exist [15–17]. Most recent studies have demonstrated a correlation between BBB leakage and advanced-stage AD [14] as well as medial temporal atrophy [13]. Animals receiving a peripheral Aβ injection [18] and transgenic AD mouse models [19] display increased BBB permeability. Aβ peptides have also been demonstrated to increase the permeability of non-human endothelial cell cultures [20, 21]. Changes in TJ organization may in part be responsible for the increased BBB permeability found in AD. Indeed, morphological alterations of BEC TJs, suggestive of a leaky BBB, have been observed in AD patients’ brain biopsies [22], and occludin down-regulation is found in APP/PS1 transgenic mice [23]. Aβ 1–42 has also been noted to transiently decrease occludin expression and cause cldn-5 translocation from TJs in rat primary BECs [24].
In this study, we have investigated the effects of Aβ on the permeability of human BECs in vitro. To do so, we used a well-established immortalized human BEC line, hCMEC/D3 cells [25], to determine whether Aβ peptides could induce increased BEC permeability and to explore the signalling pathways that could mediate such changes.
Materials and methods
Materials
Aβ 1–40 and 1–42 were obtained from Bachem (Weil am Rhein, Germany), whereas scrambled Aβ (sAβ) 1–40 and 1–42 were purchased from Covance (San Diego, CA, USA). SP600125, TAT-TI-JIP153-163, PD98059 and SB203580 were purchased from Calbiochem (Gibbstown, NJ, USA). All other reagents were from Sigma-Aldrich (Gillingham, Dorset, UK), unless stated.
Cell culture and treatments
hCMEC/D3 cells [25] were cultured on collagen-coated tissue culture plastic or collagen-coated glass cover slips (0.005%[w/v] collagen, 1 hr). Cultures were maintained in a humidified atmosphere at 37°C in 5% CO2 in EGM-2 medium (Lonza, Slough, Wokingham, UK), which was supplemented with VEGF, IGF, bFGF, hydrocortisone, ascorbate, gentamycin and 2.5% foetal bovine serum (FBS). The concentrations of the supplements are not made available by the supplier; however, one fourth of the supplied volume of VEGF, IGF, bFGF and the total volume of hydrocortisone, ascorbate, gentamycin was added. For all experiments, hCMEC/D3 cells were grown to confluence (approximately 1 × 105 cells/cm2) with the EGM-2 media changed every 2–3 days. At confluence, hCMEC/D3 cells were left for 2–3 days without media change and then incubated with Aβ or sAβ peptides at the concentrations and times indicated. For inhibition studies, hCMEC/D3 cells were pre-treated for 30 min. prior to Aβ or sAβ treatment with the inhibitors shown in Table 1. Soluble monomeric Aβ form was applied to the culture medium. Following 48 hrs of incubation, no Aβ oligomers could be detected as assessed by Western blotting (Fig. S1). In addition to Aβ treatment as described earlier, for some experiments, hCMEC/D3 cells were incubated under UV light for 30 min. as a positive control of MAPK activation.
Table 1.
Inhibitors and concentrations used
| Modulator | Pathway inhibited | Concentration | Source |
|---|---|---|---|
| SP600125 | JNK 1, 2 and 3 | 50 μM | Calbiochem, NJ |
| TAT-TI-JIP153–163 | JNK 1, 2 and 3 | 10 μM | Calbiochem |
| SB203580 | p38MAPK | 20 μM | Calbiochem |
| PD98059 | MEK | 50 μM | Calbiochem |
| pp2 | Src family | 10 μM | Calbiochem |
| Y-27632 | ROCK inhibitor | 700 nM | Calbiochem |
| Bisindolylmaleimide -1 (Bis-1) | PKC | 10 μM | Calbiochem |
| LY294002 | PI3-kinase | 300 μM | Calbiochem |
| Lithium chloride (LiCl) | GSK-3? | 5 mM | Sigma-Aldrich, Dorset, UK |
| sn50 | NF-kβ? | 200 μM | Calbiochem |
| sn50-m | Control sn-50 | 200 μM | Calbiochem |
MTT assay
hCMEC/D3 cells were incubated with 500 μg/ml of MTT for 4 hrs at 37°C, washed twice in PBS and the reaction stopped by the addition of isopropanol. The colour intensity of the dissolved formazan salts was then measured at a wavelength of 550 nm and a background reading was taken at 695 nm using a BMG plate reader (Offenberg, Germany). After subtracting background readings, cell viability was expressed as a percentage of control vehicle–treated cells.
Permeability studies
hCMEC/D3 cells were grown to confluence on transwell polyester membrane inserts (Corning Costar, High Wycombe, Buckinghamshire, UK [0.4 μm pore, 12 mm diameter]), which were first coated with collagen as before and then with fibronectin (5 μg/ml for 1 hr). The paracellular permeability to 70 kD FITC-dextran of the hCMEC/D3 cell monolayers was then investigated as previously described [25]. Briefly, 2 mg/ml of 70 kD FITC-dextran was added to the apical chamber, and the fluorescence that crossed to the basolateral chamber was determined over 30 min., in successive 5-min. periods, using a BMG plate reader. The volume cleared was plotted against time, and the slopes of the curves used to calculate the permeability coefficients (_P_e, cm/min.) of the endothelial cell monolayer:
_P_e= PS/s, where PS (clearance) is the permeability surface area of the endothelial monolayer and s is the surface area of the filter (1.1 cm2).
PS is given by 1/PS = 1/_m_e– 1/_m_f, where _m_e and _m_f are the slopes of the curves corresponding to endothelial cells on filters and to filters only, respectively. _m_e and _m_f were calculated by plotting the cleared volume against time.
The cleared volume was calculated by (AUa– AUb)/_F_i, where AUa is the total fluorescence (arbitrary units) in the basal compartment, AUb is the background fluorescence and _F_i is the fluorescence of the initial solution (AU/ml).
Western blotting
Confluent hCMEC/D3 cells were lysed in 500 μl of RIPA buffer (Sigma-Aldrich) (150 mM NaCl, 1.0%[w/v] IGEPAL® CA-630, 0.5%[w/v] sodium deoxycholate, 0.1%[w/v] SDS, 50 mM Tris, pH 8.0) containing 1 mM PMSF and 10 ng/ml of aprotinin, leupeptin and pepstatin A. Protein levels were measured by a Biorad DC protein assay (Hemel Hempstead, UK). Samples were denatured in 1 × Laemmli’s buffer and 20 μg of total protein in the whole cell lysates was loaded per lane, resolved on SDS-PAGE gels, appropriate for the molecular weight of each protein under investigation and transferred to a nitrocellulose membrane. After incubation with one of the primary antibodies shown in Table 2 for 2 hrs at room temperature (RT), membranes were incubated for 1 hr at RT with a species-specific secondary antibody conjugated to horseradish peroxidase (Pierce Biotechnology, Tattenhoe, UK) (1 in 1000 dilution), and were visualized by enhanced chemiluminescence detection (ECL, GE Healthcare, Little Chalfont, UK). To confirm equal loading, membranes were stripped in 2% (w/v) SDS, 0.0625 M Tris, pH 6.8, 0.008% (v/v) β-mercaptoethanol for 40 min. at 50°C and re-probed with one of the antibodies indicated in Table 1. For the detection of MAPK activation, membranes were first probed with antibodies for the phosphorylated form of each MAPK member, then stripped as above and re-probed with antibodies for total MAPK levels. MAPK activation following Aβ treatment was calculated using the following formula:
Table 2.
List of antibodies used for immunocytochemistry, flow cytometry and Western blotting*
| Anti- | Species | Concentration | Source |
|---|---|---|---|
| Phospho- Thr183/Tyr185 JNK | Rabbit polyclonal | 1 in 1000 dilution | Cell Signaling, Danvers, MA |
| Phospho- Thr180/Tyr182 p38MAPK | Rabbit polyclonal | 1 in 1000 dilution | Cell Signaling |
| Phospho- Thr185/Tyr187 ERK1/2 | Mouse IgG1 monoclonal | 1 in 1000 dilution | Biosource, San Jose, CA |
| ZO-1 | Rabbit polyclonal | 1 μg/ml | Zymed, San Francisco, CA |
| Cldn-5 | Mouse IgG1 monoclonal | 1 μg/ml | Zymed |
| Occludin | Rabbit polyclonal | 1 μg/ml | Zymed |
| JNK | Rabbit polyclonal | 1 in 1000 dilution | Cell Signaling |
| p38MAPK | Rabbit polyclonal | 1 in 1000 dilution | Cell Signaling |
| ERK2 | Mouse IgG2a monoclonal | 1 in 1000 dilution | Upstate, Temecula, CA |
| β Actin | Mouse IgG1 monoclonal | 1 μg/ml | Sigma-Aldrich, Dorset, UK |
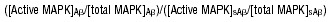
RNA extraction and qPCR
RNA was extracted from hCMEC/D3 cells using Trizol reagent (Invitrogen, Paisley, Renfrewshire, UK), and 1 μg of RNA was then converted to cDNA using a reaction-ready first-strand synthesis kit (Superarray, Frederick, MD) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was then carried out using SYBR green and primers for cldn-5, occludin, actin and ZO-1 according to the manufacturer’s protocol (Superarray). Amplification was performed in a thermal cycler (DNA engine Opticon2, Bio-Rad, Hercules, CA) using a 40-cycle program (95°C for 30 sec., 55°C for 30 sec. and 72°C for 30 sec.). The expression of each target mRNA in hCMEC/D3 cells treated with Aβ or sAβ was compared with that of the housekeeping gene, actin, using the following formula:

Immunocytochemistry
hCMEC/D3 cells were fixed and permeabilized in ice-cold methanol for 10 min. at –20°C, followed by incubation in blocking buffer (3%[w/v] normal goat serum [NGS] in phosphate buffered saline [PBS]) for 30 min. Cells were then incubated with antibodies for cldn-5, ZO-1, occludin (Table 1) or rabbit IgG as an isotype-matched control at a concentration of 2.5 μg/ml overnight at 4°C, washed three times in PBS and incubated with Alexa Fluor-488 anti-rabbit polyclonal IgG (10 μg/ml, 1 hr, RT [Invitrogen]). After three further washes in PBS, the slides were mounted and viewed with an Olympus BX61 fluorescent microscope (Watford, UK).
Flow cytometry
hCMEC/D3 cells were washed in HBSS without Ca2+ and Mg2+ and then detached with 0.25% w/v porcine trypsin and 0.2% w/v EDTA. EGM-2 (containing supplements as described earlier) was then added to neutralize trypsin activity and the cells were fixed in 4%_p_-formaldehyde (PAF) for 10 min. After one wash in PBS, cells were blocked and permeabilized for 30 min. (0.1%[v/v] Triton X-100, 3%[w/v] NGS in PBS), then incubated for 2 hrs at RT with occludin or cldn-5 antibodies (2.5 μg/ml in PBS). hCMEC/D3 cells were then washed three times in PBS and incubated with a species-specific Alexa-Fluor-488 or FITC-conjugated secondary antibody (10 μg/ml). After two final washes with PBS, the cells were suspended in 500 μl PBS and analyzed using a FACSCalibur flow cytometer with Cellquest software (488 nm λexcitation, 530 nm λemission[Becton Dickinson, Oxford, UK]). For each sample, the median fluorescence of 10,000 cells was determined. After background subtraction, the data were represented as percentage median fluorescence of cells treated with the sAβ peptide.
Statistical analysis
All data are represented as means ± S.E.M. and the number of experiments, n, indicated. Statistical significance was calculated using anova followed by Student’s t-test, comparing each treatment to the sAβ control (*P < 0.05, **P < 0.01). For the Western blotting, qPCR and permeability experiments, a paired t-test was employed because of variability in control values between experiments. For qPCR, analysis was carried out using ΔCT values, as the 2−ΔΔCT method standardized all data so that the sAβ value is always 1.
Results
High concentrations of Aβ peptides reduce hCMEC/D3 cell viability
An aim of this study was to investigate the effect of non-cytotoxic concentrations of Aβ peptides on hCMEC/D3 cell permeability. We therefore measured hCMEC/D3 cell viability in the presence of Aβ 1–42 or Aβ 1–40 or their scrambled counterparts using an MTT assay. As shown in Fig. 1, hCMEC/D3 cell viability was unaffected by 48 hrs of incubation with Aβ 1–42 or Aβ 1–40 at concentrations up to 5 μM. However, at 10 μM, both Aβ 1–40 and Aβ 1–42, but not sAβ peptides, reduced cell viability compared with vehicle-treated cells (70 and 74%, respectively). Therefore, for subsequent investigations, to avoid cytotoxic effects, treatments with Aβ peptides were at 5 μM for 48 hrs. It should be noted that the 4 kD form of Aβ was added initially to the culture medium, and no soluble oligomers were detected by Western blotting over 48 hrs of incubation (Suppl. Fig. 1).
Fig 1.
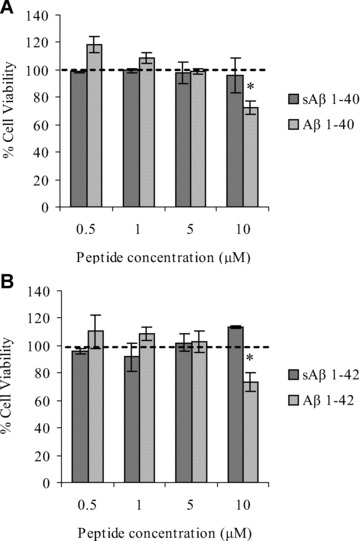
Cell viability of hCMEC/D3 cells incubated with Aβ 1–40 and Aβ 1–42 peptides for 48 hrs. hCMEC/D3 cells were incubated with (A) Aβ 1–40 or sAβ 1–40 and (B) Aβ 1–42 or sAβ 1–42 for 48 hrs at the concentrations indicated. hCMEC/D3 cell viability was measured using an MTT assay and expressed as a percentage of untreated cells. Data represent mean ± S.E.M., _n_= 3 experiments with quintuplet samples. *P < 0.05 compared with control.
Aβ 1–40 increases hCMEC/D3 cell paracellular permeability
Increased BBB permeability has been demonstrated in AD patient brain tissue and in vivo AD models [13, 14, 19]. We therefore investigated the effect of Aβ peptides on the paracellular permeability of hCMEC/D3 cells to 70 kD FITC-dextran. As shown in Fig. 2, Aβ 1–40 or Aβ 1–42 incubation increased the paracellular permeability of hCMEC/D3 cells by 50 and 27% respectively, compared with cells treated with sAβ peptides. However, only Aβ 1–40-mediated increased BEC permeability was statistically significant. For subsequent studies, we focussed our attention on the increased paracellular permeability induced by Aβ 1–40, because, compared with Aβ 1–42, this peptide is found at higher concentrations in the plasma [26] and in cerebrovascular deposits [27].
Fig 2.
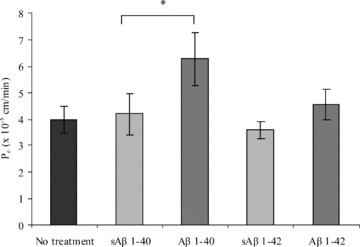
Permeability coefficient (_P_e) of hCMEC/D3 cells to 70-kD FITC-dextran after 48 hrs of incubation with Aβ or sAβ peptides. hCMEC/D3 cells were incubated with 5 μM of Aβ 1–40, Aβ1–42, sAβ 1–40 or sAβ 1–42 for 48 hrs and the _P_e of the hCMEC/D3 cell monolayer to 70-kD FITC-dextran was investigated. Data represent mean ± S.E.M., _n_= 3 experiments with duplicate samples. *P < 0.05 comparing _P_e values of cells treated with Aβ with those treated with sAβ.
Aβ 1–40 specifically decreases levels of occludin at the protein and mRNA levels
The mechanisms behind Aβ 1–40-induced increases in hCMEC/D3 cell paracellular permeability were investigated next. TJ proteins mediate BEC cell–cell contacts at the BBB, and changes in their expression are associated with increased BEC permeability. We therefore investigated the expression of occludin, cldn-5 and ZO-1, the most extensively investigated TJ proteins expressed by BECs, following Aβ 1–40 or sAβ 1–40 treatment. As shown in Fig. 3A and B, 48 hrs of incubation with Aβ 1–40 caused a decrease in occludin levels, but not in cldn-5 or ZO-1 levels, as assessed by Western blotting. After normalizing for equal loading with actin, occludin expression was 50% in cells treated with Aβ 1–40 compared with those treated with sAβ 1–40. Alterations in occludin protein expression were preceded by a decrease in occludin mRNA levels in hCMEC/D3 cells. As can be seen in Fig. 3C, after 24 hrs of incubation, Aβ 1–40 induced a significant decrease in mRNA levels for occludin but not for cldn-5 or ZO-1. Interestingly, after 48 hrs of incubation, the mRNA levels of ZO-1 were increased with Aβ 1–40 treatment compared with treatment with sAβ 1–40.
Fig 3.
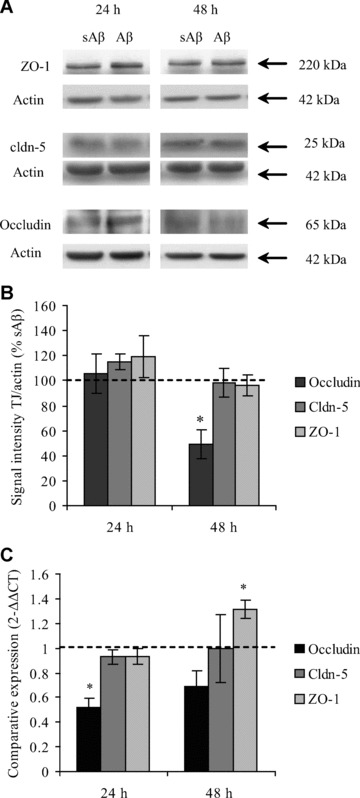
Tight junctional (TJ) protein and mRNA expression by hCMEC/D3 cells after incubation with Aβ 1–40. hCMEC/D3 cells were incubated for 24 hrs or 48 hrs with 5 μM of Aβ 1–40 or sAβ 1–40. (A) The protein levels of TJ proteins occludin, cldn-5 and ZO-1 were assessed by Western blotting, (B) normalized to actin levels and expressed as a percentage of expression of cells treated with the sAβ 1–40 peptide. (C) mRNA levels of occludin, cldn-5 and ZO-1 were assessed by qPCR. Data represent mean ± S.E.M., _n_= at least three experiments with duplicate samples. *P < 0.05 comparing values for Aβ- treated versus sAβ-treated cells.
Occludin expression is often found at TJ, where it acts to reduce BEC paracellular permeability. As demonstrated in Fig. 4E, after sAβ 1–40 incubation, occludin expression was strongly detected at the cell–cell borders between confluent hCMEC/D3 cells, indicative of TJ localization, with a lower level of intracellular staining. In comparison, in hCMEC/D3 cells incubated with Aβ 1–40 (Fig. 4F), staining was reduced both at the cell borders and intracellularly and in some instances, a complete loss of occludin at the cell junctions was observed. In contrast, there were no changes in either overall expression levels or in the sub-cellular localization of ZO-1 (Fig. 4C and D) or cldn-5 (Fig. 4A and B) in hCMEC/D3 cells after Aβ 1–40 incubation, compared with sAβ 1–40 incubation. These data suggest that the Aβ 1–40-induced increase in hCMEC/D3 cell paracellular permeability may be a consequence of reduced occludin levels at cell junctions.
Fig 4.
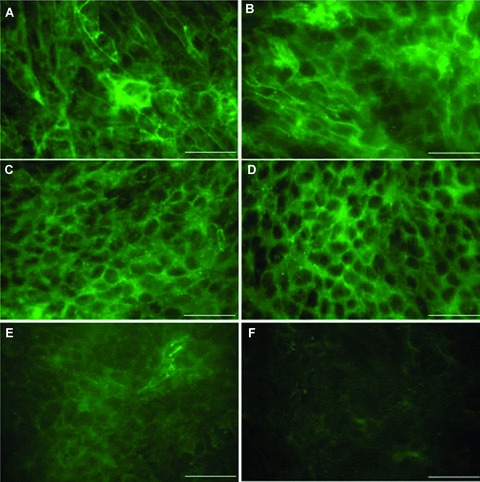
The expression of TJ proteins by hCMEC/D3 cells after 48 hrs of Aβ 1–40 incubation as assessed by immunocytochemistry. hCMEC/D3 cells were incubated with 5 μM (A, C, E) sAβ 1–40 or (B, D, F) Aβ 1–40 for 48 hrs, fixed in methanol and the expression of cldn-5 (A, B), ZO-1, (C, D) or occludin (E, F) visualized by fluorescence microscopy. Image shown is of one experiment representative of three. Scale bar = 50 μM.
Occludin down-regulation is mediated by the JNK and p38MAPK signalling pathways
We next sought to identify the contribution of various signalling pathways in Aβ-mediated occludin down-regulation, using flow cytometry as a quantitative measure of protein expression analysis. We found that inhibitors for pathways that have been previously shown to regulate TJ expression (ROCK, Src, PKC, PI3K) [28] and those known to be activated by Aβ in neurons (NF-κβ, GSK-3β) [29] did not prevent occludin down-regulation in hCMEC/D3 cells mediated by Aβ 1–40 (Suppl. Fig. 2).
MAPK family members ERK [30], JNK [31] and p38MAPK [32] have been reported to be up-regulated in neurons from AD patients. Aβ 1–40 treatment induced a time-dependent activation of JNK (46-kD form), ERK (42-kD form) and p38MAPK (42–kD form) in hCMEC/D3 cells (Fig. 5A and B). JNK activation was detected as early as 6 hrs post-incubation with Aβ 1–40 and increased with time up to 48 hrs, whereas ERK2 and p38MAPK were also activated by Aβ 1–40 but from 24 hrs onwards.
Fig 5.
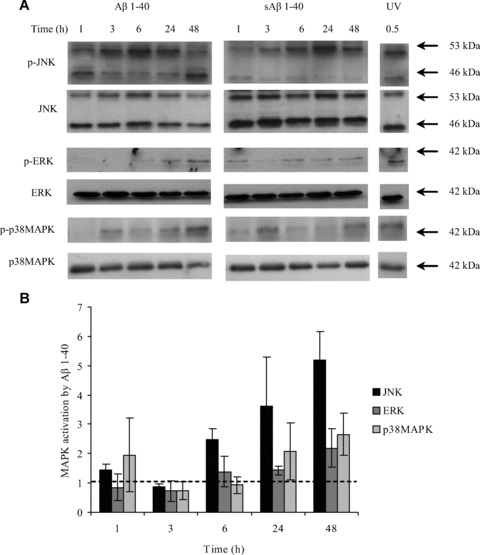
MAPK activation by Aβ 1–40 in hCMEC/D3 cells. (A) hCMEC/D3 cells were incubated with 5 μM sAβ 1–40 or Aβ 1–40 for 1, 3, 6, 24 and 48 hrs, or under UV light for 30 min. as a positive control. The cells were then lysed in RIPA buffer, and the protein levels of phosphorylated and non-phosphorylated MAPK family members were investigated by Western blotting. (B) The optical densities of the active JNK (46 kD), ERK2 and p38MAPK bands were normalized, respectively, to total JNK, ERK and p38MAPK levels. The normalized values for Aβ 1–40 were divided by those obtained for sAβ 1–40. Data represent mean ± S.E.M., _n_= 3 experiments.
Inhibitors for each MAPK family member were used in order to investigate the signalling pathways mediating Aβ 1–40-induced occludin down-regulation (Fig. 6A). In this set of experiments, hCMEC/D3 occludin levels were decreased to 71% in Aβ 1–40-treated cells compared with those treated with sAβ 1–40, and pre-incubation with 50 μM of the MEK inhibitor PD98059, which inhibits ERK, did not prevent this down-regulation. In contrast, 20 μM SB203580 (a p38MAPK inhibitor), and 50 μM SP600125 or 10 μM TAT-TI-JIP153–163 (both JNK inhibitors), prevented Aβ-induced occludin down-regulation in hCMEC/D3 cells. In addition, pre-treatment of hCMEC/D3 cells with SB203580, SP600125 or TAT-TI-JIP153–163 prevented the Aβ 1–40-mediated decrease in occludin mRNA levels after 24 hrs of incubation (Fig. 6B). These results suggest that Aβ 1–40 induces brain endothelial occludin down-regulation in a p38MAPK- and JNK-dependent manner.
Fig 6.
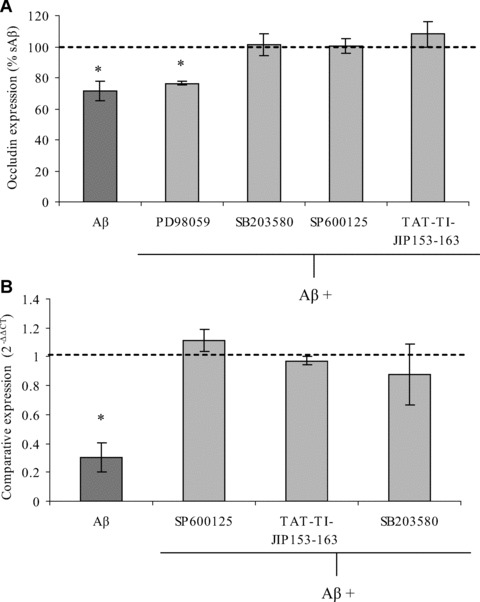
The effect of MAPK inhibitors on Aβ 1–40-mediated occludin down-regulation by hCMEC/D3 cells. hCMEC/D3 cells were pre-incubated for 30 min. with 50 μM SP600125, 10 μM TAT-TI-JIP153–163, 20 μM SB203580, 50 μM PD98059 or a vehicle control, and then treated with Aβ 1–40 or sAβ 1–40 for 48 hrs. The expression of occludin was then measured (A) at the protein level by flow cytometry or (B) at the mRNA level using qPCR. Data represent mean ± S.E.M., _n_= 3 experiments with duplicate samples. *P < 0.05 comparing cells treated with Aβ 1–40 with those of cells treated with sAβ 1–40.
JNK and p38MAPK inhibition attenuates Aβ 1–40-induced increased paracellular hCMEC/D3 cell permeability
Since JNK and p38MAPK inhibitors all attenuated occludin down-regulation mediated by Aβ 1–40, we investigated whether they could also prevent Aβ 1–40-increased hCMEC/D3 cell permeability (Fig. 7). Aβ 1–40 treatment increased the _P_e values of hCMEC/D3 cells to 70-kD FITC-dextran by 70% compared with sAβ 1–40-treated cells. Pre-incubation with SB203580, TAT-TI-JIP153–163 and SP600125 prevented this increase in brain endothelial permeability. However, it should be noted that pre-incubation with SB203580 slightly increased the _P_e values for hCMEC/D3 cells when incubated with both sAβ 1–40 and Aβ 1–40 (by 32% and 25%± 12.6, respectively, when expressed as a percentage of vehicle–pre-incubated sAβ 1–40 controls). Our results show that JNK and p38MAPK inhibition can prevent Aβ 1–40-mediated increase in hCMEC/D3 permeability to 70-kD FITC-dextran, most likely by preventing occludin down-regulation.
Fig 7.
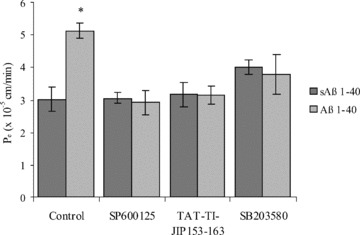
The effect of p38MAPK and JNK inhibition on the permeability coefficient (_P_e) of hCMEC/D3 cells to 70-kD FITC-dextran. hCMEC/D3 cells were pre-incubated with 50 μM SP600125, 10 μM TAT-TI-JIP153–163, 20 μM SB203580 or a vehicle control for 30 min. and then treated with Aβ 1–40 or sAβ 1–40 for 48 hrs. The _P_e of the hCMEC/D3 cell monolayer to 70-kD FITC-dextran was determined. Data represent mean ± S.E.M., _n_= 3 experiments with duplicate samples. *P < 0.05 comparing _P_e values of cells treated with Aβ with those treated with sAβ.
Discussion
In this study, we initially determined the cytotoxicity of Aβ peptides to hCMEC/D3 cells. We then selected non-cytotoxic concentrations to further investigate specific Aβ effects on BBB permeability, rather than Aβ-induced gross permeability changes due to BEC death. The lowest dose of Aβ 1–40 or Aβ 1–42 to elicit cytotoxic effects to human BECs after 48 hrs of incubation was, in both cases, 10 μM. Results from other research groups have found a large variation in Aβ concentrations needed to elicit toxic effects, ranging from 100 nM to greater than 20 μM in studies using different EC types and culture conditions. Multiple factors may affect the cytotoxicity of Aβ to ECs in vitro, including whether cultured ECs are primary or immortalized cell lines, their confluent state, tissue and species of origin and Aβ aggregation in the culture medium. Whether Aβ oligomers or aggregates were responsible for Aβ toxicity to hCMEC/D3 cells at high concentrations remains to be determined. In the current study, we observed that incubation with non-cytotoxic concentrations of Aβ 1–40, but not of Aβ 1–42, increased the permeability of human BECs. Aβ 1–40 was added in soluble monomeric form and, at the concentrations used here, no aggregate and/or oligomer formation was detected by Western blotting over the duration of the experiment (Fig. S1). Although Aβ 1–40 aggregates and/or oligomers may still induce EC death at high concentrations, as previously shown in neurons [4], the soluble monomeric form of Aβ 1–40 may be responsible for subtler changes in BBB function that do not involve cell death.
Our in vitro findings are consistent with observations in AD patients, where a leaky BBB could potentially disrupt the CNS homeostatic environment and lead to neuronal degeneration. Increased CSF/plasma protein ratio of albumin and IgG has been noted in AD patients, suggestive of a leaky BBB [13, 14, 33–36]. However, other studies have not confirmed this finding [15–17]. The most compelling evidence for increased BBB permeability in AD patients was provided by Zipser _et al._[14], who found that prothrombin leakage from the blood into the CNS was significant in advanced-stage AD. In addition, increased cerebrospinal fluid levels of albumin are associated with medial temporal atrophy as assessed by MRI measurements in AD patients [13]. Aβ could play a central role in mediating increased BBB permeability. Indeed, Tg2576 mice, which harbour the human Swedish mutation of APP, display increased BBB permeability after 4 months of age, prior to plaque deposition and disease onset, and these effects can be reversed via Aβ peptide immunization [19, 37]. Further evidence comes from studies showing that intravascular jugular vein injections of Aβ 1–40 can induce a leaky BBB in rats [18]. In vitro endothelial cells of non-human origin also display increased permeability after Aβ treatment [20, 21]. In the present study, Aβ 1–40 was added to the luminal compartment, which corresponds to the blood side in vivo. Strazielle et al. compared the effects of luminal and abluminal application of 5 μM Aβ 25–35 on the permeability of bovine BECs to the paracellular tracer PEG [21]. They observed that although both luminal and abluminal Aβ increased BEC permeability, the rise in PEG efflux was not observed until much later when Aβ 25–35 was applied abluminally. These authors speculated that the lag period might be explained as the time required for the peptide to diffuse from the lower compartment through the BEC monolayer and subsequently reach high enough levels on the luminal interface to induce barrier changes. Because of the polarized nature of BECs, it is possible that the receptor(s) activated by Aβ (e.g. RAGE, megalin, P-gp or an unknown receptor) mediating permeability effects might be differentially expressed between the luminal and abluminal membranes. Whether Aβ acts on the luminal or abluminal membranes or on both to increase BEC permeability remains to be determined.
Measurements of plasma Aβ levels have been inconclusive as to whether Aβ levels are increased or unaltered in AD [26], although Van Oijen _et al._[38] found that high Aβ 1–40 levels in plasma in early life corresponded with an increased risk of developing dementia in later life. Increased plasma levels of Aβ, acting at the luminal surface, might induce subtle changes in BBB permeability and lead to the leakage of plasma proteins observed in AD. On the abluminal side of the BBB, increased Aβ production by neurons, stiffening of arteries during aging and/or increased plasma Aβ leakage (as a result of occludin down-regulation in BECs) in AD could potentially reduce the clearance of Aβ from the brain via drainage of interstitial fluid and lead to cerebral amyloid angiopathy (CAA), a frequent occurrence in AD patients [39, 40]. In CAA, cerebrovascular deposits of Aβ (of both 1–40 and 1–42 amino acids in length) are found aggregated with other proteins, including apoE and α-2 macroglobulin. CAA occurs in both capillaries and small arteries and, to a lesser extent, veins. With respect to capillaries, Aβ deposition in CAA occurs on the BEC basement membrane and protrudes into the neuropil, and can result in capillary occlusion. Whether luminal or abluminal, soluble Aβ may induce occludin down-regulation in BECs prior to or at early stages of CAA, thereby inducing an increased BBB permeability. Plasma proteins that cross the endothelium could in turn bind to and exacerbate the deposition of Aβ around cerebrovascular vessels.
Occludin is an important TJ protein and its down-regulation is associated with increased permeability in BECs [2]. Indeed, the importance of occludin in conferring restrictive TJs has been demonstrated in vitro and in vivo. Transfection of truncated occludin or siRNA knockdown in MDCK cells [41] or Xenopus embryo cells [42] results in increased paracellular permeability. In addition, occludin down-regulation and increased permeability has been observed in models of inflammatory pain [43] and oxidative stress [44], and CNS disorders associated with BBB breakdown such as multiple sclerosis [45]. Here, we found that occludin down-regulation was the most likely cause of the Aβ-induced increase in hCMEC/D3 cell permeability. Marco and Skaper [24] have observed that 20 μM Aβ 1–42 can induce a decrease in occludin protein levels after 24 hrs, but not 48 hrs, of incubation in rat BECs. In agreement with our study, the authors did not detect any alterations in overall cldn-5 or ZO-1 levels, although cldn-5 appeared to translocate to the cytoplasm. Cerebral extravasation of IgG in APP/PS1 mice fed on a high saturated-fat diet has also been associated with decreased occludin expression at the BBB [23]. A disruption in TJ organization has also been noted in AD patient’s biopsy samples [22]. Intriguingly, neuronal occludin over-expression has been detected in brains of AD individuals [46], although the significance of this observation is unclear.
Changes in the levels of occludin protein at 48 hrs were preceded by a decrease in occludin mRNA at 24 hrs. At earlier time-points, Aβ may still induce changes in BBB permeability by directly acting on TJ organization, either by post-translational modifications of TJ proteins such as phosphorylation or by inducing TJ protein degradation and/or sub-cellular reorganization. Whether Aβ induces post-translational modifications of the occludin protein requires further investigation. Our observation that inhibition of JNK restores occludin mRNA levels to those of controls would indicate that Aβ acts on occludin expression either by suppression of occludin gene transcriptional activity or by decreasing occludin mRNA stability. Aβ-mediated occludin down-regulation in BECs could therefore represent an important mechanism of increased BBB permeability in AD, as suggested by the present study.
The observation that p38MAPK or JNK inhibition could prevent the Aβ-induced increase in BEC permeability suggests that these signalling pathways are potential targets for therapeutic treatments in AD. p38MAPK activation has been associated with occludin down-regulation in response to different stimuli, including combined tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) incubation [47], transforming growth factor-β (TGF-β) [48] and alcohol [49]. Indeed, Lui _et al._[48] have demonstrated that CdCl2-induced TGF-β3 production, and occludin down-regulation, can be prevented by p38MAPK inhibition at the rat blood–testis barrier. Crucially, in a study by Munoz and co-workers [50], a novel p38MAPK inhibitor was observed to improve synaptic dysfunction and behavioural deficits after Aβ 1–42 intracerebroventricular injections into mice. Although they did not investigate the permeability of the BBB, the authors did find that p38MAPK inhibition suppressed an Aβ 1–42-induced increase in TNF-α and IL-1β levels in the hippocampus of these mice. Aβ-mediated activation of astrocytes and microglia can induce cytokine production _in vitro_[7, 51–53] and astrocytic end feet swelling [54] and microglia activation [51] have been observed in AD tissue sections. IL-1β, TNF-α and IL-6 levels are found increased in the CSF or serum of AD patients [55] and cytokines can induce a leaky TJ _in vitro_[56–59]. p38MAPK inhibition could therefore act as a dual target in AD, preventing cytokine production and Aβ-induced increased BBB permeability.
The JNK pathway also represents a potential novel potential therapeutic target for AD, with its inhibition not only preventing Aβ-induced increase in BBB permeability, as demonstrated here, but also neuronal atrophy [60]. There are three genes that encode JNK in mammalian systems, jnk1, jnk2 and jnk3, and each gene is alternatively spliced to create 46 and 55-kD forms of the proteins [61, 62]. The significance of each variant is not fully understood. However, JNK1 and -2 are expressed ubiquitously, whereas JNK3 is largely restricted to the brain (reviewed in [63]). Morishima _et al._[64] have demonstrated that Aβ 25–35 toxicity is significantly reduced in cortical neurons derived from JNK3 knock-out mice compared with the controls; however, they also state that JNK1-deficient neurons are also resistant to apoptosis. Using antisense technology, JNK1-depleted rat cortical neurons are found to be resistant to Aβ 1–40-induced apoptosis [65]. Aβ in this study was found to activate the 46-kD JNK isoform, which in some studies was found to correspond to JNK1 [65]. Regardless of the isoform, JNK inhibition protects SH-SY5Y neuroblastoma, rat cortical neurons and PC12 arrhenoblastoma cells from Aβ-induced apoptosis _in vitro_[66–68]. A further interesting study has demonstrated that JNK inhibition can prevent H2O2-induced γ-secretase–mediated APP cleavage and Aβ production in SH-SY5Y cells [69]. The JNK pathway might therefore represent a very attractive therapeutic target, to prevent Aβ production, neuronal atrophy and BBB dysfunction.
In summary, we have found that Aβ 1–40 can increase the paracellular permeability of human BECs and this correlates with occludin down-regulation. p38MAPK and JNK inhibition prevented both occludin down-regulation and increased paracellular permeability of BECs, as induced by Aβ. The p38MAPK and JNK pathway therefore potentially represent interesting therapeutic targets to ameliorate cerebrovascular dysfunction in AD.
Acknowledgments
We would like to thank Dr. Hilary McQueen for her advice during this study. This work was supported by the MRC UK.
Supporting Information
Fig. S1. Aβ 1-40 after 48 hr incubation in thepresence (right) or absence (left) of hCMEC/D3 cells. **(A)**Soluble Aβ 1-40 monomer was added to culture medium and thenincubated either in a collagen-coated tissue culture plate or withconfluent monolayers of hCMEC/D3 cells grown on a collagen-coatedtissue culture plate for 1, 3, 6, 24 or 48 hr. The supernatant wascollected and the Aβ peptide levels (6E10, mouse monoclonalIgG1) was investigated using western blotting techniques and a 15 %acrylamide gel. (B) Aβ peptide levels were measured as above in whole cell lysates or in scraped collagen-coated dishes at each time point on 15 % acrylamide gels.
Fig. S2. The role of signalling pathway inhibition onoccludin expression by hCMEC/D3 cells following Aβ 1-40incubation for 48 hr. Fully confluent hCMEC/D3 cells werepre-incubated for 30 min with inhibitors for the PI-3K,NF-κB, GSK-3β, Src, ROCK, PKC and JNK pathways (Table 1)and subsequently incubated for 48 hr with either 5 μM Aβ1-40 or 5 μM sAβ 1-40, and the expression of occludinassessed via flow cytometry. Data represents mean ± SEM.n = 3 experiments with duplicate samples. *P <0.05 using a Student's t test comparing each treatment to the control.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not) Trends Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res. Commun. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 4.Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging. 2004;25:569–80. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Avila J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006;580:2922–7. doi: 10.1016/j.febslet.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger KA. Cell death mechanisms in neurodegeneration. J Cell Mol Med. 2001;5:1–17. doi: 10.1111/j.1582-4934.2001.tb00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rojo LE, Fernandez JA, Maccioni AA, et al. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J. Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006;9:151–3. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 9.De La Torre JC. Pathophysiology of neuronal energy crisis in Alzheimer’s disease. Neurodegener Dis. 2008;5:126–32. doi: 10.1159/000113681. [DOI] [PubMed] [Google Scholar]
- 10.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Mooradian AD, Chung HC, Shah GN. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging. 1997;18:469–74. doi: 10.1016/s0197-4580(97)00111-5. [DOI] [PubMed] [Google Scholar]
- 12.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto Y, Yanase D, Noguchi-Shinohara M, et al. Blood-brain barrier permeability correlates with medial temporal lobe atrophy but not with amyloid-beta protein transport across the blood-brain barrier in Alzheimer’s disease. Dement Geriatr. Cogn Disord. 2007;23:241–5. doi: 10.1159/000100019. [DOI] [PubMed] [Google Scholar]
- 14.Zipser BD, Johanson CE, Gonzalez L, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging. 2007;28:977–86. doi: 10.1016/j.neurobiolaging.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 15.Frolich L, Kornhuber J, Ihl R, et al. Integrity of the blood-CSF barrier in dementia of Alzheimer type: CSF/serum ratios of albumin and IgG. Eur Arch Psychiatry Clin Neurosci. 1991;240:363–6. doi: 10.1007/BF02279767. [DOI] [PubMed] [Google Scholar]
- 16.Kay AD, May C, Papadopoulos NM, et al. CSF and serum concentrations of albumin and IgG in Alzheimer’s disease. Neurobiol Aging. 1987;8:21–5. doi: 10.1016/0197-4580(87)90053-4. [DOI] [PubMed] [Google Scholar]
- 17.Leonardi A, Gandolfo C, Caponnetto C, et al. The integrity of the blood-brain barrier in Alzheimer’s type and multi-infarct dementia evaluated by the study of albumin and IgG in serum and cerebrospinal fluid. J Neurol Sci. 1985;67:253–61. doi: 10.1016/0022-510x(85)90121-2. [DOI] [PubMed] [Google Scholar]
- 18.Su GC, Arendash GW, Kalaria RN, et al. Intravascular infusions of soluble beta-amyloid compromise the blood-brain barrier, activate CNS glial cells and induce peripheral hemorrhage. Brain Res. 1999;818:105–17. doi: 10.1016/s0006-8993(98)01143-3. [DOI] [PubMed] [Google Scholar]
- 19.Ujiie M, Dickstein DL, Carlow DA, et al. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10:463–70. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 20.Blanc EM, Toborek M, Mark RJ, et al. Amyloid beta-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J Neurochem. 1997;68:1870–81. doi: 10.1046/j.1471-4159.1997.68051870.x. [DOI] [PubMed] [Google Scholar]
- 21.Strazielle N, Ghersi-Egea JF, Ghiso J, et al. In vitro evidence that beta-amyloid peptide 1–40 diffuses across the blood-brain barrier and affects its permeability. J Neuropathol Exp Neurol. 2000;59:29–38. doi: 10.1093/jnen/59.1.29. [DOI] [PubMed] [Google Scholar]
- 22.Stewart PA, Hayakawa K, Akers MA, et al. A morphometric study of the blood-brain barrier in Alzheimer’s disease. Lab Invest. 1992;67:734–42. [PubMed] [Google Scholar]
- 23.Takechi R, Galloway S, Pallebage-Gamarallage MM, et al. Chylomicron amyloid-beta in the aetiology of Alzheimer’s disease. Atherosclerosis. 2008;9:19–25. doi: 10.1016/j.atherosclerosissup.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Marco S, Skaper SD. Amyloid beta-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–24. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 25.Weksler BB, Subileau EA, Perriere N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–4. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 26.Lichtlen P, Mohajeri MH. Antibody-based approaches in Alzheimer’s research: safety, pharmacokinetics, metabolism, and analytical tools. J Neurochem. 2008;104:859–74. doi: 10.1111/j.1471-4159.2007.05064.x. [DOI] [PubMed] [Google Scholar]
- 27.Gravina SA, Ho L, Eckman CB, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J Biol Chem. 1995;270:7013–6. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–56. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Small DH, Mok SS, Bornstein JC. Alzheimer’s disease and Abeta toxicity: from top to bottom. Nat Rev. 2001;2:595–8. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- 30.Hyman BT, Elvhage TE, Reiter J. Extracellular signal regulated kinases. Localization of protein and mRNA in the human hippocampal formation in Alzheimer’s disease. Am J Pathol. 1994;144:565–72. [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu X, Raina AK, Rottkamp CA, et al. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem. 2001;76:435–41. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhu X, Rottkamp CA, Boux H, et al. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–8. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- 33.Berzin TM, Zipser BD, Rafii MS, et al. Agrin and microvascular damage in Alzheimer’s disease. Neurobiol Aging. 2000;21:349–55. doi: 10.1016/s0197-4580(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 34.Elovaara I, Palo J, Erkinjuntti T, et al. Serum and cerebrospinal fluid proteins and the blood-brain barrier in Alzheimer’s disease and multi-infarct dementia. Eur Neurol. 1987;26:229–34. doi: 10.1159/000116341. [DOI] [PubMed] [Google Scholar]
- 35.Skoog I, Wallin A, Fredman P, et al. A population study on blood-brain barrier function in 85-year-olds: relation to Alzheimer’s disease and vascular dementia. Neurology. 1998;50:966–71. doi: 10.1212/wnl.50.4.966. [DOI] [PubMed] [Google Scholar]
- 36.Wada H. Blood-brain barrier permeability of the demented elderly as studied by cerebrospinal fluid-serum albumin ratio. Intern Med (Tokyo, Japan) 1998;37:509–13. doi: 10.2169/internalmedicine.37.509. [DOI] [PubMed] [Google Scholar]
- 37.Dickstein DL, Biron KE, Ujiie M, et al. Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 2006;20:426–33. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- 38.Van Oijen M, Hofman A, Soares HD, et al. Plasma Abeta(1–40) and Abeta(1–42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5:655–60. doi: 10.1016/S1474-4422(06)70501-4. [DOI] [PubMed] [Google Scholar]
- 39.Weller RO, Subash M, Preston SD, et al. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol (Zurich, Switzerland) 2008;18:253–66. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thal DR, Griffin WS, De Vos RA, et al. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- 41.Balda MS, Whitney JA, Flores C, et al. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–49. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Merzdorf C, Paul DL, et al. COOH terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J Cell Biol. 1997;138:891–9. doi: 10.1083/jcb.138.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brooks TA, Hawkins BT, Huber JD, et al. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. Am J Physiol Heart Circ Physiol. 2005;289:H738–43. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H1485–94. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wosik K, Cayrol R, Dodelet-Devillers A, et al. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci. 2007;27:9032–42. doi: 10.1523/JNEUROSCI.2088-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romanitan MO, Popescu BO, Winblad B, et al. Occludin is overexpressed in Alzheimer’s disease and vascular dementia. J Cell Mol Med. 2007;11:569–79. doi: 10.1111/j.1582-4934.2007.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patrick DM, Leone AK, Shellenberger JJ, et al. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma modulate epithelial barrier function in Madin-Darby canine kidney cells through mitogen activated protein kinase signaling. BMC Physiol. 2006;6:2. doi: 10.1186/1472-6793-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lui WY, Wong CH, Mruk DD, et al. TGF-beta3 regulates the blood-testis barrier dynamics via the p38 mitogen activated protein (MAP) kinase pathway: an in vivo study. Endocrinology. 2003;144:1139–42. doi: 10.1210/en.2002-0211. [DOI] [PubMed] [Google Scholar]
- 49.Singh AK, Jiang Y, Gupta S. Effects of chronic alcohol drinking on receptor-binding, internalization, and degradation of human immunodeficiency virus 1 envelope protein gp120 in hepatocytes. Alcohol. 2007;41:591–606. doi: 10.1016/j.alcohol.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Munoz L, Ranaivo HR, Roy SM, et al. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bamberger ME, Landreth GE. Microglial interaction with beta-amyloid: implications for the pathogenesis of Alzheimer’s disease. Microsc Res Tech. 2001;54:59–70. doi: 10.1002/jemt.1121. [DOI] [PubMed] [Google Scholar]
- 52.Franciosi S, Ryu JK, Choi HB, et al. Broad-spectrum effects of 4-aminopyridine to modulate amyloid beta1–42-induced cell signaling and functional responses in human microglia. J Neurosci. 2006;26:11652–64. doi: 10.1523/JNEUROSCI.2490-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pyo H, Jou I, Jung S, et al. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. Neuroreport. 1998;9:871–4. doi: 10.1097/00001756-199803300-00020. [DOI] [PubMed] [Google Scholar]
- 54.Higuchi Y, Miyakawa T, Shimoji A, et al. Ultrastructural changes of blood vessels in the cerebral cortex in Alzheimer’s disease. Jpn J Psychiatry Neurol. 1987;41:283–90. doi: 10.1111/j.1440-1819.1987.tb00414.x. [DOI] [PubMed] [Google Scholar]
- 55.Weisman D, Hakimian E, Ho GJ. Interleukins, inflammation, and mechanisms of Alzheimer’s disease. Vitam Horm. 2006;74:505–30. doi: 10.1016/S0083-6729(06)74020-1. [DOI] [PubMed] [Google Scholar]
- 56.Abe T, Sugano E, Saigo Y, et al. Interleukin-1beta and barrier function of retinal pigment epithelial cells (ARPE-19): aberrant expression of junctional complex molecules. Invest Ophthalmol Vis Sci. 2003;44:4097–104. doi: 10.1167/iovs.02-0867. [DOI] [PubMed] [Google Scholar]
- 57.Afonso PV, Ozden S, Prevost MC, et al. Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J Immunol. 2007;179:2576–83. doi: 10.4049/jimmunol.179.4.2576. [DOI] [PubMed] [Google Scholar]
- 58.Al-Sadi R, Ye D, Dokladny K, et al. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol. 2008;180:5653–61. doi: 10.4049/jimmunol.180.8.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazzon E, Cuzzocrea S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir Res. 2007;8:75. doi: 10.1186/1465-9921-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okazawa H, Estus S. The JNK/c-Jun cascade and Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2002;17:79–88. doi: 10.1177/153331750201700209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs) Int J Biochem Cell Biol. 2001;33:1047–63. doi: 10.1016/s1357-2725(01)00093-0. [DOI] [PubMed] [Google Scholar]
- 62.Bogoyevitch MA, Arthur PG. Inhibitors of c-Jun N-terminal kinases: JuNK no more. Biochim Biophys Acta. 2008;1784:76–93. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): differences revealed by gene targeting. Bioessays. 2006;28:923–34. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- 64.Morishima Y, Gotoh Y, Zieg J, et al. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–60. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fogarty MP, Downer EJ, Campbell V. A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the beta-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem J. 2003;371:789–98. doi: 10.1042/BJ20021660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bozyczko-Coyne D, O’Kane TM, Wu ZL, et al. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–63. doi: 10.1046/j.1471-4159.2001.00294.x. [DOI] [PubMed] [Google Scholar]
- 67.Troy CM, Rabacchi SA, Xu Z, et al. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–64. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- 68.Wei W, Wang X, Kusiak JW. Signaling events in amyloid beta-peptide-induced neuronal death and insulin-like growth factor I protection. J Biol Chem. 2002;277:17649–56. doi: 10.1074/jbc.M111704200. [DOI] [PubMed] [Google Scholar]
- 69.Shen C, Chen Y, Liu H, et al. Hydrogen peroxide promotes Abeta production through JNK-dependent activation of gamma-secretase. J Biol Chem. 2008;283:17721–30. doi: 10.1074/jbc.M800013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Aβ 1-40 after 48 hr incubation in thepresence (right) or absence (left) of hCMEC/D3 cells. **(A)**Soluble Aβ 1-40 monomer was added to culture medium and thenincubated either in a collagen-coated tissue culture plate or withconfluent monolayers of hCMEC/D3 cells grown on a collagen-coatedtissue culture plate for 1, 3, 6, 24 or 48 hr. The supernatant wascollected and the Aβ peptide levels (6E10, mouse monoclonalIgG1) was investigated using western blotting techniques and a 15 %acrylamide gel. (B) Aβ peptide levels were measured as above in whole cell lysates or in scraped collagen-coated dishes at each time point on 15 % acrylamide gels.
Fig. S2. The role of signalling pathway inhibition onoccludin expression by hCMEC/D3 cells following Aβ 1-40incubation for 48 hr. Fully confluent hCMEC/D3 cells werepre-incubated for 30 min with inhibitors for the PI-3K,NF-κB, GSK-3β, Src, ROCK, PKC and JNK pathways (Table 1)and subsequently incubated for 48 hr with either 5 μM Aβ1-40 or 5 μM sAβ 1-40, and the expression of occludinassessed via flow cytometry. Data represents mean ± SEM.n = 3 experiments with duplicate samples. *P <0.05 using a Student's t test comparing each treatment to the control.