Inherited IL-12p40 Deficiency: Genetic, Immunologic, and Clinical Features of 49 Patients From 30 Kindreds (original) (raw)
Supplemental digital content is available in the text.
Abstract
Autosomal recessive interleukin (IL)-12 p40 (IL-12p40) deficiency is a rare genetic etiology of Mendelian susceptibility to mycobacterial disease (MSMD). We report the genetic, immunologic, and clinical features of 49 patients from 30 kindreds originating from 5 countries (India, Iran, Pakistan, Saudi Arabia, and Tunisia). There are only 9 different mutant alleles of the IL12B gene: 2 small insertions, 3 small deletions, 2 splice site mutations, and 1 large deletion, each causing a frameshift and leading to a premature stop codon, and 1 nonsense mutation. Four of these 9 variants are recurrent, affecting 25 of the 30 reported kindreds, due to founder effects in specific countries. All patients are homozygous and display complete IL-12p40 deficiency. As a result, the patients lack detectable IL-12p70 and IL-12p40 and have low levels of interferon gamma (IFN-γ). The clinical features are characterized by childhood onset of bacille Calmette-Guérin (attenuated Mycobacterium bovis strain) (BCG) and Salmonella infections, with recurrences of salmonellosis (36.4%) more common than recurrences of mycobacterial disease (25%). BCG vaccination led to BCG disease in 40 of the 41 patients vaccinated (97.5%). Multiple mycobacterial infections were rare, observed in only 3 patients, whereas the association of salmonellosis and mycobacteriosis was observed in 9 patients. A few other infections were diagnosed, including chronic mucocutaneous candidiasis (n = 3), nocardiosis (n = 2), and klebsiellosis (n = 1). IL-12p40 deficiency has a high but incomplete clinical penetrance, with 33.3% of genetically affected relatives of index cases showing no symptoms. However, the prognosis is poor, with mortality rates of up to 28.6%. Overall, the clinical phenotype of IL-12p40 deficiency closely resembles that of interleukin 12 receptor β1 (IL-12Rβ1) deficiency.
In conclusion, IL-12p40 deficiency is more common than initially thought and should be considered worldwide in patients with MSMD and other intramacrophagic infectious diseases, salmonellosis in particular.
INTRODUCTION
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare clinical syndrome that was probably first described in 1951 (OMIM 209950).2,8,9 MSMD is clinically characterized by susceptibility to poorly virulent mycobacteria, such as bacillus Calmette-Guérin (attenuated Mycobacterium bovis strain) (BCG) vaccines and environmental mycobacteria (EM), as well as nontyphoidal Salmonella.6,19 The clinical features of MSMD patients are diverse, ranging from disseminated, overwhelming disease to localized, recurrent disease.3,19 The patients are also vulnerable to more virulent mycobacteria (Mycobacterium tuberculosis) and typhoidal Salmonella.1,12,29,33,41 Other infections, with viruses or intramacrophagic microorganisms, are rare.12,40 Various modes of inheritance have been reported in multiplex kindreds, with autosomal recessive, autosomal dominant, and X-linked recessive patterns.3,19,40 Unsurprisingly, MSMD has been reported to be genetically heterogeneous.7,20 Since 1996, MSMD-causing mutations have been found in 7 autosomal (IFNGR1, IFNGR2, STAT1, IL12RB1, IL12B, IRF8, and ISG15) and 2 X-linked (NEMO, CYBB) genes.4a,5,19,24 Allelic heterogeneity further contributes to the definition of up to 17 genetic disorders, with complete and partial defects, dominant and recessive traits, and complete defects with and without protein production.5,19,24,41,47 With the possible exception of CYBB, MSMD-causing genes encode molecules involved in the IL-12-IFN-γ circuit, either in the IL-12-dependent induction of IFN-γ or cellular responses to IFN-γ.6,10,19 An account of the first patient with IL-12p40 deficiency was reported in 1998.4 Since then, to our knowledge there have been 4 other reports describing a total of 20 patients with IL-12p40 deficiency.14,30,33,38 IL-12p40 deficiency is therefore thought to be a very rare genetic etiology of MSMD, estimated to account for less than 9% of cases.19
In mice and humans, IL-12p70 is a heterodimeric cytokine composed of a 35-kDa light chain (p35) encoded by IL12A and a 40-kDa heavy chain (p40) encoded by IL12B.26,44 IL-12p70 is produced mostly by myeloid macrophages and dendritic cells44 and plays a major role in inducing the production of IFN-γ by NK and T lymphocytes. IL-12p70 signals through a specific IL-12 heterodimeric receptor (IL-12R) consisting of IL-12Rβ1 and IL-12Rβ2 chains and is expressed principally on activated NK and T cells.34 The binding of IL-12 to IL-12R in NK and T cells leads to the janus kinase 2- and tyrosine kinase 2-dependent activation of signal transducer and activator of transcription factor (STAT) 4, leading to the translocation of this molecule to the nucleus, where it induces the transcription of target genes, including the IFN-γ and Furin genes, in particular.26,32,43 In mice, IL-12R is also expressed by some myeloid cells, in which the IL-12 signaling pathway differs from that operating in lymphocytes, making use of nuclear factor kB instead of STAT.23 IL-12p40 is not specific for IL-12p70, as it can also associate with the IL-23p19 subunit to form IL-23, which is involved in the induction of IL-17-producing T cells.44 The heterodimeric IL-23 receptor also includes the IL-12Rβ1 chain. Individuals with IL12B mutations therefore have defects in both IL-12 and IL-23 immunity.26 Likewise, patients with IL-12Rβ1 deficiency do not respond to either IL-12 or IL-23.12 Patients with IL-12p40 or IL-12Rβ1 deficiency suffer from MSMD because of impaired IL-12-dependent IFN-γ immunity; some suffer from Candida albicans infections because of impaired IL-23-dependent IL-17 immunity.11,12,35 We recently reviewed the clinical and genetic data from a large series of patients with IL-12Rβ1 deficiency.12 We conducted the current study to describe the genetic, immunologic, and clinical features of a large cohort of IL-12p40-deficient patients.
PATIENTS AND METHODS
Subjects and Kindreds
Patients and their families, including asymptomatic relatives, were recruited to this study through a large, worldwide network of collaborating clinicians and immunologists, from 1988 to 2012. These patients presented with a history of unusual infections, such as disseminated disease caused by weakly virulent mycobacteria and Salmonella, corresponding to the description of MSMD and other similar conditions. Healthy volunteers were also recruited. The study was conducted in accordance with the Helsinki Declaration, with informed consent obtained from each patient or the patient’s family, as well as the healthy volunteers, as required, and with the approval of the institutional review boards of the various institutions involved.
The kindreds are described in the Supplemental Patients and Methods section (see Supplemental Digital Content, http://links.lww.com/MD/A17).
Whole-Blood Activation Assay
Venous blood samples were collected into heparinized tubes and were transported by express mail, at room temperature. We received a blood sample from a healthy blood donor together with each blood sample from a patient. Blood samples from healthy donors were collected at the same time and at the same institution as the patient’s blood sample and were transported with the patient’s blood sample. These samples are identified as “travel control” samples in the text. Blood was diluted 1/2 in RPMI 1640 medium (Invitrogen). We activated 1 mL of blood dilution per well of a 48-well plate as follows: with medium alone, with live BCG (Mycobacterium bovis BCG, Pasteur strain) at a multiplicity of infection of 20:1, with BCG and IFN-γ (5000 IU/mL, Imukin, Boehringer Ingelheim) or with BCG and IL-12p70 (20 ng/mL, R&D Systems).15 All cells were incubated at 37°C, under an atmosphere containing 5% CO2. Supernatants were collected after 48 hours and centrifuged at 1800 g for 10 minutes. The resulting supernatants were stored at −20°C until analysis.
Determination of Cytokine Levels by ELISA
IL-12p40, IL-12p70, and IFN-γ (in the 48-h supernatant) were determined by enzyme-linked immunosorbent assay (ELISA). We used the capture antibodies, detection antibodies and standards supplied in the R&D Systems kits for IL-12p40 (Quantikine SP400) and IL-12p70 (Quantikine HS120) and in the Sanquin kit for IFN-γ (M9333), diluted in HPE dilution buffer. Milk was used for blocking and antibody binding was detected with streptavidin-conjugated horseradish peroxidase (M2032, Sanquin) and TMB microwell peroxidase substrate (50-76-00, KPL). The reaction was stopped by adding H2SO4 (1.8 M). Absorbance was determined with an MRX microplate reader (Thermolab Systems), at 490 nm for IL-12p70 and at 450 nm for IL-12p40 and IFN-γ. The detection limits of the assays were 0.625 pg/mL for IL-12p70, 15.6 pg/mL for IL-12p40, and 5 pg/mL for IFN-γ. Quantitative analysis was carried out with a nonlinear, 4-parameter logistic (4PL) calibration model, with in-house software based on the Microsoft Excel application language developed for this purpose (a gift from Max Feinberg). Intermediate results for each cytokine are expressed in pg/mL per 106 peripheral blood mononuclear cells (PBMC).15
Activation of Cell Lines
Epstein-Barr virus-transformed lymphoblastoid cell lines (EBV-B cell lines) were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen). Cells were incubated in 24-well plates at a density of 1×106 cells/mL with 2 × 10–7 M phorbol-12,13-dibutyrate (Sigma) for 18 h. EBV-B cell lines derived from a healthy individual and from a patient previously diagnosed with IL-12 deficiency4,33 were used as positive and negative controls, respectively. All cells were incubated at 37°C, under an atmosphere containing 5% CO2.
Genetic Analysis
Human genomic DNA was isolated from the Ficoll gradient pellets obtained during PBMC purification and/or from the cell lines. The cells were lysed by incubation in extraction buffer (10 mM Tris, 0.1 M EDTA, 0.5% SDS, and 20 mg/mL proteinase K) overnight at 37°C. DNA was isolated by phenol/chloroform extraction, precipitated in isopropanol and ethanol, and resuspended in autoclaved H2O. RNA was isolated from EBV-B cell lines with Trizol reagent (Invitrogen), according to the manufacturer’s instructions. RNA was reverse-transcribed with oligo-dT primers and Superscript II reverse transcriptase (Invitrogen). The first-strand cDNA was then stored at −20°C. Polymerase chain reaction (PCR) amplification was performed with the AmpliTaq DNA polymerase (Applied Biosystems) and the GeneAmp 9700 PCR system (Applied Biosystems). The primers and conditions used for PCR amplification of the coding exons, including the flanking intronic sequences or the IL12B cDNA are available on request. Amplified PCR products were checked by gel electrophoresis in a 1% agarose gel and purified by centrifugation through Sephadex G-50 Superfine resin (Amersham GE) on a MAHV-N45 filter-plate multiscreen (Millipore). PCR products were sequenced by dideoxynucleotide termination, with the BigDye Terminator kit v1.1 or v3.1 (Applied Biosystems) and the PCR primers. Sequencing products were purified by centrifugation through Sephadex G-50 Superfine resin and analyzed on an ABI Prism 3100 or 3130xl apparatus (Applied Biosystems). Sequence files and chromatograms were analyzed with GENALYS Software from the CNG, France.42
Polymorphic Marker Genotyping and Dating of Mutations
The age of the 2 founder mutations, 315insA and 526del2, was estimated from a subset of 8 available unrelated 315insA patients from Saudi Arabia and 2 available unrelated 526del2 patients from Iran. Approximately 250,000 SNPs, from the Affymetrix GeneChip Human Mapping 250K, were genotyped. SNPs with 100% call rates were scanned for continuous stretches of homozygosity up- and downstream from the IL12B locus. Pairwise comparisons within each mutation group revealed the limits of the longest shared haplotype and the positions of subsequent recombination breakpoints. The likelihood-based ESTIAGE method22 was used to estimate the age of the most recent common ancestor for each mutation from the observed shared haplotypes, together with recombination rates and haplotype frequencies obtained from the HapMap Project.21
Statistical Methods
Infection-free status, survival, and penetrance curves as a function of age were estimated by the Kaplan-Meier method. Penetrance curves for IL-12p40 deficiency were obtained from the data for 15 symptomatic relatives of index cases. The production of IL-12p40, IL-12p70, and IFN-γ by whole blood cells was compared between controls and patients, in various stimulation conditions, by nonparametric Kruskal-Wallis rank sum tests. All calculations were carried out and curves plotted with R software (http://cran.r-project.org/).
RESULTS
Clinical Features and Mutation Analysis of 30 Index Cases
We identified 30 kindreds, comprising a total of 49 IL12p40-deficient patients (30 index cases and 19 sibs), including 44 symptomatic and 5 asymptomatic subjects (Table 1, Figure 1). Detailed pedigrees were available for 29 families, 25 (86.2%) of which were consanguineous (mostly due to first-cousin marriages). Sequencing of the 6 IL12B coding exons and flanking intron regions in the 30 index cases from 5 different countries (India, Iran, Saudi Arabia, Pakistan, and Tunisia) revealed only 9 different mutant IL12B alleles (Figure 2), 5 of which had not previously been reported (Figure 3). The known mutations were 2 small deletions (526del2, 297del8),16,32 1 insertion (315insA),14,30 and 1 large deletion (g.482+82_856-854del).33 All 4 known mutations induced frameshifts, resulting in the generation of premature termination codons (526del2, 297del8, 35del10, 315insA) or the excision of 2 coding exons (g.482+82_856-854del). The 5 newly identified IL12B mutations comprised 1 deletion (35del10), 1 insertion (909insA), 1 nonsense mutation (W60X), and 2 splice-site mutations (697+2T>C and 697+5G>A). Each of these mutations was found in a single kindred. These previously unknown mutations had a major impact on the structure of the IL12B mRNA. The splice-site mutations (697+2T>C and 697+5G>A) led to the excision of exon 6 and an absence of full-length IL12 mRNA, as shown by RT-PCR (data not shown). The 909insA mutation resulted in the insertion of a premature stop codon at amino acid 307. The most prevalent IL12B mutation was the previously reported 315insA mutation, which was detected only in patients from Saudi Arabia (n = 24; 13 kindreds). The next most frequent mutation was the 297del8 mutation, which was identified exclusively in kindreds originating from Tunisia (n = 8; 5 kindreds).14,33
TABLE 1.
Genetic and Clinical Features of Patients With IL-12B Deficiency
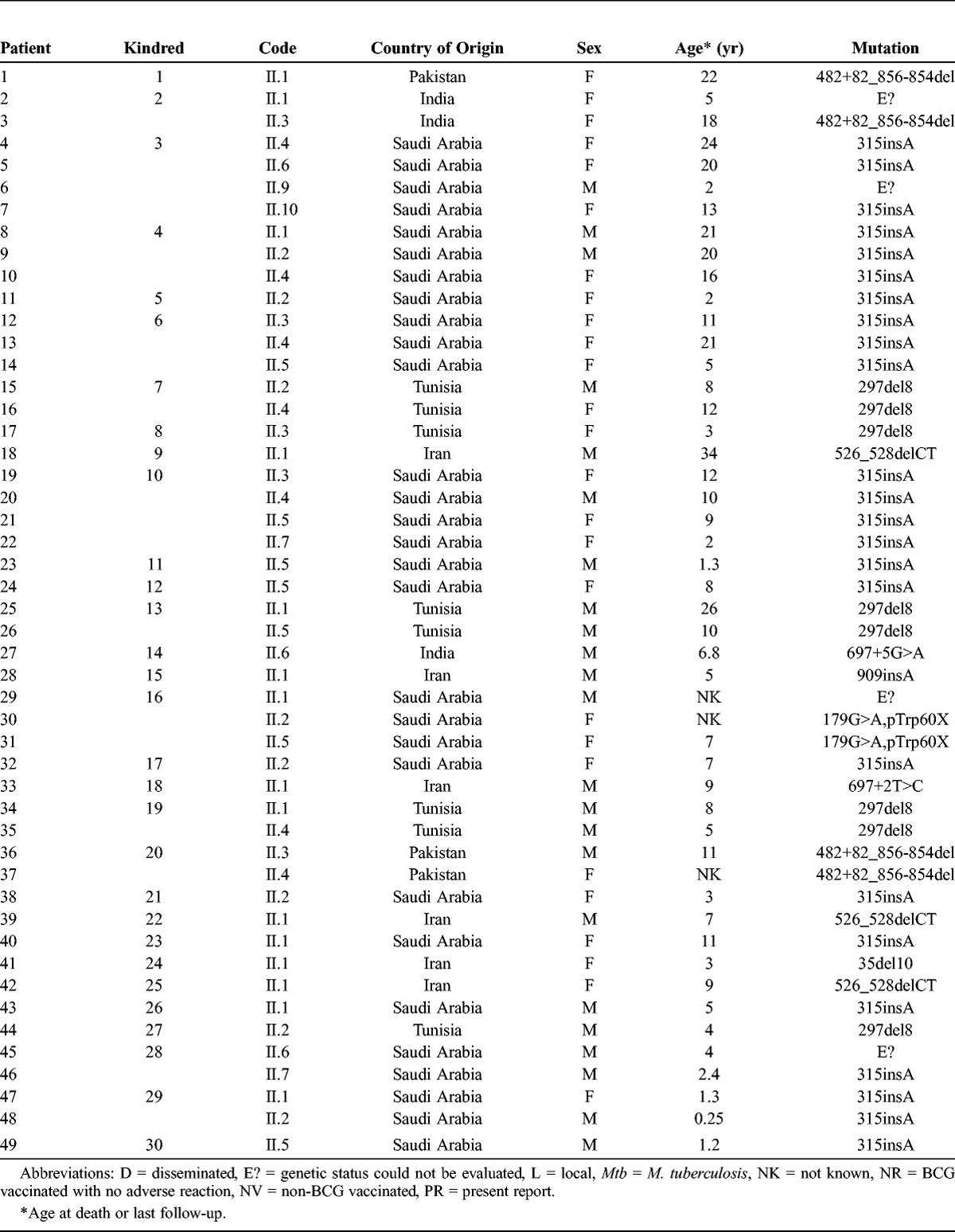
FIGURE 1.
Pedigrees of 30 families with IL-12p40 deficiency. Each kindred is designated by an integer (numbers 1–30), each generation is designated by a roman numeral (numerals I–II), and each individual by an arabic numeral (each individual studied is identified by these 3 numbers, organized from left to right). Symbols are split in 2 by a horizontal line. The upper part of the symbol indicates mycobacterial infection (black for BCG or atypical mycobacteriosis, gray for tuberculosis); the lower part of the symbol indicates salmonellosis status (black indicating the patient has had salmonellosis). The probands are indicated by an arrow. Individuals whose genetic status could not be evaluated are indicated by the symbol “E?” Asymptomatic individuals carrying 2 mutant IL12B alleles are represented by a vertical line.
FIGURE 2.
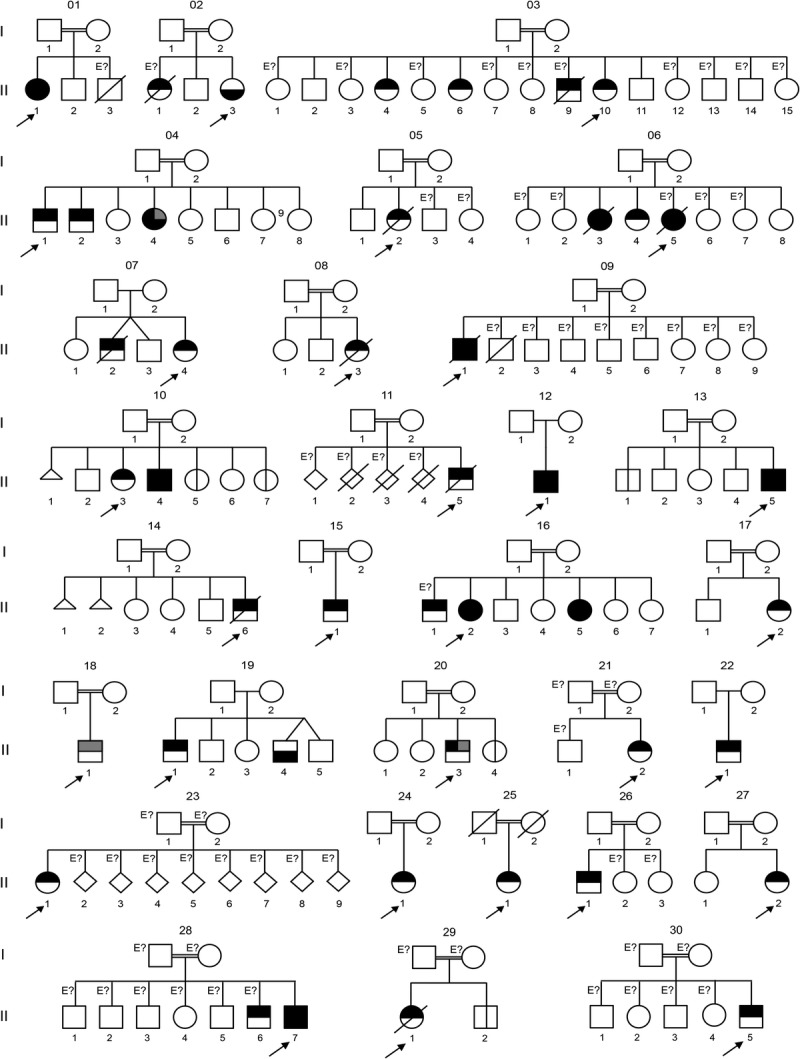
Origin of the kindreds. Geographic origin of the 49 patients with complete IL-12p40 deficiency. These patients originated from 5 countries (India, Iran, Pakistan, Saudi Arabia, and Tunisia). The numbers indicate the number of patients originating from a given country. The numbers of kindreds and mutations for each country are shown. [This figure can be viewed in color online at http://www.md-journal.com].
FIGURE 3.
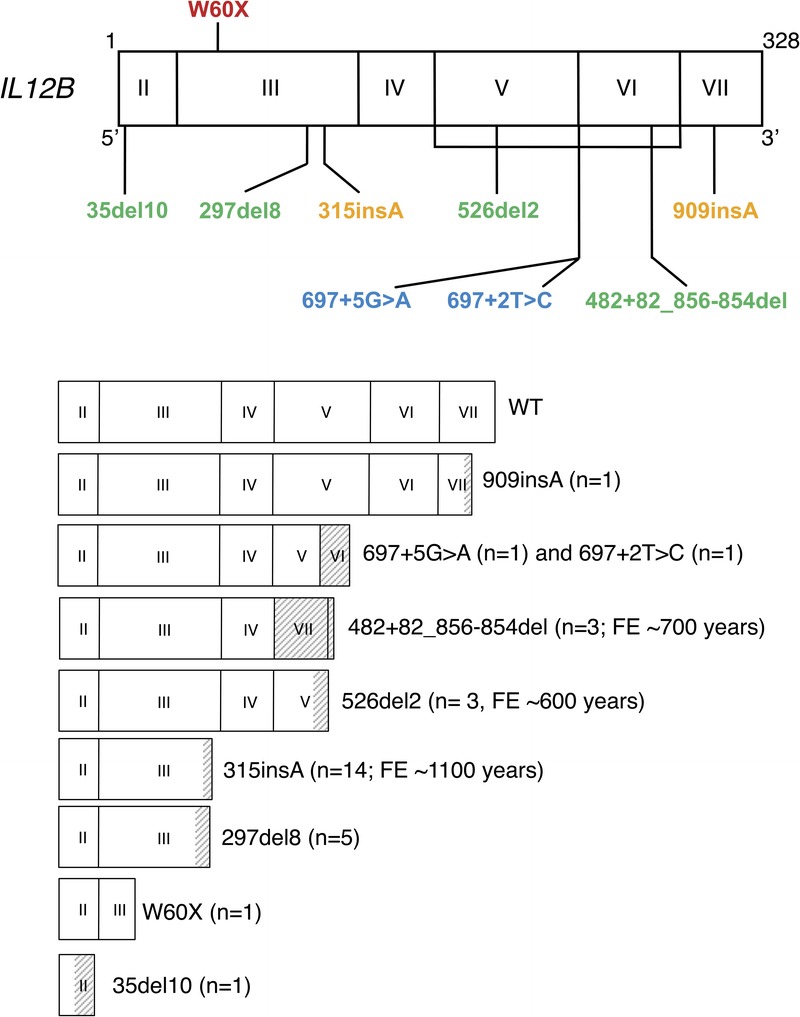
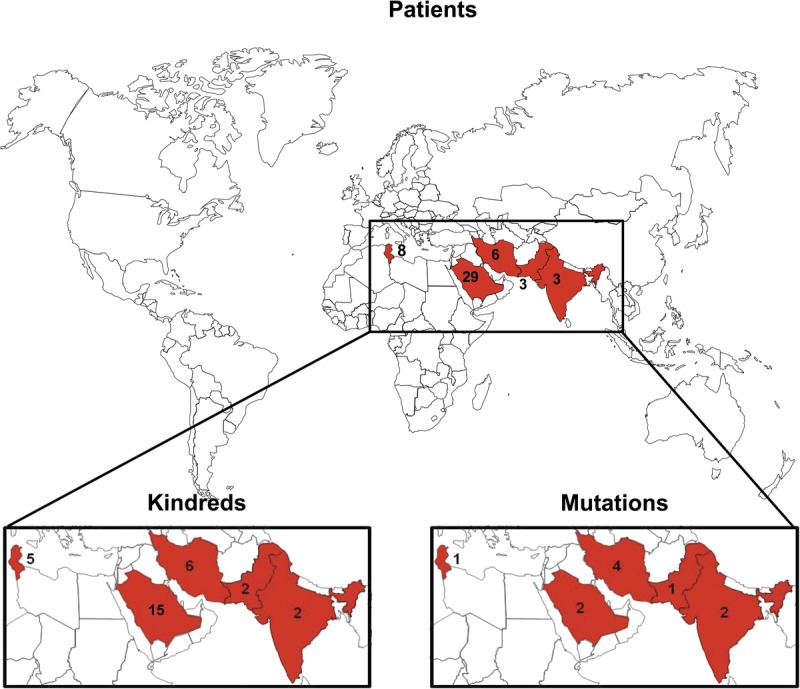
Mutated alleles of the IL12B gene. Schematic representation of the coding region of the IL12B gene containing 6 coding exons and encoding a 328-amino acid protein. Exons I and VIII are not translated. The coding region for each mutant IL12B allele is presented, together with the number of kindreds (n) and the date of the founder effect (FE). Gray regions correspond to stretches of new amino acids resulting from frameshift mutation. [This figure can be viewed in color online at http://www.md-journal.com].
TABLE 1.
Continued
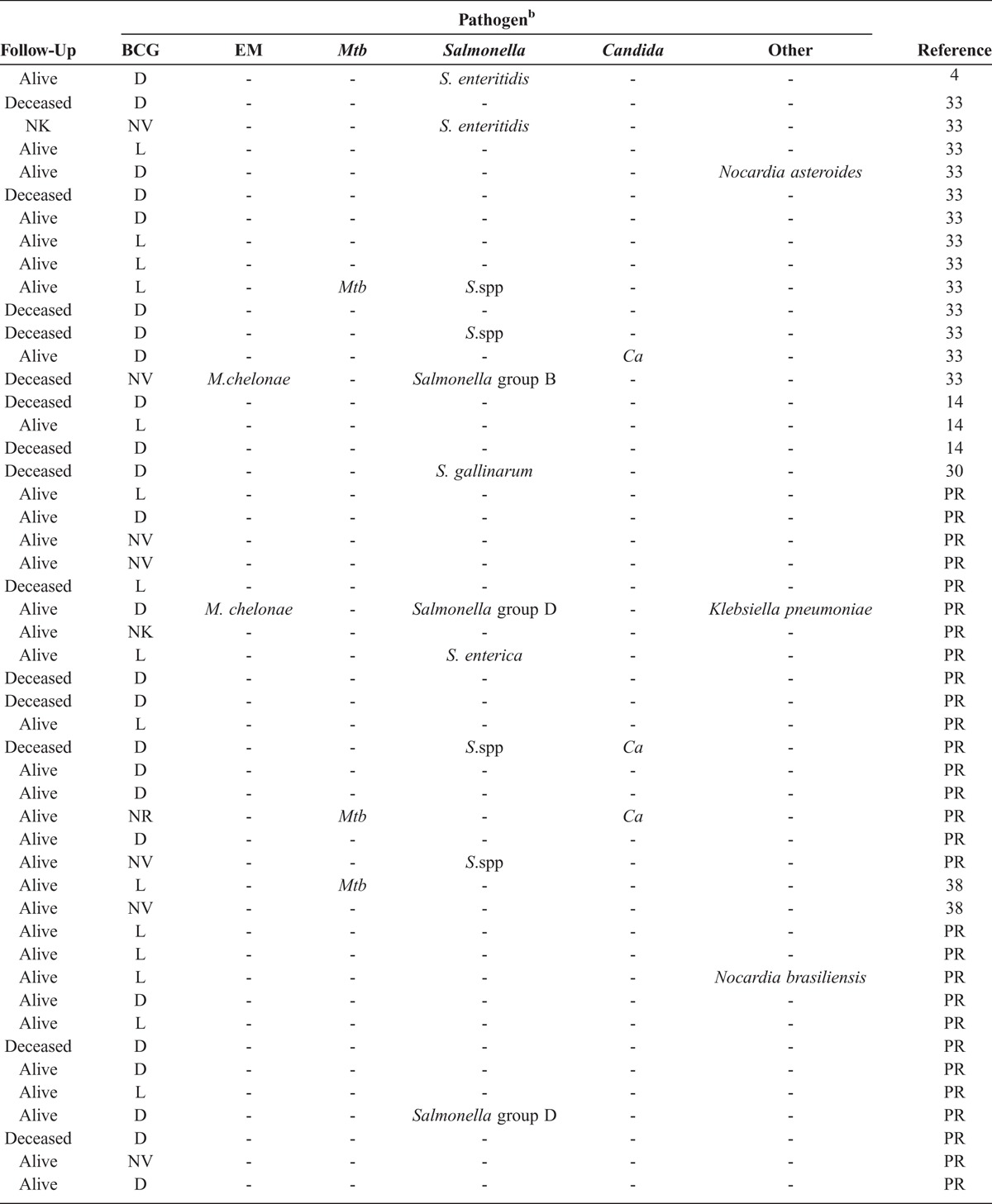
Figure 4 indicates the clinical features of the 30 IL-12p40-deficient index cases. The first clinical symptoms occurred in childhood (mean age, 1.1 yr; SD, 1.76 yr; range, 1 mo–7.6 yr). BCG was, by far, the most frequent infectious agent, causing the first clinical manifestation of MSMD (n = 27). Disseminated BCG disease (BCG-osis) was the most frequent presentation (19 of 27 cases), the other patients displaying regional BCG disease (BCG-itis; 8 of 27). Salmonella (2.II.3), EM (6.II.5) and M. tuberculosis (18.II.1) were responsible for the first clinical manifestation in 1 index case each. Patients 2.II.3 and 6.II.5 were not vaccinated with BCG. Patient 18.II.1 was vaccinated with BCG at the age of 3 years, with no adverse reaction. Twenty of 30 index cases (66.7%) presented with BCG disease as the only MSMD-related infection during their lifetime. After BCG disease, 5 patients developed salmonellosis, 1 patient developed tuberculosis (TB) (20.II.3), and another patient developed both salmonellosis and EM disease (12.II.1). Salmonellosis and TB were the only relevant infections observed during the lifetimes of patients 2.II.3 and 18.II.1, respectively. Patient 6.II.5 developed salmonellosis soon after EM infection.
FIGURE 4.
Clinical phenotypes for IL-12p40-deficient index cases. Each patient is classified as a function of mycobacterial infection status (BCG, TB, EM) and Salmonella infection status, as labeled. [This figure can be viewed in color online at http://www.md-journal.com].
Immunologic Phenotype of IL-12p40-Deficient Patients at the Whole-Blood and Cellular Levels
We assessed the IL-12 and IFN-γ responses of whole-blood cells from IL-12p40-deficient patients. We evaluated the production of IFN-γ, IL-12p40, and IL-12p70, after stimulation with BCG, BCG plus IL-12, and BCG plus IFNγ, as previously described.15–17 We tested blood from 22 patients from 19 different kindreds. All 9 mutant IL12B alleles were found among the 22 patients studied here. We compared the results obtained with cytokine determinations in the same stimulation conditions for whole blood from 44 healthy travel controls. The whole-blood cells of patients stimulated with live BCG alone or BCG plus exogenous recombinant IFN-γ produced no IL-12p40, whereas IL-12p40 was generated by the cells of healthy controls (p < 0.001 in both conditions), as shown by ELISA (Figure 5 A). IL-12p70 production by cells from the patients was more severely impaired upon stimulation with BCG plus exogenous recombinant IFN-γ (p < 0.001) than upon stimulation with BCG alone (p = 0.12). Furthermore, following stimulation with BCG, the patients’ cells produced significantly less IFN-γ than the cells of healthy travel controls (p < 0.001) (Figure 5 B). Stimulation with a combination of IL-12 plus BCG increased IFN-γ levels in the whole blood of IL-12p40-deficient patients, albeit to levels that remained significantly lower than those in travel controls (p < 0.001) (Figure 5 C). The cellular defect of IL-12p40 production in IL-12p40-deficient patients was further confirmed by the stimulation with PDBu of EBV-B cells lines derived from patients’ PBMCs.33 We have shown that IL-12p40-deficient patients have a lower than normal percentage of CD3+IL-17A+ cells ex vivo.11 However, their T-cell blasts responded to IL-23 stimulation by producing IL-17.11 The leukocytes of the patients described here displayed complete IL-12p40 and IL-12p70 deficiencies and impaired, but not abolished, IFN-γ production.
FIGURE 5.
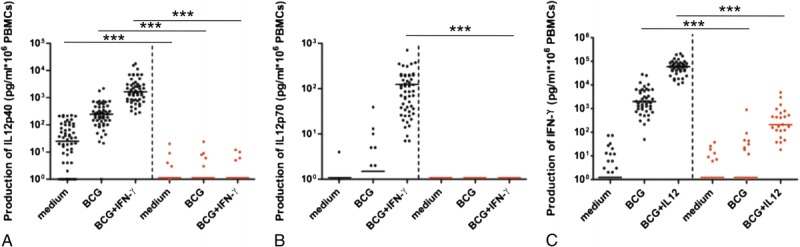
Impaired cellular response in IL-12p40-deficient patients. A logarithmic scale depicting the production of IL-12p40 (A), IL-12p70 (B), and IFN-γ (C) by whole blood cells from 44 healthy travel control patients (left side of each graph [black circles in color representation]), and from 22 IL-12p40-deficient patients (right side of each graph [red circles in color representation]), either unstimulated (medium) or stimulated with BCG alone or with BCG plus recombinant IFN-γ or IL-12. The horizontal bars indicate the mean. We calculated p values for differences between healthy travel controls and IL-12p40-deficient patients in the nonparametric Kruskal-Wallis rank sum test. ***p < 0.001. [This figure can be viewed in color online at http://www.md-journal.com].
Founder Effects Account for the Recurrence of Mutations
The distribution of the 9 different mutations observed in this cohort of IL-12p40-deficient patients did not overlap between different ethnic groups. A founder effect has been documented for g.482+82_856-854del, in 2 patients from Pakistan and India.33 This effect is thought to have occurred ∼700 years ago (95% confidence interval [CI], 216–2760 yr) based on analyses with a method developed in our laboratory.22,33 In the same study, the 315insA mutation, which is found only in patients from Saudi Arabia, was estimated to have first occurred ∼1100 years ago (528–2640 yr).33 This study added 10 new families, all from Saudi Arabia, to the original 4 families found to carry the 315insA mutation. Again, we estimated the date of the founder effect with a larger sample of 8 independent patients to ∼1100 years ago, with a smaller 95% CI, extending from 650 to 1850 years (Figure 6). We also found that the 526del2 deletion segregating exclusively in 3 Iranian families resulted from a founder effect. We were able to date this founder event from the data for 2 independent individuals to ∼600 years ago (175–2175 years). Finally, the 297del8 deletion is found, to our knowledge, only in Tunisian patients and is also the result of a founder effect that will be reported elsewhere (Barbouche et al, unpublished data [in preparation]). In conclusion, we identified 4 variants specific to 4 regions that resulted from founder effects and segregated in 25 of the 30 kindreds reported here.
FIGURE 6.
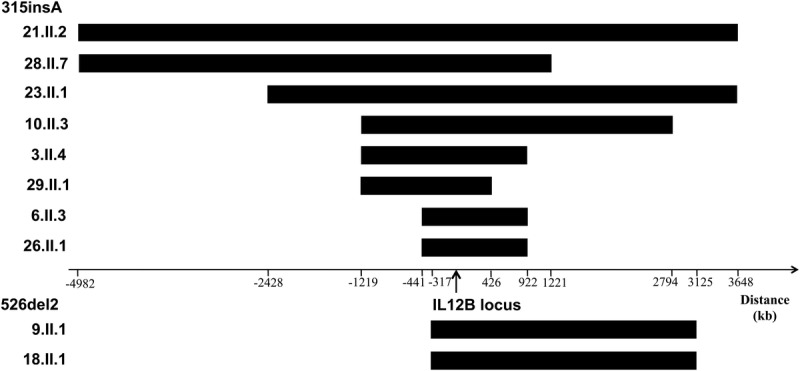
Haplotype sharing in the IL12B region. Long contiguous stretches of homozygosity were observed around the gene, consistent with its mode of inheritance. Mutations 315_insA and 526del2 were studied.
Relatives of Index Cases
The 30 probands had a total of 104 sibs. Genotyping was carried out for 51 of 104 sibs. Fifteen of 51 genotyped sibs were homozygous for mutations in IL12B. Five of these 15 sibs were asymptomatic at last follow-up (1.II.1, 10.II.7, 13.II.5, 21.II.4), at the ages of 9 years (1.II.1), 2 years (10.II.7), 26 years (13.II.5), 7 years (21.II.4), and 3 months (29.II.2). All had the same cellular phenotype as their clinically affected IL-12p40-deficient sibs. Four of the asymptomatic sibs had not been vaccinated with BCG vaccine, and BCG vaccination status is unknown for the fifth (13.II.5). Nine of the 10 genetically affected and symptomatic sibs had been vaccinated with BCG vaccination and all developed BCG disease. The only genetically affected, symptomatic sib not to have been vaccinated presented with nontyphoidal salmonellosis at the age of 2 years as the first and only symptom of MSMD (2.II.3). Five genetically affected sibs had displayed BCG disease only as of the last follow-up (3.II.4, 4.II.2, 7.II.4, 10.II.4, 16.II.5) (Figure 7). One sib developed disease caused by Nocardia asteroides (3.II.6). One patient presented with multiple infections, with BCG disease, nontyphoidal salmonellosis, and TB (4.II.4). One patient displayed BCG disease and candidiasis (6.II.4). Only 1 of the genetically affected sibs died (6.II.3); she presented with BCG disease and salmonellosis at the ages of 3 and 18 months, respectively. She died at the age of 11 years from meningoencephalitis that had not been confirmed bacteriologically. The infectious phenotype of these 15 genetically affected symptomatic sibs was thus very similar to that of the 30 index cases (see Figure 7). Four of the 53 nongenotyped sibs displayed MSMD (2.II.1, 3.II.9, 16.II.1, 28.II.1). Three (2.II.1, 3.II.9, 28.II.1) died of disseminated BCG disease, whereas the fourth (16.II.1) recovered from localized BCG infection. Eight of the remaining 53 nongenotyped sibs died from unknown causes. The 4 nongenotyped MSMD patients were considered to be genetically affected in subsequent analyses relating to clinical descriptions, giving a total of 44 symptomatic patients (30 index cases, 10 genotyped sibs, and 4 nongenotyped sibs). However, only genotyped subjects were used for penetrance estimation, as we do not know how many of the nongenotyped sibs were genetically affected but asymptomatic.
FIGURE 7.
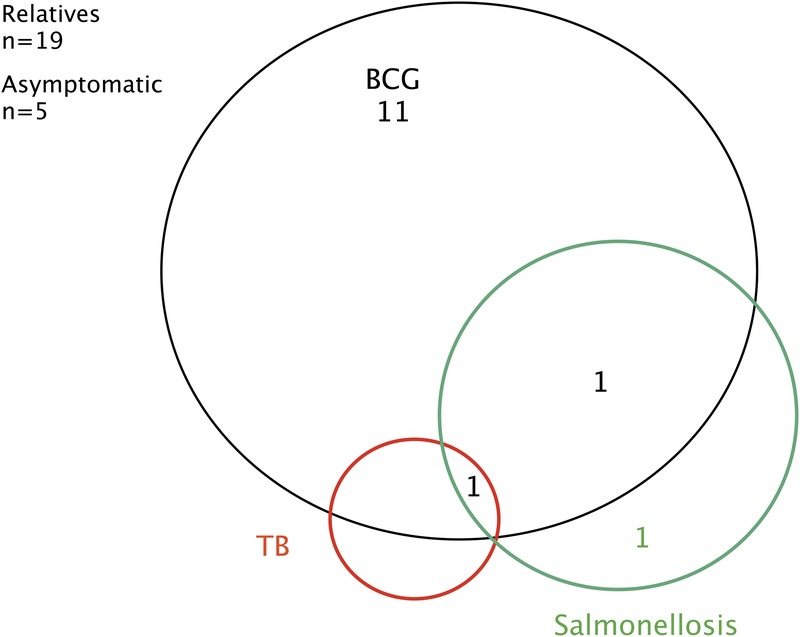
Clinical phenotypes for IL-12p40-deficient relatives. Each patient is classified as a function of mycobacterial infection status (BCG and TB) and Salmonella infection status, as labeled. [This figure can be viewed in color online at http://www.md-journal.com].
Mycobacterial Diseases in 44 Symptomatic Patients
Mycobacterial diseases were the most frequent infections, diagnosed in 42 of 44 symptomatic patients (95.45%). Infections caused by BCG vaccine accounted for 95.23% of all mycobacterial diseases. Thirty-seven of the genotyped patients had been vaccinated with BCG, and 36 (97.3%) developed BCG disease (localized, n = 13; disseminated, n = 23). Only 1 genotyped patient was vaccinated with BCG but did not develop an adverse reaction (18.II.1), and this patient was vaccinated later than the others, at the age of 3 years. Adverse reactions to BCG vaccination were significantly more frequent (p = 0.002) in patients with IL-12p40 deficiency (97.3%) than in patients with IL-12Rβ1 deficiency (76.4%, 84 of 110 vaccinated patients). Far fewer patients (2 of 44) developed EM disease, due to Mycobacterium chelonae in both cases. One of these patients had an EM infection followed by salmonellosis (6.II.5); the other developed BCG, EM, and Salmonella infections, as well as a surgical site infection due to Klebsiella species (12.II.5). Disseminated TB was found in 2 patients. One of these patients developed TB in combination with BCG disease (21.II.3). The second (18.II.1) was not vaccinated with BCG until the age of 3 years and developed disseminated TB at the age of 5 years. Patient 18.II.1 also presented with Candida infection. Recurrence was defined as a subsequent episode of disease with the same microorganism after a period free from clinical symptoms and treatment. Eleven cases (BCG, n = 10; TB, n = 1) presented with recurrent mycobacterial disease. Eight of the 10 deaths among the 44 symptomatic patients were due to mycobacterial disease. BCG disease was the only type of mycobacterial infection resulting in death.
Salmonellosis in the 44 Symptomatic Patients
Salmonellosis occurred in 11 patients (25%). It was the only infectious disease in 2 patients (2.II.3, 19.II.4), neither of whom had been vaccinated with BCG. The other 9 patients with salmonellosis also had BCG disease (n = 6), EM disease (n = 1), EM and BCG disease (n = 1), or TB and BCG disease (n = 1). Several serotypes of nontyphoidal Salmonella were identified in our patients: S. enteritidis, n = 3; S. gallinarum, n = 1; S. group B, n = 1; S. group C, n = 1; S. group D, n = 2. Salmonella serotype was not determined in 3 patients (1.II.1, 2.II.3, 13.II.1). Disseminated disease was the most common presentation (n = 5), followed by adenitis (n = 4) and gastroenteritis (n = 1). Recurrent salmonellosis was found in 36.4% of cases and was more frequent than BCG disease recurrence (25%).
Infections Caused by Other Agents
Nocardia, an intramacrophagic pathogen closely phylogenetically related to Mycobacterium, caused disease in 2 patients (N. brasiliensis, n = 1; N. asteroides, n = 1). Both these patients also displayed BCG disease. Klebsiella pneumoniae, a gram-negative enterobacterium closely related to Salmonella, caused disease in 1 patient (12.II.5), who also had several other infections (BCG, Salmonella, and EM). Three patients displayed Candida albicans infections (6.II.4, oral thrush; 16.II.2, disseminated; and 18.II.1, oral thrush). The incidence of Candida infections was significantly lower (p = 0.006) in IL-12p40-deficient patients (6.7%) than in IL-12Rβ1-deficient patients (24%). Mean age did not differ significantly between the IL-12p40-deficient patients (mean, 9.7 yr; SD, 7.74 yr; range, 3 mo–34 yr) and the IL-12Rβ1-deficient patients (mean, 11.2 yr; SD, 9.7 yr; range, 6 mo–46.4 yr), and therefore cannot account for this observation. We and others have reported the presence of autoantibodies against IL-17A, IL-17F, and IL-22 in patients suffering from chronic mucocutaneous candidiasis (CMC) and autoimmune polyendocrine syndrome type I.27,36 We also identified autosomal dominant IL-17F deficiency, autosomal recessive IL-17RA deficiency, and dominant gain-of-function STAT1 mutations as responsible for disease in patients with CMC.28,35,45 The impaired development of IL-17-producing T cells in at least some IL-12p40- and IL-12Rβ1-deficient patients11 probably accounts for their susceptibility to candidiasis.37 Finally, patient 7.II.2 presented with fulminant varicella zoster virus (VZV) infection, which may or may not result from IL-12p40 deficiency.39
Age at Onset of Infection in the 44 Symptomatic Patients
Consistent information concerning the onset of clinical symptoms was available for 42 of the 44 symptomatic patients: 30 index cases and 12 sibs. First infection occurred early in childhood, at a mean age of 1 year (range, 1 mo–7.6 yr; SD, 1.59 yr) (Figure 8). BCG was the most frequent causal agent (n = 40) and disseminated disease occurred as the first clinical manifestation of MSMD in 25 patients. BCG disease occurred in children as young as 1 month, with a mean age at onset of 9 months (range, 1 mo–7.6 yr; SD, 1.39 yr). In 35 cases (83.3%), BCG disease occurred within 1 year of vaccination. The mean time from BCG vaccination to disease was 6.37 months (range, 1 mo-4.16 yr; SD, 7.95 mo). The onset of salmonellosis (n = 11) occurred over a larger range of ages, extending from 3 months to 33 years (mean, 5.39 yr; SD, 9.78 yr). EM infection was found in 2 patients, at the ages of 3 and 4 years, respectively. Mean age at the onset of TB was 2.8 years (range, 6 mo–5 yr; SD, 2.25 yr). We can conclude that BCG disease was not only the most frequent infection in IL-12p40-deficient patients, but also the infection with the earliest age at onset.
FIGURE 8.
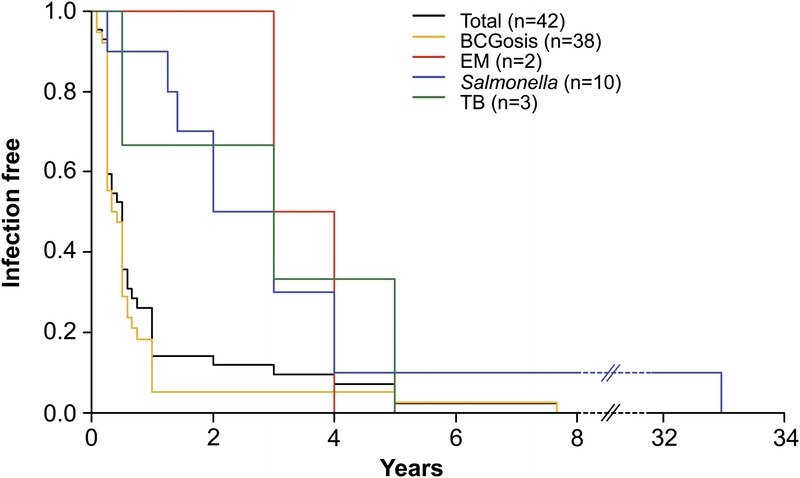
First onset of MSMD-related infections in IL-12p40-deficient patients (BCG, EM, Salmonella, and TB).
Survival Analysis
The mortality rate among symptomatic patients was 31.8% (14 of 44 symptomatic patients) (see Table 1). This is close to the rate of 38% reported in a series of 13 IL-12p40-deficient patients.33 Global mortality for all patients, including the 5 asymptomatic patients, was 28.6%. The mean age at death was 7.1 years for the 14 IL-12p40-deficient patients who died (range, 1.25–34 yr; SD, 8.33 yr; Figure 9). Most of the deceased patients died from disseminated BCG infection (n = 10). One patient survived multiple relapses of disseminated BCG disease (9.II.1) but succumbed to disseminated salmonellosis at the age of 34 years. Fulminant VZV infection caused the death of a second patient (7.II.2) who survived at least 5 relapses of BCG disease. Patient 6.II.5 was not vaccinated with BCG and died from disseminated EM infection (M. chelonae). For the remaining patient (6.II.3), the pathogen causing the central nervous system infection that resulted in death could not be identified. Only 12 of 44 symptomatic patients received human recombinant IFN-γ together with specific antibiotic treatment during infection; 5 of these patients have since died. One patient developed biliary cirrhosis during human recombinant IFN-γ treatment and underwent liver transplantation.38 Rarely, patients underwent surgical resection of the affected areas (lymph nodes, n = 2; bone, n = 1). We are currently collecting data to evaluate the impact of current treatment options and preventive management on outcome for IL-12p40-deficient patients.
FIGURE 9.
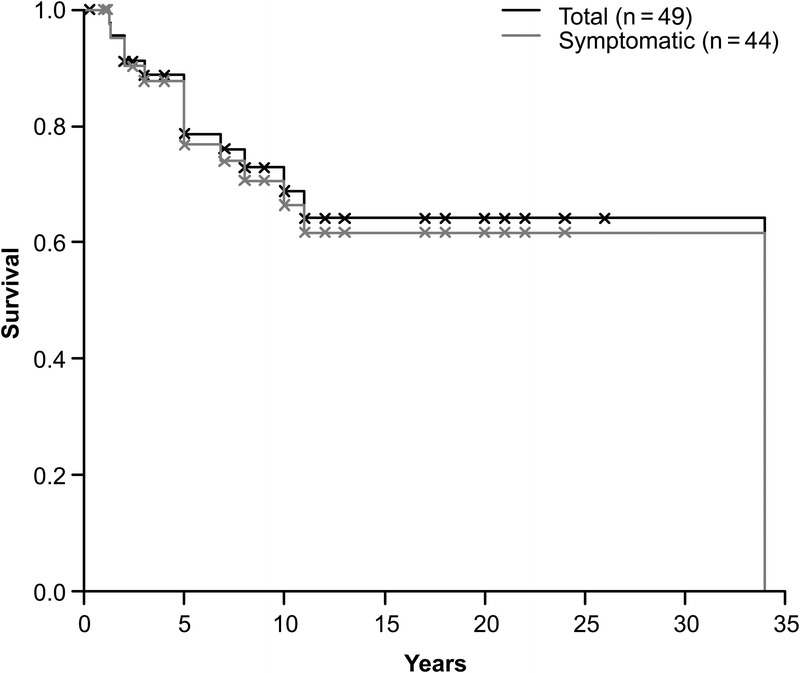
Survival curve for IL-12p40-deficient patients. Each death is indicated as a downward step in the curve. The crosses indicate the age at last follow-up for living patients. The continuous line represents survival for the total patient population (n = 49). The dashed line represents survival for the 44 symptomatic patients.
Incomplete Clinical Penetrance
Five of the 15 genetically affected sibs did not present with MSMD-related or other unusual infections at last follow-up (follow-up range, 3 mo–26 yr; mean, 8.85 yr; SD, 10.23 yr). For the estimation of clinical penetrance, we excluded 1 symptomatic patient about whom we had no information concerning age at first infection. The overall clinical penetrance of infections (Figure 10) increased rapidly from 0.45 (95% CI, 0.1–0.65) at the age of 7 months, to 0.71 (95% CI, 0.29–0.88) by the age of 48 months. All 3 sibs over the age of 5 years are asymptomatic. Ten of 11 genotyped relatives who had been vaccinated with BCG presented with BCG infection, of the disseminated form in 5 of these individuals. Two of these patients also presented with salmonellosis or salmonellosis and TB. One patient, who had not been vaccinated with BCG, presented with salmonellosis only.
FIGURE 10.
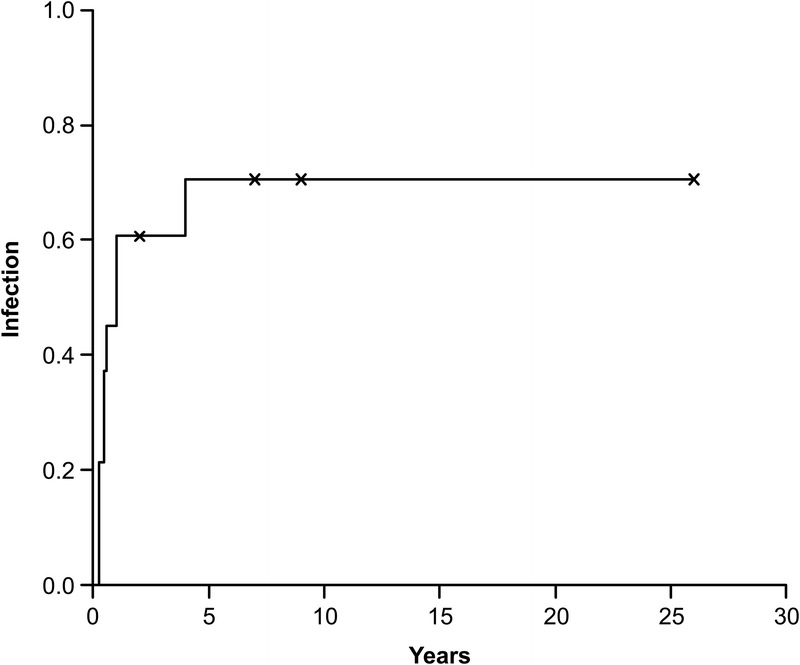
Penetrance of infection in patients with IL-12p40 deficiency. Estimation was based on findings for 14 genetically affected relatives with complete follow-up data. The onset of the first infection is indicated as an upward step in the curve. The crosses indicate the age at last follow-up for asymptomatic relatives.
DISCUSSION
IL-12p40 deficiency is considered a very rare immunodeficiency, but may occur more frequently than was previously thought. To our knowledge, the last IL12B mutation to be identified was reported in 2005.4,14,30,33,38 We have since identified 5 other mutations resulting in IL-12p40 deficiency. All patients with a given mutation have the same ethnic origin (Saudi Arabia, Tunisia, India/Pakistan, Iran), providing evidence for underlying founder effects. These founder effects have been studied for 3 of the 4 recurrent mutations (482+92_856-854del, 315insA, 526del2). The possible date of origin of the mutations varies from 600 years ago in Iran to 875 years ago in Saudi Arabia. Despite genotypic heterogeneity, with 9 mutant alleles, the immunologic phenotype remains homogeneous, with a complete deficiency of IL-12p40 and IL-12p70. To our knowledge no patients with IL-12p40 deficiency have yet been diagnosed in countries with a low prevalence of consanguinity, even in countries with national BCG vaccination programs, such as France. This suggests that mutant IL12B alleles are very rare and that heterozygosity for these alleles may be subject to negative selection at the population level. In any case, IL-12p40 deficiency remains the most common inborn error of cytokines, leading to primary immunodeficiency, as mutations in IL17F have been found in only 4 patients.35 IL-12p40 deficiency is the first AR cytokine defect identified in humans followed by the description of IL-10 deficiency causing infant colitis, in 2010.22a
Like the genotype, the clinical phenotype is more heterogeneous than the cellular phenotype. Clinical signs begin early in childhood, at a mean age of 1 year, as reported for IL-12Rβ1-deficient patients12 and patients with the complete form of IFN-γR deficiency,13 but earlier than suggested by recently published data for partial recessive IFN-γR1 deficiency (mean, 11.25 yr).41 With an increase in the number of identified IL-12-p40-deficient patients from 13 to 49 cases, we observed a decrease in overall mortality rate from 38%35 to 28.6%, with a mean age at death of 7.1 years. These data contrast with those for IL-12Rβ1-deficient patients,12 for whom an increase in mortality rate (from 15% to 32%) was observed together with increases in the number of cases.12,18 Mortality rates and clinical penetrance are similar for these 2 diseases, with clinical penetrance reaching 50% before the age of 12 months for both IL-12p40 and IL-12Rβ1 deficiencies.12 Overall clinical penetrance is about 80% for both diseases.12 BCG infection was the most frequent disease and the main cause of death in IL-12p40-deficient patients. Only 1 of the patients vaccinated with BCG (18.II.1) did not present with BCG disease, and this patient was not vaccinated until the age of 3 years. Salmonellosis was the sole clinical manifestation in 2 patients not vaccinated with BCG vaccine and another 9 patients developed salmonellosis in combination with other MSMD-related pathogens. This observation, together with the high incidence of salmonellosis as the only infection in IL-12Rβ1-deficient patients (21 of 57 cases of salmonellosis),12 highlights the importance of IL-12p40 and IL-12Rβ1 for protective immunity against Salmonella, via IL-12 and/or IL-23. Nontyphoidal, extraintestinal salmonellosis should lead physicians to check for IL-12p40 and IL-12Rβ1 deficiencies.
Other infectious agents related to the classical MSMD pathogens were found in our cohort of patients. For at least 3 of these pathogens, IL-12 is known to be required for the generation of an appropriate immune response. As expected, due to the impaired IL-17 immunity previously reported in patients with IL-12p40 deficiency,12 our patients were susceptible to mucocutaneous Candida infections. K. pneumoniae was found in patients with IL-12p40 and IL-12Rβ1 deficiencies. Both IL-17 and IL-23 have been shown to be important for the immune responses to Klebsiella and Salmonella.25,29 This may account for the higher frequency of Salmonella and Klebsiella infections in patients with IL-12p40 or IL-12Rβ1 deficiencies than in patients with IFN-γR deficiency.12,29 Nocardia asteroides, an intramacrophagic pathogen, was previously described in an IL-12p40-deficient patient.33 We report here an additional patient presenting with Nocardia brasiliensis infection. In both these cases, Nocardia infection was associated with BCG disease. Infection with a third Nocardia species (N. nova) was the only clinical sign in 1 IL-12Rβ1-deficient patient.12 Other intracellular pathogen infections found in IL-12Rβ1-deficient patients, such as paracoccidioidomycosis,31 coccidioidomycosis,46 histoplasmosis, toxoplasmosis, and leishmaniasis,12 have yet to be diagnosed in patients with IL-12p40 deficiency. However, IL-12Rβ1 deficiency has been diagnosed in 30 countries, whereas IL-12p40-deficient patients have been diagnosed in only 5 countries to date, to our knowledge.
Overall, IL-12p40 deficiency is not as rare as previously thought, and its outcome is more favorable than suggested by the description of a smaller group of patients. We can also conclude that IL-12p40 deficiency is clinically indistinguishable from IL-12Rβ1 deficiency; the modest differences documented probably reflect the differences in size and ethnic backgrounds of the 2 population samples. The current description of IL-12p40 deficiency in a large series of patients should help to increase awareness, improving the accuracy of diagnosis and patient care.
ACKNOWLEDGMENTS
The authors thank the patients, their families, and their physicians, whose trust, support, and cooperation were essential for collection of the data used in this study. We thank all members of the laboratory for helpful discussions. We thank M. Courat, C. Bidalled, M. N’Guyen, T. Leclerc, Y. Nemirovskaya, T. Nivare, and T. Kochetkov for secretarial and technical assistance.
Abbreviations
BCG
bacille Calmette-Guérin
CI
confidence interval
CMC
chronic mucocutaneous candidiasis
EBV
Epstein-Barr virus
ELISA
enzyme-linked immunosorbent assay
EM
environmental mycobacteria
FE
founder effect
IFN-γ
interferon gamma
IL
interleukin
IL-12R
interleukin 12 receptor
MSMD
Mendelian susceptibility to mycobacterial disease
PBMC
peripheral blood mononuclear cells
PCR
polymerase chain reaction
STAT
signal transducer and activator of transcription
TB
tuberculosis
VZV
varicella zoster virus
Footnotes
*These 3 authors contributed equally to this work.
†These 2 authors contributed equally to this work and share senior coauthorship.
Financial support and conflicts of interest: The Laboratory of Human Genetics of Infectious Diseases is supported by institutional grants to INSERM and The Rockefeller University, and grants from the Agence Nationale de la Recherche (ANR), the European Union HOMITB (E08153KK) and NEOTIM (018736), the St. Giles Foundation, the Thrasher Research Fund, the Jeffrey Modell Foundation, Talecris Biotherapeutics, National Institutes of Health (1R01AI089970-01), and the Rockefeller University Center for Clinical and Translational Science (5UL1RR024143-04). A. Samarina was supported by an Institut Pasteur fellowship. D. S Kumararatne was supported by NIHR Cambridge Biomedical Centre. J. L. Casanova was an International Scholar of the Howard Hughes Medical Institute from 2005 to 2008. The authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s website (www.md-journal.com).
REFERENCES
- 1.Alcais A, Fieschi C, Abel L, Casanova JL. Tuberculosis in children and adults: two distinct genetic diseases.J Exp Med. 2005; 202: 1617– 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity?Ann N Y Acad Sci. 2010; 1214: 18– 33. [DOI] [PubMed] [Google Scholar]
- 3.Al-Muhsen S, Casanova JL. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases.J Allergy Clin Immunol. 2008; 122: 1043– 1051. [DOI] [PubMed] [Google Scholar]
- 4.Altare F, Lammas D, Revy P, Jouanguy E, Doffinger R, Lamhamedi S, Drysdale P, Scheel-Toellner D, Girdlestone J, Darbyshire P, Wadhwa M, Dockrell H, Salmon M, Fischer A, Durandy A, Casanova JL, Kumararatne DS. Inherited interleukin 12 deficiency in a child with bacille Calmette-Guerin and Salmonella enteritidis disseminated infection.J Clin Invest. 1998; 102: 2035– 2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a. 4a. Bogunovic D, Byun M, Durfee LA, Abhyanker A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong FX, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency.Science. 2012; 337: 1684– 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, Casanova JL. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease.Nat Immunol. 2011; 12: 213– 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, Casanova JL. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases.Curr Opin Immunol. 2008; 20: 39– 48. [DOI] [PubMed] [Google Scholar]
- 7.Bustamante J, Picard C, Fieschi C, Filipe-Santos O, Feinberg J, Perronne C, Chapgier A, de Beaucoudrey L, Vogt G, Sanlaville D, Lemainque A, Emile JF, Abel L, Casanova JL. A novel X-linked recessive form of Mendelian susceptibility to mycobaterial disease.J Med Genet. 2007; 44: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model.Annu Rev Immunol. 2002; 20: 581– 620. [DOI] [PubMed] [Google Scholar]
- 9.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy.Science. 2007; 317: 617– 619. [DOI] [PubMed] [Google Scholar]
- 10.Cooper AM, Solache A, Khader SA. Interleukin-12 and tuberculosis: an old story revisited.Curr Opin Immunol. 2007; 19: 441– 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells.J Exp Med. 2008; 205: 1543– 1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, Al-Muhsen S, Janniere L, Rose Y, de Suremain M, Kong XF, Filipe-Santos O, Chapgier A, Picard C, Fischer A, Dogu F, Ikinciogullari A, Tanir G, Al-Hajjar S, Al-Jumaah S, Frayha HH, AlSum Z, Al-Ajaji S, Alangari A, Al-Ghonaium A, Adimi P, Mansouri D, Ben-Mustapha I, Yancoski J, Garty BZ, Rodriguez-Gallego C, Caragol I, Kutukculer N, Kumararatne DS, Patel S, Doffinger R, Exley A, Jeppsson O, Reichenbach J, Nadal D, Boyko Y, Pietrucha B, Anderson S, Levin M, Schandene L, Schepers K, Efira A, Mascart F, Matsuoka M, Sakai T, Siegrist CA, Frecerova K, Bluetters-Sawatzki R, Bernhoft J, Freihorst J, Baumann U, Richter D, Haerynck F, De Baets F, Novelli V, Lammas D, Vermylen C, Tuerlinckx D, Nieuwhof C, Pac M, Haas WH, Muller-Fleckenstein I, Fleckenstein B, Levy J, Raj R, Cohen AC, Lewis DB, Holland SM, Yang KD, Wang X, Wang X, Jiang L, Yang X, Zhu C, Xie Y, Lee PP, Chan KW, Chen TX, Castro G, Natera I, Codoceo A, King A, Bezrodnik L, Di Giovani D, Gaillard MI, de Moraes-Vasconcelos D, Grumach AS, da Silva Duarte AJ, Aldana R, Espinosa-Rosales FJ, Bejaoui M, Bousfiha AA, Baghdadi JE, Ozbek N, Aksu G, Keser M, Somer A, Hatipoglu N, Aydogmus C, Asilsoy S, Camcioglu Y, Gulle S, Ozgur TT, Ozen M, Oleastro M, Bernasconi A, Mamishi S, Parvaneh N, Rosenzweig S, Barbouche R, Pedraza S, Lau YL, Ehlayel MS, Fieschi C, Abel L, Sanal O, Casanova JL. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries.Medicine (Baltimore). 2010; 89: 381– 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, Rosenzweig SD, Newport M, Levin M, Roesler J, Kumararatne D, Casanova JL, Holland SM. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies.Lancet. 2004; 364: 2113– 2121. [DOI] [PubMed] [Google Scholar]
- 14.Elloumi-Zghal H, Barbouche MR, Chemli J, Bejaoui M, Harbi A, Snoussi N, Abdelhak S, Dellagi K. Clinical and genetic heterogeneity of inherited autosomal recessive susceptibility to disseminated Mycobacterium bovis bacille calmette-guerin infection.J Infect Dis. 2002; 185: 1468– 1475. [DOI] [PubMed] [Google Scholar]
- 15.Feinberg J, Fieschi C, Doffinger R, Feinberg M, Leclerc T, Boisson-Dupuis S, Picard C, Bustamante J, Chapgier A, Filipe-Santos O, Ku CL, de Beaucoudrey L, Reichenbach J, Antoni G, Balde R, Alcais A, Casanova JL. Bacillus Calmette Guerin triggers the IL-12/IFN-gamma axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes.Eur J Immunol. 2004; 34: 3276– 3284. [DOI] [PubMed] [Google Scholar]
- 16.Fieschi C, Bosticardo M, de Beaucoudrey L, Boisson-Dupuis S, Feinberg J, Santos OF, Bustamante J, Levy J, Candotti F, Casanova JL. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors.Blood. 2004; 104: 2095– 2101. [DOI] [PubMed] [Google Scholar]
- 17.Fieschi C, Casanova JL. The role of interleukin-12 in human infectious diseases: only a faint signature.Eur J Immunol. 2003; 33: 1461– 1464. [DOI] [PubMed] [Google Scholar]
- 18.Fieschi C, Dupuis S, Catherinot E, Feinberg J, Bustamante J, Breiman A, Altare F, Baretto R, Le Deist F, Kayal S, Koch H, Richter D, Brezina M, Aksu G, Wood P, Al-Jumaah S, Raspall M, Da Silva Duarte AJ, Tuerlinckx D, Virelizier JL, Fischer A, Enright A, Bernhoft J, Cleary AM, Vermylen C, Rodriguez-Gallego C, Davies G, Blutters-Sawatzki R, Siegrist CA, Ehlayel MS, Novelli V, Haas WH, Levy J, Freihorst J, Al-Hajjar S, Nadal D, De Moraes Vasconcelos D, Jeppsson O, Kutukculer N, Frecerova K, Caragol I, Lammas D, Kumararatne DS, Abel L, Casanova JL. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: medical and immunological implications.J Exp Med. 2003; 197: 527– 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, Jouanguy E, Boisson-Dupuis S, Fieschi C, Picard C, Casanova JL. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features.Semin Immunol. 2006; 18: 347– 361. [DOI] [PubMed] [Google Scholar]
- 20.Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, Frucht DM, Christel K, von Bernuth H, Jouanguy E, Feinberg J, Durandy A, Senechal B, Chapgier A, Vogt G, de Beaucoudrey L, Fieschi C, Picard C, Garfa M, Chemli J, Bejaoui M, Tsolia MN, Kutukculer N, Plebani A, Notarangelo L, Bodemer C, Geissmann F, Israel A, Veron M, Knackstedt M, Barbouche R, Abel L, Magdorf K, Gendrel D, Agou F, Holland SM, Casanova JL. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production.J Exp Med. 2006; 203: 1745– 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe’er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Tsunoda T, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Gibbs RA, Belmont JW, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L’Archeveque P, Bellemare G, Saeki K, Wang H, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J. A second generation human haplotype map of over 3.1 million SNPs.Nature. 2007; 449: 851– 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genin E, Tullio-Pelet A, Begeot F, Lyonnet S, Abel L. Estimating the age of rare disease mutations: the example of Triple-A syndrome.J Med Genet. 2004; 41: 445– 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22a. 22a. Glocker EO, Frede N, Perro M, Sebire N, Elawad M, Shah N, Grimbacher B. Infant colitis–it’s in the genes.Lancet. 2010; 376: 1272. [DOI] [PubMed] [Google Scholar]
- 23.Grohmann U, Belladonna ML, Bianchi R, Orabona C, Ayroldi E, Fioretti MC, Puccetti P. IL-12 acts directly on DC to promote nuclear localization of NF-kappaB and primes DC for IL-12 production.Immunity. 1998; 9: 315– 323. [DOI] [PubMed] [Google Scholar]
- 24.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, Menon G, Trouillet C, McDonald D, Carey P, Ginhoux F, Alsina L, Zumwalt TJ, Kong XF, Kumararatne D, Butler K, Hubeau M, Feinberg J, Al-Muhsen S, Cant A, Abel L, Chaussabel D, Doffinger R, Talesnik E, Grumach A, Duarte A, Abarca K, Moraes-Vasconcelos D, Burk D, Berghuis A, Geissmann F, Collin M, Casanova JL, Gros P. IRF8 mutations and human dendritic-cell immunodeficiency.N Engl J Med. 2011; 365: 127– 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae.J Exp Med. 2005; 202: 761– 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation.Annu Rev Immunol. 2007; 25: 221– 242. [DOI] [PubMed] [Google Scholar]
- 27.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJ, Pura M, Schalke B, Strobel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, Meager A. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines.J Exp Med. 2010; 207: 299– 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis.J Exp Med. 2011; 208: 1635– 1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacLennan C, Fieschi C, Lammas DA, Picard C, Dorman SE, Sanal O, MacLennan JM, Holland SM, Ottenhoff TH, Casanova JL, Kumararatne DS. Interleukin (IL)-12 and IL-23 are key cytokines for immunity against Salmonella in humans.J Infect Dis. 2004; 190: 1755– 1757. [DOI] [PubMed] [Google Scholar]
- 30.Mansouri D, Adimi P, Mirsaeidi M, Mansouri N, Khalilzadeh S, Masjedi MR, Adimi P, Tabarsi P, Naderi M, Filipe-Santos O, Vogt G, de Beaucoudrey L, Bustamante J, Chapgier A, Feinberg J, Velayati AA, Casanova JL. Inherited disorders of the IL-12-IFN-gamma axis in patients with disseminated BCG infection.Eur J Pediatr. 2005; 164: 753– 757. [DOI] [PubMed] [Google Scholar]
- 31.Moraes-Vasconcelos D, Grumach AS, Yamaguti A, Andrade ME, Fieschi C, de Beaucoudrey L, Casanova JL, Duarte AJ. Paracoccidioides brasiliensis disseminated disease in a patient with inherited deficiency in the beta1 subunit of the interleukin (IL)-12/IL-23 receptor.Clin Infect Dis. 2005; 41: e31– e37. [DOI] [PubMed] [Google Scholar]
- 32.Pesu M, Muul L, Kanno Y, O’Shea JJ. Proprotein convertase furin is preferentially expressed in T helper 1 cells and regulates interferon gamma.Blood. 2006; 108: 983– 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picard C, Fieschi C, Altare F, Al-Jumaah S, Al-Hajjar S, Feinberg J, Dupuis S, Soudais C, Al-Mohsen IZ, Genin E, Lammas D, Kumararatne DS, Leclerc T, Rafii A, Frayha H, Murugasu B, Wah LB, Sinniah R, Loubser M, Okamoto E, Al-Ghonaium A, Tufenkeji H, Abel L, Casanova JL. Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds.Am J Hum Genet. 2002; 70: 336– 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gately MK, Gubler U. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits.Proc Natl Acad Sci U S A. 1996; 93: 14002– 14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity.Science. 2011; 332: 65– 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I.J Exp Med. 2010; 207: 291– 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puel A, Picard C, Cypowyj S, Lilic D, Abel L, Casanova JL. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines?Curr Opin Immunol. 2010; 22: 467– 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pulickal AS, Hambleton S, Callaghan MJ, Moore CE, Goulding J, Goodsall A, Baretto R, Lammas DA, Anderson ST, Levin M, Pollard AJ. Biliary cirrhosis in a child with inherited interleukin-12 deficiency.J Trop Pediatr. 2008; 54: 269– 271. [DOI] [PubMed] [Google Scholar]
- 39.Roesler J, Hedrich C, Laass MW, Heyne K, Rosen-Wolff A. Meningoencephalitis caused by varicella-zoster virus reactivation in a child with dominant partial interferon-gamma receptor-1 deficiency.Pediatr Infect Dis J. 2011; 30: 265– 266. [DOI] [PubMed] [Google Scholar]
- 40.Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways.Immunol Rev. 2005; 203: 38– 47. [DOI] [PubMed] [Google Scholar]
- 41.Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernandez-Perez L, Chapgier A, Cardenes M, Feinberg J, Garcia-Laorden MI, Picard C, Santiago E, Kong X, Janniere L, Colino E, Herrera-Ramos E, Frances A, Navarrete C, Blanche S, Faria E, Remiszewski P, Cordeiro A, Freeman A, Holland S, Abarca K, Valeron-Lemaur M, Goncalo-Marques J, Silveira L, Garcia-Castellano JM, Caminero J, Perez-Arellano JL, Bustamante J, Abel L, Casanova JL, Rodriguez-Gallego C. Partial recessive IFN-gammaR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds.Hum Mol Genet. 2011; 20: 1509– 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi M, Matsuda F, Margetic N, Lathrop M. Automated identification of single nucleotide polymorphisms from sequencing data.Proc IEEE Comput Soc Bioinform Conf. 2002; 1: 87– 93. [PubMed] [Google Scholar]
- 43.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity.Nat Rev Immunol. 2003; 3: 133– 146. [DOI] [PubMed] [Google Scholar]
- 44.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses.Immunity. 2003; 19: 641– 644. [DOI] [PubMed] [Google Scholar]
- 45.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis.N Engl J Med. 2011; 365: 54– 61. [DOI] [PubMed] [Google Scholar]
- 46.Vinh DC, Schwartz B, Hsu AP, Miranda DJ, Valdez PA, Fink D, Lau KP, Long-Priel D, Kuhns DB, Uzel G, Pittaluga S, Hoover S, Galgiani JN, Holland SM. Interleukin-12 receptor beta1 deficiency predisposing to disseminated coccidioidomycosis.Clin Infect Dis. 2011; 52: e99– e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, Mahlaoui N, Gineau L, Alcais A, Lamaze C, Puck JM, de Saint Basile G, Khayat CD, Mikhael R, Casanova JL. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation.J Exp Med. 2008; 205: 1729– 1737. [DOI] [PMC free article] [PubMed] [Google Scholar]