Toll-like Receptor 3-mediated Necrosis via TRIF, RIP3, and MLKL (original) (raw)
Background: RIP3-dependent programmed necrosis is an alternative to apoptosis.
Results: When caspase-8 is compromised, TRIF-dependent TLRs directly activate RIP3 kinase through RHIM-dependent interactions.
Conclusion: TRIF mediates direct RHIM-dependent signaling, triggering necrosis via RIP3 and MLKL.
Significance: Programmed necrosis eliminates cells following stimulation of either MyD88 or TRIF signaling pathways that converge on RIP3.
Keywords: Apoptosis, Caspase, Necrosis (Necrotic Death), Serine/Threonine Protein Kinase, Toll-like Receptors (TLR), RIPK3
Abstract
Toll-like receptor (TLR) signaling is triggered by pathogen-associated molecular patterns that mediate well established cytokine-driven pathways, activating NF-κB together with IRF3/IRF7. In addition, TLR3 drives caspase 8-regulated programmed cell death pathways reminiscent of TNF family death receptor signaling. We find that inhibition or elimination of caspase 8 during stimulation of TLR2, TLR3, TLR4, TLR5, or TLR9 results in receptor interacting protein (RIP) 3 kinase-dependent programmed necrosis that occurs through either TIR domain-containing adapter-inducing interferon-β (TRIF) or MyD88 signal transduction. TLR3 or TLR4 directly activates programmed necrosis through a RIP homotypic interaction motif-dependent association of TRIF with RIP3 kinase (also called RIPK3). In fibroblasts, this pathway proceeds independent of RIP1 or its kinase activity, but it remains dependent on mixed lineage kinase domain-like protein (MLKL) downstream of RIP3 kinase. Here, we describe two small molecule RIP3 kinase inhibitors and employ them to demonstrate the common requirement for RIP3 kinase in programmed necrosis induced by RIP1-RIP3, DAI-RIP3, and TRIF-RIP3 complexes. Cell fate decisions following TLR signaling parallel death receptor signaling and rely on caspase 8 to suppress RIP3-dependent programmed necrosis whether initiated directly by a TRIF-RIP3-MLKL pathway or indirectly via TNF activation and the RIP1-RIP3-MLKL necroptosis pathway.
Introduction
Pathogen sensors recognize pathogen-associated molecular patterns during viral or microbial infection, initiating well recognized host defense pathways through transcriptional activation of immunomodulatory cytokines, chemokines, and interferons. These innate host defense pathways restrict pathogens and sculpt the adaptive immune response. Pattern recognition receptors (PRRs2 or sensors) also facilitate antigen presentation to produce an optimal adaptive immune response with memory to protect from reinfection. Although important in host defense, PRRs also facilitate inflammation and allied processes leading to autoimmunity that may depend on cytokine activation, cell death, or a mixture of both. Growing evidence implicates sensors in infected cell fate decisions via regulated cell death pathways. Apoptosis, pyroptosis, and necrosis all contribute to first line elimination of infected cells (1). The importance of cell death in host defense is evident because these pathways have the ability to stop a pathogen from infecting a host. An enormous variety of pathogen-encoded cell death suppressors contribute to virulence (1, 2). Toll-like receptors (TLRs) were the first PRRs to be identified (3), sensing pathogen-associated peptidoglycan (TLR2), double-stranded (ds)RNA (TLR3), lipopolysaccharide (LPS) (TLR4), flagellin (TLR5), unmethylated CpG DNA motifs (TLR9), as well as other pathogen-associated molecular patterns (3). TLRs recruit Toll/IL-1R (TIR) domain-containing adapters to activate gene expression via transcription factors, such as NF-κB and IRF3/IRF7. This leads to the expression of inflammatory cytokines that include TNF, interferons, and many others. TLR3 and TLR4 are unique in employing the adapter TRIF to signal. TLR4 and all other TLRs signal via MyD88. TLRs, like TNF family death receptors, mediate cytokine and interferon activation (3), while also holding rein over cell fate decisions, including apoptosis (4) and programmed necrosis (5).
Viral infection triggers apoptosis or necrosis via death receptors (6–8) and other infection-associated signals (9–11), to cut short infection. Apoptosis depends on a caspase-dependent proteolytic cascade that dismantles cells in an orderly fashion while maintaining membrane integrity (12, 13), whereas programmed necrosis leads to cell leakage through mechanisms that are currently being defined. Death receptor-induced programmed necrosis, also called necroptosis (14), depends on an association of the receptor interacting protein kinase (RIP)1 with RIP3 (6–8, 10, 15). Virus-induced programmed necrosis depends on the interaction of the DNA sensor DAI and RIP3 (11) independent of RIP1 (9, 10). Furthermore, TLR3 and TLR4 can induce necrotic death via TRIF (5), although the relative contribution of RIP1 to this process has not been fully dissected. These diverse studies resulted in the recognition of RIP3 as the key common mediator of programmed necrosis (10), with adapters such as MLKL and PGAM5 implicated downstream through as yet undefined mechanisms (16–18). The entwined nature of these distinct death processes has been most extensively studied in the context of TNFR1 signaling (6–8, 10, 15).
Death receptor activation drives the assembly of a cytosolic caspase-8 (Casp8) signaling platform (called complex IIB) that includes RIP1, Casp8, Fas-associated via death domain (FADD), and cellular FLICE/Casp8 inhibitory protein (cFLIP). This complex maintains control over Casp8-dependent apoptosis as well as RIP3-dependent necroptosis. A comparable death receptor-independent signaling platform (called a ripoptosome) forms downstream of TLR3 activation and is likely dependent on TRIF (10, 19, 20). Either complex regulates dimerization and autocleavage that can drive Casp8-mediated apoptosis and suppress RIP3-dependent death. This relationship became very clear when the midgestational death of Casp8-deficient mice was reversed by the elimination of RIP3 (21, 22). In the face of either Casp8 or FADD compromise, RIP1 and RIP3 oligomerize via a common RIP homotypic interaction motif (RHIM)-dependent process to drive necroptosis (6–8, 14, 15). Thus, Casp8 prevents programmed necrosis, possibly by cleaving RIP1 and/or RIP3 directly, separating the kinase and RHIM domains (23–26), or by targeting some other component in the pathway. The long form of cFLIP (cFLIPL), an NF-κB-inducible noncatalytic paralog that dimerizes with Casp8, is best known for its ability to blunt apoptosis by preventing maturation of Casp8 into a fully active pro-apoptotic form (27). Recently, cFLIPL has been directly implicated in maintaining basal Casp8 catalytic activity within a cytosolic complex that prevents the unleashing of necroptosis mediated by RIP1 and RIP3 (22, 28).
TLR3 signaling may result in any of three distinct cellular outcomes that are triggered by TRIF via a C-terminal RHIM domain interaction with RIP1 or RIP3 (4, 29) as follows: 1) activation of NF-κB (29, 30), partly dependent on RIP1; 2) initiation of apoptosis via Casp8, which is influenced by RIP1 engagement (4, 19); and 3) initiation of programmed necrosis dependent on RIP1 and RIP3 when Casp8 activity is compromised (5, 31). These outcomes are all analogous to TNFR1 signaling (10), where RIP1 complexes with FADD via a death domain-dependent interaction and deploys protein kinase activity following RHIM-dependent oligomerization, recruiting RIP3 to execute necroptosis. In this process, the kinase activities of both RIP1 and RIP3 contribute to the necrotic death. In binding to TRIF, however, RIP3 has been reported to out-compete RIP1 to disrupt NF-κB activation (29), raising the question of which RHIM-dependent interactions are most relevant to natural settings.
Although little is known about the hierarchy of RHIM-dependent interactions dictating cell fate decisions, a precedent has recently emerged from studies of the cytosolic DNA-sensor protein DAI (also called ZBP1). Necrotic death in response to murine cytomegalovirus (MCMV) infection depends on a complex between DAI and RIP3 that is RHIM-dependent but completely independent of RIP1, NF-κB activation, and interferon signaling (9, 11). MCMV, similar to other large DNA viruses, depends on an array of cell death suppressors to block apoptotic and necrotic death during infection. M36-encoded viral inhibitor of Casp8 activation (vICA) impairs the full maturation of Casp8 possibly with an impact on basal Casp8 catalytic activity necessary to prevent necrotic cell death (21). MCMV has evolved a dedicated suppressor to counteract regulated cell death pathways (32–35). This viral inhibitor of RIP activation (vIRA) acts as a competitor of RHIM-dependent interactions able to block apoptosis and NF-κB activation (32, 33) that naturally prevents association of DAI and RIP3 during infection (9, 11). vIRA prevents all forms of RHIM signaling and, independent of RHIM interactions, interferes with NF-κB essential modulator-dependent NF-κB activation (35, 36).
In this study, we demonstrate that cell survival following TLR engagement requires caspase activity to suppress RIP3-dependent necrosis. TLRs rely on either MyD88 or TRIF for signal transduction. Those using the adapter protein MyD88 trigger RIP1–RIP3 activation indirectly by inducing intermediate TNF that triggers necroptosis via TNFR1, whereas TLR3 and TLR4 drive RIP3 activation directly via the adapter protein TRIF. In this manner, competing RHIM-dependent cell death and survival signals radiate from TRIF via RIP1 and RIP3 as well as Casp8. This pathway parallels death receptor signaling, where Casp8 compromise by virus-encoded suppressors unleashes necrotic death and curtails infection (9, 10). Here, we show that the TRIF, RIP3, and MLKL promote death via a pathway that is analogous to the RIP1-RIP3-MLKL (16–18) axis. Likewise, we implicate DAI-RIP3-MLKL in virus-induced necrosis (9, 10). A picture emerges that the host defense arsenal employs programmed necrosis as a trap door that may be initiated by RIP1, DAI, or TRIF whenever caspase 8 activity is compromised. RHIM-dependent oligomerization of RIP3 followed by RIP3 kinase-dependent recruitment of MLKL emerge as a common execution step in programmed necrosis.
MATERIALS AND METHODS
Mice
TRIF mutant (C57BL/6J-Ticam1_Lps2_) mice (37) were from The Jackson Laboratory. _Rip3_−/− mice (38), Rip1+/− mice (39), _Tnf_−/− (40), and Casp8+/− mice (41) were described previously (21). Mice were bred and maintained by Emory University Division of Animal Resources. Procedures were approved by the Emory University Institutional Animal Care and Use Committee.
Cell Culture, Plasmids, Viruses, Transfections, and Transductions
L929, NIH3T3, 3T3-SA, SVEC4-10, J774, and primary MEFs were maintained in DMEM containing 4.5 g/ml glucose, 10% FBS (Atlanta Biologicals), 2 mm l-glutamine, 100 units/ml penicillin, and 100 units/ml streptomycin (Invitrogen). For bone marrow-derived macrophage (BMDM) culture, pooled bone marrow cells from flushed tibias and femurs were harvested into PBS containing 0.5 mm EDTA, placed in culture for at least 18 h in DMEM containing 10% FBS, and then differentiated for 5–7 days in DMEM containing 20% FBS and 20% L929-conditioned medium. Where indicated, cells were stimulated with murine IFNβ (Chemicon) or TNF (PeproTech). The following compounds were employed: necrostatin (Nec)-1 (Calbiochem); Z-VAD-fmk (Enzo Life Sciences); bafilomycin A1 and cycloheximide (Sigma); poly(I:C) (GE Healthcare); and LPS, Pam3CysK, and CpG DNA (Invivogen). Flagellin was kindly provided by Andrew Gewirtz (Georgia State University). Selective small molecule RIP3 kinase inhibitors GSK'843 and GSK'872 were identified through compound screening and optimization efforts.3 Control, RIP1, and MLKL siRNA ON-TARGET SMARTpools were obtained from Thermo Scientific, and transfection employed Lipofectamine RNAi Max (Invitrogen). The pLKO.1-based RIP3 shRNA constructs were obtained from Open Biosystems (TRCN0000022535). The pLKO.1 control scramble shRNA vector, lentiviral/retroviral vector production, infection, and selection of transduced cells as well as all other MCMV strains and plasmids have been described (4, 9, 21, 32, 42).
Immunoblot and Immunoprecipitations
Following preparation of cell extracts, immunoprecipitation, and electrophoretic separation on denaturing polyacrylamide gels followed by transfer (4, 9), immunoblot analysis was performed on the following antibodies: mouse anti-β-actin (clone AC-74; Sigma); rabbit anti-Casp8 (Cell Signaling); rabbit anti-MLKL (Abgent); mouse anti-RIP1 (clone 38; BD Biosciences); rabbit anti-RIP3 (Imgenex); goat anti-RIP3 (clone C-16; Santa Cruz Biotechnology); rabbit anti-IκBα (Santa Cruz Biotechnology); rabbit anti-phospho-IκBα (Cell Signaling Technology); and anti-mouse IgG-HRP and anti-rabbit IgG-HRP (Vector Laboratories). For immunoprecipitation analyses, goat anti-RIP3 anti-body and protein A/G-agarose (Santa Cruz Biotechnology) were used.
Cell Viability Assays
L929 cells (5000 cells/well), BMDM (30,000 cells/well), NIH3T3 (10,000 cells/well), 3T3-SA (10,000 cells/well), and SVEC4-10 (10,000 cells/well) were seeded into Corning 96-well tissue culture plates (3610). In most experiments, cell viability was assessed by measuring the intracellular levels of ATP using the Cell Titer-Glo luminescent cell viability assay kit (Promega) according to the manufacturer's instructions, with results graphed relative to control cultures. Luminescence was measured on a Synergy HT Multi-Detection Microplate Reader (BioTek).
Quantitative Real Time PCR
Total RNA was prepared from siRNA-treated 3T3-SA cells at 48 h post-siRNA transfection using Ambion's miRVana miRNA isolation kit. SYBR Green-based quantitative real time assays for MLKL mRNA used the following primers: MLKL forward, GGATTGCCCTGAGTTGTTGC, and reverse, AACCGCAGACAGTCTCTCCA; β-actin forward, CTGTATTCCCCTCCATCGTG, and reverse, CTTCTCCATGTCGTCCCAGT. Experiments were carried out in triplicate and normalized to β-actin mRNA.
RESULTS
Macrophage Survival Following TLR Stimulation Requires Caspase Activity
TLR3 and TLR4 stimulation in the presence of the pan-caspase inhibitor Z-VAD-fmk drives RIP1-RIP3 complex-dependent necrotic death in macrophages (5), following a well established pathway downstream of TNF death receptor activation (6–8, 10, 15). We dissected the contribution of TRIF and MyD88 to necrosis in murine BMDM cultures stimulated with a panel of TLR agonists. In the presence of the pan-caspase inhibitor Z-VAD-fmk, cell death was uniformly induced by each TLR agonist tested, including Pam3CysK (TLR2), poly(I:C) (TLR3), LPS (TLR4), flagellin (TLR5), and CpG DNA (TLR9) as shown in Fig. 1A. TLR3 and TLR4 both activated cell death pathways via TRIF (5). TNF, a cytokine that is produced following TLR activation (3), is not involved in TLR3-dependent necrosis (5) but mediates apoptotic as well as necrotic cell death pathways downstream of TNFR1 (14). To determine whether TNF contributes to TLR-induced death in this setting, we stimulated TNF-deficient BMDM. Mutant cells survived stimulation with TLR2, -5, or -9 agonists, indicating that TNF autocrine or paracrine signaling was required for cell death in these contexts (Fig. 1A). Consistent with He et al. (5), two TLR agonists, poly(I:C) and LPS, triggered death independent of TNF, correlating with the use of the adapter protein TRIF. TLR3-induced death was unaffected by elimination of TNF but depended on TRIF for signal transduction (3), whereas TLR4 showed an intermediate response in agreement with the ability of TLR4 to use MyD88 as well as TRIF. The kinetics depended on the class of TLR engaged, such that TLR3 and TLR4 agonists induced cell death rapidly, within 4–6 h (Fig. 1B). In contrast, death induced by TLR2, -5, or -9 was apparent between 12 and 18 h after stimulation (Fig. 1A). From these data, it appears that TRIF-dependent TLRs may signal directly, in contrast to MyD88-dependent TLRs, where a two-stage process employs TNF as an intermediary. Thus, all of the TLRs tested have the biological potential to initiate necrotic death when caspase activity is blocked, consistent with the role of this pathway in host defense (10).
FIGURE 1.

TLR stimulation in the presence of caspase inhibitor triggers cell death. A, viability of WT and _TNF_−/− BMDM at 18 h after stimulation with Pam3CysK (1 μg/ml), poly(I:C) (25 μg/ml), LPS (500 ng/ml), flagellin (500 ng/ml), or CpG (1 μg/ml) in the presence of Z-VAD-fmk (25 μm) or vehicle (DMSO) control. B, viability of WT BMDM at 6 h after stimulation with the indicated TLR ligands in the presence of Z-VAD-fmk. C, viability of WT or TRIF mutant (Lps2/Lps2) BMDM at 18 h after stimulation with poly(I:C) or LPS in the presence or absence of Z-VAD-fmk. D, CellTiter-Glo assay was used to assess viability of BMDM after infection with either WT or M45_mut_RHIM MCMV (multiplicity of infection of 5) for 18 h followed by treatment with either LPS or poly(I:C) in the presence of Z-VAD-fmk. E, viability of IFNβ-primed L929 cells stably expressing a dominant negative TRIF-TIR domain-only construct (TRIF-TIR-M) or vector only control (EV). Cells were first primed with IFNβ (50 units/ml) for 24 h and then stimulated with poly(I:C) in the absence or presence of Z-VAD-fmk or with poly(I:C) and bafilomycin A1 (500 nm) for 18 h, as indicated. F, viability of WT, _Tnf_−/−, or TrifLps2/Lps2 MEFs at 18 h after stimulation with TLR3 agonist poly(I:C) in the presence of Z-VAD-fmk. Cell viability was assessed by determining ATP levels (CellTiter-Glo, Promega). Error bars, S.D.
In agreement with He et al. (5), we found that TRIF-deficient (TrifLps2/Lps2) BMDM failed to support necrotic death induced by LPS or poly(I:C). Also, death was sensitive to the RIP1 kinase inhibitor Nec-1 in TRIF-expressing cells (Fig. 1C). This RIP1 kinase-dependent death was only unveiled in the presence of Z-VAD-fmk, indicating that caspase activity suppressed programmed necrosis in macrophages similar to well defined death receptor pathways (6–8). Furthermore, RIP1 KO-immortalized fetal liver macrophages were resistant to necrosis induced by LPS and Z-VAD-fmk,4 consistent with the essential role of RIP1 in TRIF-dependent death in macrophages.
To determine whether RHIM-interactions contribute to TRIF-dependent cell death, we employed the virally encoded antagonist (vIRA) known to disrupt cellular RHIM-dependent signal transduction (32, 33). When BMDM were infected with WT or vIRA mutant (M45_mut_RHIM) MCMV for 12 h and then stimulated with either LPS or poly(I:C) in the presence of Z-VAD-fmk, WT, but not mutant virus, blocked TLR3- and TLR4-induced death. Thus, consistent with published observations (4, 5), RHIM-dependent signaling is required for TRIF-mediated programmed necrosis as well as the role of TRIF in inducing TNF (43). Although constitutively expressed in BMDM, fibroblasts and most other cell types do not respond efficiently to LPS because they lack TLR4, accessory proteins, and/or adapter proteins such as TIRAP, TRAM, or MyD88. In contrast, most cell types respond to poly(I:C) when primed with IFN to induce expression of TLR3. To further investigate TLR3-mediated cell death, we employed fibroblasts (including primary MEFs, L929, and 3T3-SA cells) and the endothelial cell line SVEC4-10 that have all contributed to dissecting death receptor and virus-induced necrotic death pathways (7, 9). Although IFN has many effects on cells and alters the sensitivity of macrophages to _Salmonella typhimurium_-induced necrosis (44), IFN stimulation alone did not result in a loss of fibroblast or endothelial cell viability (data not shown). IFN-primed (45) L929 fibroblasts died rapidly following TLR3 activation (Fig. 1E). Expression of a dominant negative TIR-only truncation (TRIF-TIR-M) (46) established the role of TRIF and reinforced the reliance on TLR3 rather than other sensors (Fig. 1E). Consistent with a requirement for endocytosis and endosomal acidification in TRIF-specific signaling (3, 47), the inhibitor bafilomycin A1 completely prevented cell death (Fig. 1E). Additionally, IFNβ-primed WT and TNF KO MEFs were susceptible to TLR3-induced necrosis, whereas TRIF-deficient MEFs were not (Fig. 1F). The observation that fibroblasts support TLR3-induced, TNF-independent, and TRIF-dependent cell death extends previous observations (5).
The TRIF-RIP3 complex that rapidly formed (Fig. 2A) following stimulation with poly(I:C) led us to focus on the contribution of RIP3 to TLR3 signaling. As expected (5), RIP3 was essential for TRIF-induced necrosis in BMDM (Fig. 2B). To define the TRIF-dependent RIP3 pathway in fibroblasts, we first generated L929 cells expressing shRNA to suppress RIP3 (9) and stimulated with poly(I:C) in the presence of Z-VAD-fmk. Cells treated to knockdown RIP3 were resistant to TLR3-induced necrosis, whereas cells with control scramble shRNA remained sensitive (Fig. 2, C and D). Similar to BMDM lacking RIP3 (5), RIP3 KO MEFs resisted TLR3-induced necrosis (Fig. 2E). Importantly, reconstitution of RIP3-deficient MEFs revealed that the RHIM and kinase domains of RIP3 were both essential for TLR3-induced necrosis (Fig. 2F).
FIGURE 2.

TLR3-induced programmed necrosis requires RIP3. A, following priming of 3T3-SA cells for 24 h, cells were stimulated with poly(I:C) (25 μg/ml). RIP3 was immunoprecipitated (IP) from cell lysates to detect an interaction with TRIF. IB, immunoblot. B–F, viability assays at 18 h after stimulation with poly(I:C) in the presence of Z-VAD-fmk, showing WT (Rip3+/+) or _Rip3_−/− BMDM (B); IFNβ-primed L929 cells expressing control scramble (Sc) or RIP3-specific shRNA (C); photomicrographs of IFNβ-primed L929 cells expressing control scramble or RIP3-specific shRNA (D); IFNβ-primed WT (Rip3+/+) or _Rip3_−/− MEFs (E); _Rip3_−/− MEFs reconstituted with WT RIP3, RIP3-KD, or RIP3-mRHIM (F). Cell viability in B–F was determined by ATP assay. G, immunoblot detection of total and phosphorylated IkBα in WT (Rip3+/+, left panel) or _Rip3_−/− (right panel) BMDM following stimulation with poly(I:C) for the indicated times (minutes).
The ability of RIP3 to partner with DNA sensor DAI (33, 42, 48) to potentiate NF-κB activation led us to evaluate the role of RIP3 in TRIF-dependent signaling. Upon stimulation with poly(I:C), IκBα was degraded with similar kinetics in WT and RIP3 KO BMDM (Fig. 2G). RIP3 did not influence the level of NF-κB-dependent IL-6 or IFNα expression following TLR3 activation (data not shown). Unlike DAI signaling (4, 33), cytokine induction via TRIF proceeds independently of RIP3. To address the role of IRF3 and NF-κB in necrosis, we showed that RHIM-containing mutant TRIF (TRIF-C) (29) induced similar levels of necrosis as full-length TRIF. TRIF-C induced necrosis even in the presence of the dominant negative IκBα super-repressor (IκBα-SR) (49) (data not shown). The observations that NF-κB- and IRF3-activated gene expression failed to influence TRIF-induced necrosis are in agreement with He et al. (5). Thus, although DAI and TRIF differ in their requirement for RIP3 to support IFN activation, both sensors trigger necrosis independent of any IRF3 or NF-κB contribution (11).
To evaluate the role of RIP3 kinase activity in death induction more directly, we identified potent and selective RIP3 kinase inhibitors, GSK'843 and GSK'872 (Fig. 3A), following optimization of hits identified by screening a small molecule library using a fluorescence polarization assay. When tested at a concentration of 1 μm, these compounds demonstrated >1000-fold specificity for RIP3 in comparison with the vast majority of the more than 300 different kinases tested, including RIP1 (data not shown). First, we confirmed the ability of these inhibitors to prevent necroptosis in 3T3-SA cells (9) by showing both compounds suppressed TNF-induced death in a dose-dependent fashion (Fig. 3B). Second, the RIP3 inhibitors prevented virus-induced necrosis, a pathway dependent on DAI-RIP3 complex formation (Fig. 3C) (11). Finally, and most relevant here, both RIP3 kinase inhibitors blocked TLR3-induced necrosis induced in fibroblasts by poly(I:C) in the presence of Z-VAD-fmk. Both virus- and TLR3-induced necrosis proceed independently of RIP1 kinase inhibition by Nec-1 but sensitive to inhibition by GSK'843 or GSK'872 (Fig. 3D). These data establish that TLR3-induced necrosis in fibroblasts requires TRIF and RIP3 kinase, although TLR3- or TLR4-induced necrosis in BMDM (see Fig. 1C) requires these plus RIP1 kinases (5).
FIGURE 3.

Role of RIP3 kinase in TLR3-induced programmed necrosis. A, chemical structure of compounds GSK'843 and GSK'872. B, viability of 3T3-SA cells at 18 h after treatment with TNF in the presence of Z-VAD-fmk in vehicle control (DMSO) or treated with the indicated concentrations of RIP3 kinase inhibitors, GSK'843 or GSK'872. C, viability of SVEC4-10 cells at 18 h post-infection with WT or M45_mut_RHIM MCMV in vehicle control (DMSO) or treated with the indicated concentrations of RIP3 kinase inhibitors. D, viability of IFNβ-primed 3T3-SA cells at 18 h after stimulation with poly(I:C) in the absence or presence of Z-VAD-fmk and treatment with Nec-1 (30 μm) or the indicated concentrations of RIP3 kinase inhibitors. E, immunoblot detecting RIP3 and β-actin present in the soluble (supernatant) and insoluble (pellet) fraction following stimulation of 3T3-SA cells with poly(I:C) for the indicated times (hours) in the absence or presence of the caspase inhibitor Z-VAD-fmk and GSK'872 (3 μm) or Nec-1 (30 μm). Cell viability was determined by the ATP assay. Inquiries about RIP3 kinase inhibitors GSK'843 and GSK'872 should be directed to P. Gough (peter.j.gough@gsk.com).
We performed an immunoblot analysis to evaluate RIP3 behavior during necrosis in fibroblasts. Following poly(I:C) stimulation in the presence of Z-VAD-fmk, RIP3 was rapidly eliminated from the soluble fraction and accumulated in the detergent-insoluble fraction (Fig. 3E). The partitioning of RIP3 into the insoluble fraction did not depend on the induction of necrosis or the kinase activities of either RIP3 or RIP1 kinase (Fig. 3E and data not shown). Caspase suppression, rather than death, correlated with partitioning of RIP3 into the pellet. In addition to the changes in solubility, low mobility forms of RIP3 accumulated in the pellet when Z-VAD-fmk was included (Fig. 3E), consistent with post-translational modifications during necrosis (4, 5, 29, 50). Treatment with GSK'872 prevented the accumulation of these altered forms at the stacking gel interface, implicating RIP3 kinase activity in their formation.
The differential impact of RIP3 and RIP1 kinase inhibitors on TLR3-induced death in fibroblasts led us to evaluate TLR3 signaling in J774 macrophages, 3T3-SA fibroblasts, and SVEC4-10 endothelial cells, the latter two cell lines have been key to dissecting virus-induced necrosis (11). When RIP1 was suppressed using siRNA, 3T3-SA cells became more sensitive to poly(I:C)-induced death relative to scramble control siRNA-treated cells. Furthermore, reduction in RIP1 levels did not diminish necrosis induced by poly(I:C) and Z-VAD-fmk or alter the kinetics of death as most cells treated succumbed to necrosis within 4 h following stimulation. Similar to 3T3-SA fibroblasts, SVEC4-10 cells also remained sensitive to necrosis induced by poly(I:C) when RIP1 levels were suppressed by siRNA (Fig. 4B). Death in SVEC4-10 cells was insensitive to reduced RIP1 levels as well as to RIP1 kinase inhibitor Nec-1. When IFN-primed WT and RIP1-deficient primary fibroblasts were stimulated with poly(I:C) and Z-VAD-fmk, similar levels of cell death were observed (Fig. 4C), although death in RIP1-deficient cells occurred in the absence of Z-VAD-fmk. Thus, fibroblasts and endothelial cells support TLR3-induced necrosis independent of RIP1 levels (Fig. 4C). Because RIP1 kinase inhibition prevented TLR-induced necrosis in BMDM, we next investigated whether the J774 macrophage cell line was sensitive to TLR3-induced necrosis (5). RIP1 shRNA did not prevent TLR3-induced necrosis in J774 cells; however, Nec-1 conferred modest protection to either LPS- or poly(I:C)-induced necrosis, despite diminished expression of RIP1 (Fig. 4D). These data suggest that macrophages rely on RIP1, whereas fibroblasts and endothelial cells are independent of RIP1. As expected, RIP3 inhibitor GSK'872 or RIP3 shRNA protected J774 cells from TRIF-dependent necrosis, reinforcing the central role of this protein kinase independent of the cell type. In addition, macrophages or fibroblasts from DAI-deficient mice supported necrosis (data not shown), demonstrating that the TRIF-dependent pathway does not require the participation of this RHIM-signaling DNA sensor. Thus, TLR3-induced necrosis requires TRIF and RIP3 but proceeds independently of the RIP1 or DAI when evaluated in fibroblasts or endothelial cells. In this setting, a novel RHIM-dependent association between TRIF and RIP3 appears to control death, reminiscent of the RHIM-dependent recruitment of RIP3 by either RIP1 in necroptosis (6–8) or DNA sensor DAI in virus-induced necrosis (9–11). RHIM-dependent interactions involving RIP3 kinase are therefore key in all three of these settings. Despite the central role of RIP1 in necroptosis, and its apparent contribution to TLR3- and TLR4-induced necrosis in macrophages, RIP1 kinase does not contribute to TRIF-dependent programmed necrosis in other cell types.
FIGURE 4.

Differential role of RIP1 in TLR-induced necrosis in macrophages versus other cell types. A, viability of IFNβ-primed 3T3-SA cells transfected with either RIP1 or MLKL siRNA smartpools. Cells were stimulated with poly(I:C) in the absence or presence of Z-VAD for 4 h. B, viability of SVEC4-10 cells expressing control scramble and RIP1-specific or RIP3-specific shRNA in the absence or presence of Z-VAD-fmk and Nec-1 (30 μm) for 18 h. C, WT (Rip1+/+) or _Rip1_−/− MEFs at 18 h after stimulation with poly(I:C) in the absence or presence of Z-VAD-fmk and IFNβ. D, J774 macrophages after 18 h of stimulation with LPS or poly(I:C) in the absence or presence of Z-VAD-fmk, Nec-1, and GSK'872. Cell viability was determined by the ATP assay.
TNF-induced necroptosis proceeds via a RIP3-dependent phosphorylation of MLKL (16, 17). To determine the contribution of MLKL across all three RIP3-dependent necrotic death pathways, we blocked MLKL expression levels in necrosis-sensitive 3T3-SA cells using siRNA methods (Fig. 5, A and B) prior to triggering necrotic death with different stimuli. Virus-induced (Fig. 5C), TNFR1-induced (Fig. 5D), and TLR3-induced necrosis (Fig. 5D) were uniformly sensitive to MLKL-specific siRNA knockdown but not to nontargeting siRNA treatment. These data strongly suggest that the previously identified RIP3-dependent phosphorylation of MLKL (17) following death receptor-dependent activation is likely to be involved in DAI-RIP3 and TRIF-RIP3 signal transduction. Thus, RIP3 kinase and MLKL emerge as common steps in programmed necrosis triggered by PRRs and death receptors.
FIGURE 5.

Role of MLKL in TLR3- and DAI-induced necrosis. 3T3-SA cells were transfected with either MLKL or scramble (Scr) siRNA pools. A, at 48 h post-transfection, quantitative real time PCR detected the fold change in MLKL mRNA relative to β-actin. B, immunoblot analysis of MLKL and β-actin in siRNA-transfected 3T3-SA cell. C, viability of 3T3-SA cells at 18 h post-infection with WT or M45_mut_RHIM MCMV. Cells were infected in the presence of vehicle control (DMSO) or 30 μm Nec-1. D, viability of siRNA-transfected 3T3-SA cells at 18 h after stimulation with TNF or poly(I:C) in the absence or presence of Z-VAD-fmk or cycloheximide (CHX). Cells were primed with IFNβ for 24 prior to stimulation where indicated. Cell viability was determined by the ATP assay.
Whereas L929 cells and SVEC4-10 cells are sensitive to poly(I:C)-induced necrosis even in the absence of caspase inhibition, in fibroblasts necrosis signaling induced by TLR3 only predominates when caspase activity is compromised, paralleling the requirements for TNF-induced necrosis. To directly address the role of Casp8 in suppressing necrosis, we compared WT, Casp8-deficient, and Casp8/RIP3 double-deficient MEFs for the need to inhibit caspases following IFN and poly(I:C) stimulation. Casp8-deficient, Casp8/RIP1 double KO (Fig. 6A), and RIP1 KO MEFs (Fig. 4C) were sensitive to death in the absence of Z-VAD-fmk treatment, consistent with a role for Casp8 and RIP1 in suppressing RIP3-dependent death following IFN and poly(I:C) stimulation. In contrast, _Rip3_−/− (Fig. 2E) and _Casp8−/−Rip3_−/− (data not shown) fibroblasts were resistant to poly(I:C)-induced necrosis, consistent with a need for Casp8 to prevent and RIP3 to drive necrotic death. In contrast to MEFs, necrosis-sensitive 3T3-SA cells were susceptible to knockdown of Casp8 expression by siRNA in the absence of poly(I:C) treatment (Fig. 6B) such that cells died following transfection as Casp8 levels declined. Prolonged incubation of cells in the presence of the caspase inhibitor Z-VAD-fmk also led to a marked decline in cell viability (data not shown). In this regard, 3T3-SA cells appeared to behave like necrosis-sensitive L929 cells (51) or, more importantly, embryonic vascular cells in mice (21, 22) for their requirement to sustain Casp8 levels and prevent lethal RIP3 death pathways from opening (Fig. 6).
FIGURE 6.
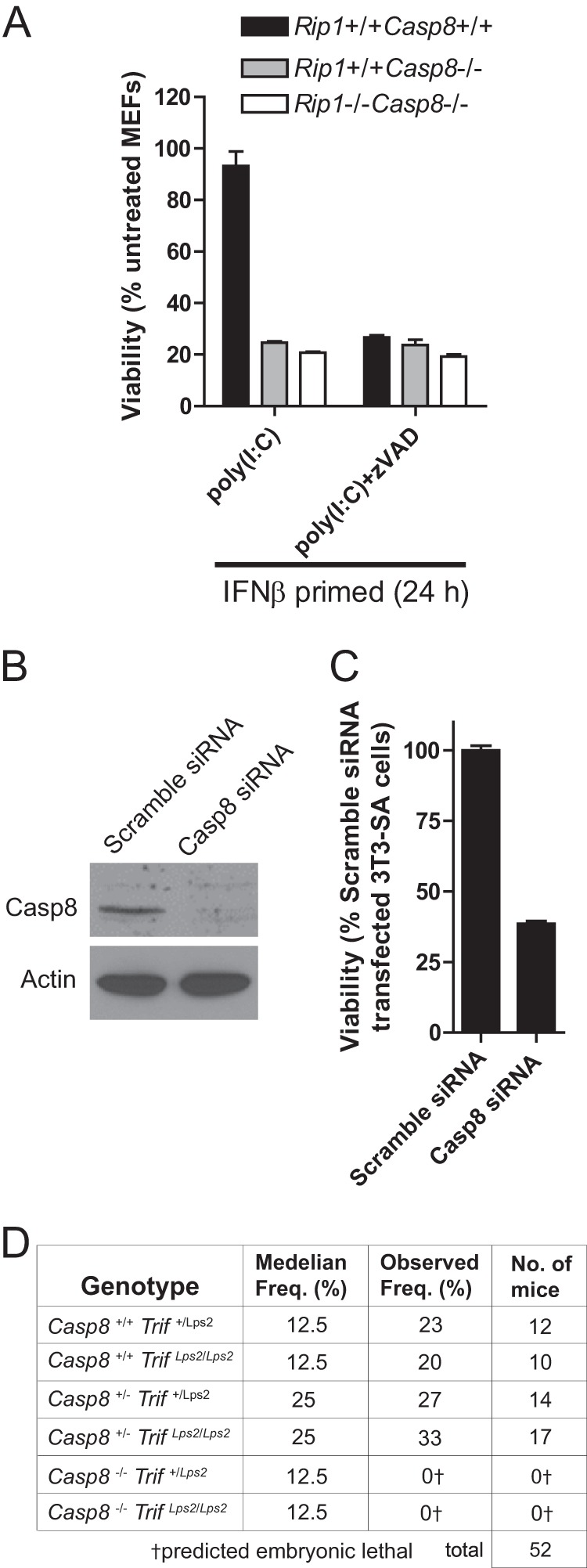
Casp8 suppression of TLR3-mediated TRIF- and RIP3-dependent programmed necrosis. A, viability of WT, Casp8_−/−, or Casp8_−/−_Rip1_−/− MEFs at 18 h after stimulation with poly(I:C) in the absence or presence of Z-VAD-fmk. B, 3T3-SA cells were transfected with either the Casp8 or Scramble siRNA pools. At 72 h post-transfection Casp8 and β-actin levels were determined by immunoblot analysis. C, cell viability was determined. A and C, cell viability was determined by ATP levels. Error bars, S.D. D, epistatic analysis of mice born following a Casp8+/−_Trif+/Lps2 × Casp8+/−_TrifLps2/Lps2 intercross with predicted and observed frequencies.
Given that the signals driving demise during midgestation (E10.5 to E11.5) have not been identified, we crossed Casp8 KO and TrifLps2/Lps2 mice to assess any contribution of TRIF. Casp8_−/−_TrifLps2/Lps2 double knock-out mice failed to develop beyond E11 (Fig. 6C) analogous to Casp8 KO embryo controls (data not shown). Thus, neither DAI (11) nor TRIF (this study) contributed to the developmental dysregulation caused by Casp8 deficiency. These data reinforce the observations demonstrating that RIP1 (52) or RIP3 (21, 22) deficiency rescues the E11 block in Casp8- or FADD-deficient mice and sustains the current model of RIP1-RIP3 complex-dependent necroptosis as the mediator of midgestational death.
DISCUSSION
Host strategies that detect and eliminate pathogens play out in evolutionarily ancient and important ways that include production of secreted proteins to control infection and initiation of regulated cell death to eliminate infected cells. Although the induction of cytokines, chemokines, and interferon following TLR stimulation molds many aspects of host defense (3), regulated cell death that directly eliminates infected cells and prevents infection of a host emerges as crucial (10). Through these diverse impacts, a range of overlapping host-encoded effector mechanisms are called up no matter the nature of the infectious insult. Plants contend with pathogens by detecting altered biochemical signatures through resistance “guard” proteins that sense perturbation of key cellular processes and trigger anti-microbial defenses that include cell death as a prominent end point (53). We have recently speculated that Casp8 may be a component of a comparable mammalian innate immune strategy (54). Casp8 sits in a powerful position; its basal activity suppresses RIP3 kinase activity that, if unleashed, eliminates the cell via programmed necrosis while also holding the reins on extrinsic apoptosis. Although this decision has been long associated with death receptor activation, sensors such as DAI (9–11), as shown here and in a previous report (5), TLRs accomplish a similar set of outcomes. Why such complexity? The combination of cytokine activation and cell death must provide an effective strategy to deal with infection. The evolutionary reason for the host to choose between cytokine activation, extrinsic apoptosis, and programmed necrosis may stem from selection pressure of pathogens (10). RIP3 necrosis likely evolved as an adaptation to pathogens that block Casp8 activity (10) hundreds of millions of years ago. This tug-of-war continues and is evident in the modulatory behavior of viruses in the poxvirus (8) and herpesvirus (9–11) families. Indeed, mammalian DNA viruses commonly encode genes products that suppress Casp8 activity to prevent apoptosis (10). RIP3 kinase-dependent programmed necrosis was first described during the investigation of TNF signaling where the anti-apoptotic cowpox Casp8 inhibitor CrmA caused death instead of blocking death (55). Later, it was shown that host control of vaccinia, which encodes B13R as well as F1L and K7L protease inhibitors, is mediated by RIP3-dependent necrosis (8). In natural settings, this ability to drive death when Casp8 activity is compromised helps to ensure pathogen-infected cells are eliminated. Highly evolved pathogens such as MCMV have acquired the ability to defuse both regulated cell death pathways (9–11), deflecting potent natural control of virus infection. This complexity enables a broad range of pathogen-sensing and death receptors to respond in an appropriate way to the varied microbes and viruses encountered throughout life. The capacity to respond to infection-associated signals, which varies with cell type but converges on common cell activation and death pathways in all cell types, defines the first line host defense.
Two general patterns emerged from our studies as follows: MyD88-dependent TLRs initiate the production of TNF as a result of NF-κB activation, with TNF then mediating conventional RIP1-RIP3 kinase-dependent necroptosis. This indirect mechanism may contribute to the apparent RIP1 role downstream of TLR3 activation in BMDMs (5) as well as to necroptosis induced by T cell receptor activation when Casp8 is compromised (10). TRIF-dependent signaling via TLR3 and TLR4 initiate a TRIF-RIP3 complex that directly triggers RIP3 kinase-dependent necrosis. The TRIF-RIP3 pathway is distinct from the MyD88-death receptor axis in that it proceeds independently of NF-κB and TNF, does not require RIP1, and follows a more rapid time course. Thus, both TLR3 and TLR4 employ the adapter protein TRIF to trigger NF-κB activation separate from the control of cell death pathways (4, 5, 29). This capacity parallels death receptor signaling as follows: 1) RIP1 controls NF-κB activation in a RIP3-independent manner; 2) basal Casp8 activity suppresses programmed necrosis; 3) autoactivation of Casp8 drives apoptosis; and 4) compromised Casp8 activity unleashes RIP3 kinase-dependent programmed necrosis. Casp8 control of death receptor and TLR necrotic death signaling depends on basal catalytic activity that suppresses the RIP3 kinase pathway.
One dramatic manifestation of this control emerged from dissecting the contribution that RIP3 makes in midgestation death of Casp8-deficient mice (21). Although the physiological changes during midgestational development that trigger RIP3 death remain unknown, the key role of RIP1 (52) and RIP3 (21, 22) are clear. Neither of the other known RHIM-containing RIP3 partners, DAI (11) or TRIF (this work), rescue the mid-gestational impact of Casp8 deficiency. The range of distinct settings where RIP3-dependent cell death becomes unleashed (10) provides evidence that homeostatic regulation via basal Casp8 activity is important in many tissues throughout life where these three RIP3 partners evolved to carry out complementary roles. Rip3−/− mice appear normal, but exhibit increased susceptibility to vaccinia (8), as well as M45-mutant MCMV (9). Elimination of RIP3 from Casp8-deficient mice rescues development, yields fertile adults that rely on other immune mechanisms to control MCMV infection (21). Clearly, the interdependency and dysregulation of Casp8-dependent control of RIP3-necrosis as well as the significant contributions viral inhibitors of these pathways continue to yield insights into how each RIP3 partner contributes to host defense.
Casp8 catalytic activity most likely regulates the formation of a signaling complex that has been varyingly called complex IIB or ripoptosome, depending on the stimulus involved. When Casp8 activity is compromised, both RIP1 and RIP3 rapidly associate with a detergent-insoluble cell fraction that is also accompanied by dramatic RHIM-dependent oligomerization (50). This process occurs concomitant with programmed necrosis. Although Casp8 can recognize and cleave both RIP1 and RIP3 as substrates (23, 24, 26), evidence of cleavage was not detected following TLR3 activation. Casp8 also targets potential regulatory proteins for cleavage, such as the deubiquitinylase CYLD (56), whose activity is necessary for RIP1-RIP3 necrotic signaling. Feoktistova et al. (19) implicated a Casp8-cFLIPL complex in preventing apoptosis following TLR3 activation. Our evidence reveals this TLR3-mediated apoptosis to be mediated by TRIF. This places TRIF at an important decision point very similar to the role of RIP1 downstream of death receptor signaling, thus metering Casp8-cFLIPL basal activity that can mediate extrinsic apoptosis or unleash necrosis (22). Additional studies will surely provide further insights into this regulation.
In TNF signaling, RIP3 is recruited via the RHIM in RIP1 to form an oligomeric complex that mediates necroptosis (57). In TLR3 signaling, TRIF is the crucial RIP3 partner and RIP1 is dispensable. Like DAI-RIP3-dependent virus-induced necrosis (11), but distinct from in necroptosis, there is no recognized upstream protein kinase such as RIP1 acting on TRIF-RIP3 complexes to initiate programmed necrosis. This situation is reminiscent of work from Meylan et al. (29), where RIP1 and RIP3 were shown to differentially compete for RHIM-dependent binding with TRIF. It is possible that high affinity TRIF-mediated RHIM-dependent interaction with RIP3 overcomes the requirement for RIP1 kinase, potentially in an oligomerization-dependent manner. This also parallels understanding of DAI recruitment of RIP3 to induce virus-induced necrosis as a trap door in host defense to eliminate virus-infected cells when Casp8 is naturally inhibited by MCMV vICA (11). Given the importance of virus-encoded caspase inhibitors in the execution of the DAI-RIP3 pathway, similar inhibitors, from vaccinia and other intracellular pathogens, may be predicted to predispose to TRIF-RIP3 or RIP1-RIP3 necrosis during natural infection. We predict that a common kinase target is involved no matter which of the three RIP3 complexes initiates oligomerization, with signaling convergent on MLKL and, possibly, PGAM5 in a serine/threonine protein kinase-dependent cascade (16, 17).
Signaling from TLR3 and TLR9 collaborate in restricting systemic MCMV infection in vivo (58). Here, we demonstrate that activation of either receptor leads directly or indirectly to Casp8 regulation of apoptotic or necrotic death decisions. This virus, like all herpesviruses, is invested in orchestrating cell fate decisions through an arsenal of cell death suppressors (10), some of which are evolutionarily conserved in mice and human relatives (59). The conserved cell death suppressor vICA binds to the prodomain of Casp8 to prevent homodimerization and autocleavage preceding apoptosis (60). Suppression of Casp8 by vICA predisposes the infected cell to TNF-driven necroptosis (21) as well as TLR-induced necrosis, as shown here. Cytomegalovirus pathogenesis in mice depends heavily upon vIRA suppression of RIP3 activity because without this suppressor the virus is completely unable to infect the host (9). Although the DAI-RIP3 pathway of programmed necrosis emerged as the predominant natural target of vIRA (9–11), this RHIM inhibitor also blocks TRIF-RIP3 and RIP1-RIP3 cell death and cytokine signaling (32, 33). In addition, vIRA blocks NF-κB essential modulator function, which appears to proceed independently of RHIM interactions (35, 36). MCMV illustrates the potentially precarious but seemingly successful balance with the entwined necrotic and apoptotic host defense pathways, whereas another large DNA virus, vaccinia, remains naturally vulnerable to RIP3-dependent death (8). Clearly, viruses have enjoyed variable success in deflecting sensor and death receptor signaling pathways that evolved to eliminate infected cells. With Casp8-dependent apoptosis frequently undermined (10), the RIP3 trap door appears to be an effective adaptation in the pathogen-host arms race.
Acknowledgment
We appreciate the technical assistance Linda Roback.
*
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 AI030363 and AI020211, Pilot Grant UL1 RR025008 from ACTSI (to E. S. M.), and OD012198 (to W. J. K.). This work was also supported by funds from the University of Texas at Austin, the Cancer Prevention Research Institute of Texas (to J. W. U.), and by GlaxoSmithKline (to P. J. G., C. A. S., R. W. M., and J. B.).
3
P. J. Gough, C. Sehon, R. Marquis, and J. Bertin, manuscript in preparation.
4
E. Lien, University of Massachusetts, personal communication.
2
The abbreviations used are:
PRR
pattern recognition receptor
TLR
Toll-like receptor
FADD
Fas-associated via death domain
RIP
receptor interacting protein
RHIM
RIP homotypic interaction motif
TIR
Toll/IL-1R
BMDM
bone marrow-derived macrophage
Z
benzyloxycarbonyl
fmk
fluoromethyl ketone
vICA
viral inhibitor of Casp8 activation
vIRA
viral inhibitor of RIP activation
MCMV
murine cytomegalovirus
cFLIP
cellular FLICE/Casp8 inhibitory protein
MEF
mouse embryo fibroblast
TRIF
TIR domain-containing adapter-inducing interferon-β
MLKL
mixed lineage kinase domain-like protein.
REFERENCES
- 1.Lamkanfi M., Dixit V. M. (2010) Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8, 44–54 [DOI] [PubMed] [Google Scholar]
- 2.Everett H., McFadden G. (1999) Apoptosis: an innate immune response to virus infection. Trends Microbiol. 7, 160–165 [DOI] [PubMed] [Google Scholar]
- 3.Kumar H., Kawai T., Akira S. (2011) Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34 [DOI] [PubMed] [Google Scholar]
- 4.Kaiser W. J., Offermann M. K. (2005) Apoptosis induced by the toll-like receptor adapter TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952 [DOI] [PubMed] [Google Scholar]
- 5.He S., Liang Y., Shao F., Wang X. (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137, 1100–1111 [DOI] [PubMed] [Google Scholar]
- 7.Zhang D. W., Shao J., Lin J., Zhang N., Lu B. J., Lin S. C., Dong M. Q., Han J. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 [DOI] [PubMed] [Google Scholar]
- 8.Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., Chan F. K. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upton J. W., Kaiser W. J., Mocarski E. S. (2010) Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mocarski E. S., Upton J. W., Kaiser W. J. (2012) Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 12, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Upton J. W., Kaiser W. J., Mocarski E. S. (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hengartner M. O. (2000) The biochemistry of apoptosis. Nature 407, 770–776 [DOI] [PubMed] [Google Scholar]
- 13.Strasser A., O'Connor L., Dixit V. M. (2000) Apoptosis signaling. Annu. Rev. Biochem. 69, 217–245 [DOI] [PubMed] [Google Scholar]
- 14.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 [DOI] [PubMed] [Google Scholar]
- 15.Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J. L., Schneider P., Seed B., Tschopp J. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 [DOI] [PubMed] [Google Scholar]
- 16.Wang Z., Jiang H., Chen S., Du F., Wang X. (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 [DOI] [PubMed] [Google Scholar]
- 17.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 [DOI] [PubMed] [Google Scholar]
- 18.Zhao J., Jitkaew S., Cai Z., Choksi S., Li Q., Luo J., Liu Z. G. (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. U.S.A. 109, 5322–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feoktistova M., Geserick P., Kellert B., Dimitrova D. P., Langlais C., Hupe M., Cain K., MacFarlane M., Häcker G., Leverkus M. (2011) cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estornes Y., Toscano F., Virard F., Jacquemin G., Pierrot A., Vanbervliet B., Bonnin M., Lalaoui N., Mercier-Gouy P., Pachéco Y., Salaun B., Renno T., Micheau O., Lebecque S. (2012) dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 19, 1482–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaiser W. J., Upton J. W., Long A. B., Livingston-Rosanoff D., Daley-Bauer L. P., Hakem R., Caspary T., Mocarski E. S. (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R. (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng S., Yang Y., Mei Y., Ma L., Zhu D. E., Hoti N., Castanares M., Wu M. (2007) Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 19, 2056–2067 [DOI] [PubMed] [Google Scholar]
- 24.Lin Y., Devin A., Rodriguez Y., Liu Z. G. (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu J. V., Weist B. M., van Raam B. J., Marro B. S., Nguyen L. V., Srinivas P., Bell B. D., Luhrs K. A., Lane T. E., Salvesen G. S., Walsh C. M. (2011) Complementary roles of FADD and RIPK3 in T cell homeostasis and antiviral immunity. Proc. Natl. Acad. Sci. U.S.A. 108, 15312–15317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan F. K., Shisler J., Bixby J. G., Felices M., Zheng L., Appel M., Orenstein J., Moss B., Lenardo M. J. (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278, 51613–51621 [DOI] [PubMed] [Google Scholar]
- 27.Thome M., Tschopp J. (2001) Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1, 50–58 [DOI] [PubMed] [Google Scholar]
- 28.Dillon C. P., Oberst A., Weinlich R., Janke L. J., Kang T. B., Ben-Moshe T., Mak T. W., Wallach D., Green D. R. (2012) Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 1, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meylan E., Burns K., Hofmann K., Blancheteau V., Martinon F., Kelliher M., Tschopp J. (2004) RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat. Immunol. 5, 503–507 [DOI] [PubMed] [Google Scholar]
- 30.Vivarelli M. S., McDonald D., Miller M., Cusson N., Kelliher M., Geha R. S. (2004) RIP links TLR4 to Akt and is essential for cell survival in response to LPS stimulation. J. Exp. Med. 200, 399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalai M., Van Loo G., Vanden Berghe T., Meeus A., Burm W., Saelens X., Vandenabeele P. (2002) Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ. 9, 981–994 [DOI] [PubMed] [Google Scholar]
- 32.Upton J. W., Kaiser W. J., Mocarski E. S. (2008) Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 283, 16966–16970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rebsamen M., Heinz L. X., Meylan E., Michallet M. C., Schroder K., Hofmann K., Vazquez J., Benedict C. A., Tschopp J. (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 10, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brune W., Ménard C., Heesemann J., Koszinowski U. H. (2001) A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291, 303–305 [DOI] [PubMed] [Google Scholar]
- 35.Mack C., Sickmann A., Lembo D., Brune W. (2008) Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. U.S.A. 105, 3094–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fliss P. M., Jowers T. P., Brinkmann M. M., Holstermann B., Mack C., Dickinson P., Hohenberg H., Ghazal P., Brune W. (2012) Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 8, e1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S. O., Goode J., Lin P., Mann N., Mudd S., Crozat K., Sovath S., Han J., Beutler B. (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748 [DOI] [PubMed] [Google Scholar]
- 38.Newton K., Sun X., Dixit V. M. (2004) Kinase RIP3 is dispensable for normal NF-κBs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 24, 1464–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelliher M. A., Grimm S., Ishida Y., Kuo F., Stanger B. Z., Leder P. (1998) The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 [DOI] [PubMed] [Google Scholar]
- 40.Kuprash D. V., Alimzhanov M. B., Tumanov A. V., Grivennikov S. I., Shakhov A. N., Drutskaya L. N., Marino M. W., Turetskaya R. L., Anderson A. O., Rajewsky K., Pfeffer K., Nedospasov S. A. (2002) Redundancy in tumor necrosis factor (TNF) and lymphotoxin (LT) signaling in vivo: mice with inactivation of the entire TNF/LT locus versus single-knockout mice. Mol. Cell. Biol. 22, 8626–8634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salmena L., Lemmers B., Hakem A., Matysiak-Zablocki E., Murakami K., Au P. Y., Berry D. M., Tamblyn L., Shehabeldin A., Migon E., Wakeham A., Bouchard D., Yeh W. C., McGlade J. C., Ohashi P. S., Hakem R. (2003) Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17, 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaiser W. J., Upton J. W., Mocarski E. S. (2008) Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 181, 6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Role of adapter TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 [DOI] [PubMed] [Google Scholar]
- 44.Robinson N., McComb S., Mulligan R., Dudani R., Krishnan L., Sad S. (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hitomi J., Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., Yuan J. (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamamoto M., Sato S., Mori K., Hoshino K., Takeuchi O., Takeda K., Akira S. (2002) Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J. Immunol. 169, 6668–6672 [DOI] [PubMed] [Google Scholar]
- 47.Kawai T., Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 48.Rebsamen M., Meylan E., Curran J., Tschopp J. (2008) The antiviral adapter proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 15, 1804–1811 [DOI] [PubMed] [Google Scholar]
- 49.Van Antwerp D. J., Martin S. J., Kafri T., Green D. R., Verma I. M. (1996) Suppression of TNF-α-induced apoptosis by NF-κB. Science 274, 787–789 [DOI] [PubMed] [Google Scholar]
- 50.Li J., McQuade T., Siemer A. B., Napetschnig J., Moriwaki K., Hsiao Y. S., Damko E., Moquin D., Walz T., McDermott A., Chan F. K., Wu H. (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu L., Alva A., Su H., Dutt P., Freundt E., Welsh S., Baehrecke E. H., Lenardo M. J. (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502 [DOI] [PubMed] [Google Scholar]
- 52.Zhang H., Zhou X., McQuade T., Li J., Chan F. K., Zhang J. (2011) Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medzhitov R. (2010) Innate immunity: quo vadis? Nat. Immunol. 11, 551–553 [DOI] [PubMed] [Google Scholar]
- 54.Kaiser W. J., Upton J. W., Mocarski E. S. (2013) Viral modulation of programmed necrosis. Curr. Opin. Virol. 3, 296–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vercammen D., Beyaert R., Denecker G., Goossens V., Van Loo G., Declercq W., Grooten J., Fiers W., Vandenabeele P. (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187, 1477–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Donnell M. A., Perez-Jimenez E., Oberst A., Ng A., Massoumi R., Xavier R., Green D. R., Ting A. T. (2011) Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vandenabeele P., Declercq W., Van Herreweghe F., Vanden Berghe T. (2010) The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 3, re4. [DOI] [PubMed] [Google Scholar]
- 58.Tabeta K., Georgel P., Janssen E., Du X., Hoebe K., Crozat K., Mudd S., Shamel L., Sovath S., Goode J., Alexopoulou L., Flavell R. A., Beutler B. (2004) Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 3516–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brune W. (2011) Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 157, 144–150 [DOI] [PubMed] [Google Scholar]
- 60.Skaletskaya A., Bartle L. M., Chittenden T., McCormick A. L., Mocarski E. S., Goldmacher V. S. (2001) A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. U.S.A. 98, 7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]