Combinational Targeting Offsets Antigen Escape and Enhances Effector Functions of Adoptively Transferred T Cells in Glioblastoma (original) (raw)
Abstract
Preclinical and early clinical studies have demonstrated that chimeric antigen receptor (CAR)-redirected T cells are highly promising in cancer therapy. We observed that targeting HER2 in a glioblastoma (GBM) cell line results in the emergence of HER2-null tumor cells that maintain the expression of nontargeted tumor-associated antigens. Combinational targeting of these tumor-associated antigens could therefore offset this escape mechanism. We studied the single-cell coexpression patterns of HER2, IL-13Rα2, and EphA2 in primary GBM samples using multicolor flow cytometry and immunofluorescence, and applied a binomial routine to the permutations of antigen expression and the related odds of complete tumor elimination. This mathematical model demonstrated that cotargeting HER2 and IL-13Rα2 could maximally expand the therapeutic reach of the T cell product in all primary tumors studied. Targeting a third antigen did not predict an added advantage in the tumor cohort studied. We therefore generated bispecific T cell products from healthy donors and from GBM patients by pooling T cells individually expressing HER2 and IL-13Rα2-specific CARs and by making individual T cells to coexpress both molecules. Both HER2/IL-13Rα2-bispecific T cell products offset antigen escape, producing enhanced effector activity in vitro immunoassays (against autologous glioma cells in the case of GBM patient products) and in an orthotopic xenogeneic murine model. Further, T cells coexpressing HER2 and IL-13Rα2-CARs exhibited accentuated yet antigen-dependent downstream signaling and a particularly enhanced antitumor activity.
Introduction
Glioblastoma (GBM) is the most common of all primary brain tumors in adults and is virtually incurable. With the combination of radical surgery, radiotherapy, and adjuvant temozolomide, the 5-year overall survival rate is <5% and treatment-related complications are debilitating.1,2 Immunotherapy is emerging as an alternative approach that could potentially overcome these limitations of the current standard therapy. Adoptive cell therapies with chimeric antigen receptor (CAR) expressing T cells have recently had substantial successes in the treatment of chronic lymphocytic leukemia, acute lymphoblastic leukemia, and neuroblastoma in first-in-man clinical trials.3,4,5,6 In preclinical models of GBM, CAR T cells have shown robust antitumor activity and are currently being investigated in phase I/II studies that target the glioma-restricted antigens IL-13Rα2, HER2, and EGFR.7,8,9
Tumors exhibit variable degrees of antigenic heterogeneity such that no single antigen could serve as a universal target that is inclusive of the whole tumor bulk. Further, tumor cells escape immune recognition by employing a number of antigen-evasion strategies including antigen mutation, downregulation/deletion of target antigens, and selective survival of antigen-negative tumor subpopulations that could well be selected by therapy.10,11,12 These concerns are particularly relevant to GBM, which is known to be heterogeneous with varying antigen expression profile within single tumors and between patients.13,14 Targeting multiple tumor-restricted antigens could therefore offset these potential escape mechanisms.
We have now studied the single-cell expression pattern of three validated glioma antigens, HER2, IL-13Rα2, and EphA2 in primary GBM samples. We constructed a mathematical model to capture the antigen expression landscape and predict the optimum cellular product with the greatest therapeutic reach in all patients studied. On the basis of the prevalence of the three antigens characterized, we generated bispecific T-cell products by modifying individual T cells to coexpress distinct CAR molecules specific for HER2 and IL-13Rα2 or by pooling unispecific CAR T cells. Further, we tested whether bispecific T cells had enhanced functionality against GBM cells and whether their ability to offset antigen escape would increase tumor control in an in vivo model of human GBM compared with unispecific CAR T cells.
Results
Selective survival and expansion of escape variants after single antigen targeting
We exposed HER2 and IL-13Rα2 expressing U373 cells (GBM cell line) in vitro to HER2-specific CAR T cells and analyzed the change in expression of these target antigens on viable tumor cells over time. At baseline, most U373 cells expressed one or both antigens on flow cytometry: 18% expressed HER2 only; 16% IL-13Rα2 only, 52% expressed both, and 14% were negative for both. Exposure to HER2-specific T cells selected a tumor cell population with dim to undetectable HER2 expression and increased IL-13Rα2 expression (Figure 1). This tumor cell population expanded despite continued exposure to HER2-specific T cells, to reach confluence in tissue culture. U373 cells exposed to nontransduced (NT) T cells retained a similar mixed phenotype to the initial tumor cell line. This prompted us to study the pattern of antigen expression in primary GBM and to quantify the odds of tumor elimination after single specific versus bispecific versus trispecific targeting.
Figure 1.
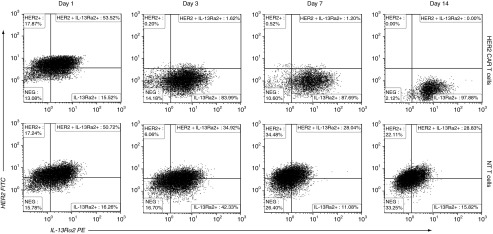
Targeting a single antigen results in selective survival and proliferation of escape tumor cell variants. U373 cells expressing HER2 and IL-13Rα2 were treated, in vitro, with HER2 chimeric antigen receptor (CAR) T cells in a 1:5 T cell to tumor cell ratio (20,000 T cells: 100,000 tumor cells). Serial analysis of viable tumor cells by flow cytometry from day 1 to 14 showed survival and expansion of a distinct HER2 negative population that was resistant to HER2 CAR T cells. These surviving tumor cells maintained positivity for IL-13Rα2. Gating strategy is shown in Supplementary Figure S1. One experiment of three is shown.
The pattern of antigen expression in GBM rationalizes targeting multiple antigens to enhance tumor control
While GBMs are known to be heterogeneous tumors with substantial histopathological and interpatient variability,15 single-cell antigen coexpression within individual tumors has not been well characterized. We therefore studied the simultaneous expression pattern of three established glioma-restricted antigens (HER2, IL-13Rα2, and EphA2) in primary GBM samples.16,17,18,19,20
To characterize the en masse single cell expression patterns in homogenized tumor cell populations, >100,000 primary GBM cells (Unique Patient Number [UPN]-1, UPN-2, and UPN-3) and U373 cells were simultaneously interrogated for HER2, IL-13Rα2 and EphA2 using multicolor flow cytometry (Figure 2 and gating strategy shown in Supplementary Figure S1). A mean of 87% (SD = 2.06) of tumor cells expressed HER2 only, 83% (SD = 15.27) IL-13Rα2 only and 73% (SD = 8.58) EphA2 only. A mean of 94% (SD = 1.48) of cells expressed either HER2 or IL-13Rα2, 93% (SD = 6.85) HER2 or EphA2 and 87% (SD = 11.5) IL-13Rα2 or EphA2 and a mean of 96% (SD = 4) of tumor cells expressed any of the three antigens. There were statistically significant differences between single antigen prevalence and permutations of two antigens for all samples (P = 0.01–0.0001).
Figure 2.

Heterogeneous antigen expression in glioblastoma (GBM). Flow cytometric analysis of single cell suspensions of primary GBM excision samples and U373-GBM cell line costained for HER2, IL-13Rα2, and EphA2 on > 100,000 gated events (Supplementary Figure S1 describes the gating strategy) shown in (a) representative histograms and (b) dot plots.
To study topographic heterogeneity within a single tumor, we used immunofluorescence costaining for HER2, IL-13Rα2, and EphA2 in six serially diagnosed primary formalin-fixed paraffin-embedded tumors. Of note, we observed substantial variability in antigen expression from different hpf's within the same tumor section (representative tumor UPN-4 is shown in Supplementary Figure S2). We therefore mapped 21-equidistant high-power fields (hpfs) of three to six sections of formalin-fixed paraffin-embedded samples from a total of six other serially-diagnosed GBM patients (UPN-4 to UPN-9) for the aforementioned three antigens (Supplementary Figure S3).
Statistically significant differences were observed between the proportion of tumor cells expressing either HER2 or IL-13Rα2 and cells expressing HER2 only (P < 0.0001 in all patients) and IL-13Rα2 only (P: 0.019 to <0.0001). Immunofluorescence from six tumors is shown in (Supplementary Figure S3) and the quantification thereof is shown in a multiaxial graph in Figure 3. Differences in the proportion of tumor cells expressing either HER2 or IL-13Rα2 and cells expressing either HER2 or IL-13Rα2 or EphA2 were statistically significant in 50% of the patients. Similar results were found on analyzing the difference between the proportion of cells expressing EphA2, either EphA2 or HER2 or IL-13Rα2 in various permutations (Table 1).
Figure 3.

Hierarchy of expression of various antigenic permutations in primary glioblastoma (GBM). Multiaxial graph depicting prevalence of target antigens in various tumor cell sub-populations studied using coimmunofluorescence for HER2, IL-13Rα2, and EphA2 of six serially diagnosed GBMs surgical specimens. Significantly, more cells fell in populations expressing any two antigen permutations (versus single antigen only) by comparing means using a one-tailed unequal variance _t_-test. Expression of any of the three antigens was not significantly more inclusive of the tumor bulk in the patient cohort studied. Representative IFC captures are shown in Supplementary Figures S2 and S3. Data are analyzed in a mathematical model in Table 1.
Table 1. Mathematical analysis of single-cell antigen expression patterns in primary glioblastoma.
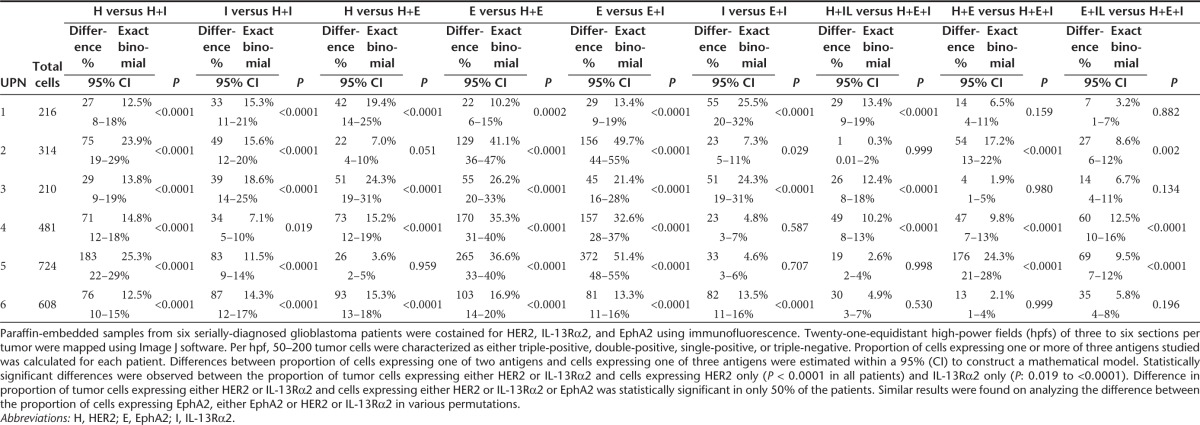
Collectively, we concluded that, in this cohort of primary tumors, the odds of capturing the bulk of tumor cells targeting two antigens simultaneously are comparable with that of targeting three antigens but are far superior to targeting a single entity; the combination of HER2 and IL-13Rα2 being the most prevalent in all patients studied.
Generation and characterization of functional bispecific T cell products
Since a dual targeting strategy is potentially advantageous, we determined whether T cells individually expressing HER2 or IL-13Rα2 CARs should be pooled or whether single lines should be prepared that coexpressed both HER2 and IL-13Rα2 CARs. We compared T cell products with single specificities to target HER2 or IL-13Rα2 (unispecific CAR T cells) with T cells coexpressing HER2 and IL-13Rα2 CARs (biCAR T cells) and with pooled products of both sets of unispecific T cells. NT T cells from the same donor were used as controls. A mean of 89% (SD = 10%) of biCAR T cells coexpressed HER2 and IL-13Rα2-specific CARs (Supplementary Figure S5a). All cellular products had similar CD4 (mean = 17%; SD = 5%) and CD8 (mean = 87%; SD = 5%) expression (Supplementary Figure S5b) and exhibited early effector T cell phenotype.
Bispecific T cell products offset antigen escape
To study if simultaneous targeting of two tumor-restricted antigens using bispecific T cell products would offset antigen escape, U373 cells were treated, in vitro, with HER2-specific T cells, biCAR T cells recognizing both HER2 and IL-13Rα2, and a pooled product of HER2-specific (50%) and IL-13Rα2-specific T cells (50%) at 1:5 T cell to tumor cell ratio. Cocultures were analyzed after 1, 3, 7, and 14 days by flow cytometry to quantify live tumor cells and characterize their antigen expression pattern (Figure 4 and Supplementary Figure S4). At 14 days, HER2-specific T cells reduced viable tumor cells by 3.4-fold, attributable to depletion of HER2-expressing tumor cells but a HER2-negative IL-13Rα2-positive (94%) tumor cell population survived. A pooled product of unispecific T cells induced more efficient (7.3-fold) and more symmetrical depletion of viable tumor cells with significantly fewer tumor escape variants (62% IL-13Rα2-positive HER2-negative; P = 0.02). An equal number of biCAR T cells, however, consistently induced the most complete depletion of tumor cells (15-fold; P < 0.04), again with minimal antigen escape (65% IL-13Rα2-positive HER2-negative; P = 0.04). Of the 6.7% tumor cells surviving biCAR T cells, ~78% (0.18% of the input tumor cells), we null for both HER2 and IL-13Rα2. The remaining ~22% expressed very modest levels of IL-13Rα2 or less prominently HER2 and had an attenuated forward and side scatter and were therefore probably of low viability. Unmanipulated tumor cells and those exposed to NT T cells had antigen expression profiles similar to U373 at baseline.
Figure 4.

Bispecific T cell products efficiently offset antigen escape resulting in symmetric depletion of HER2 and IL-13Rα2 expressing glioblastoma (GBM) cells. Composite bar diagram representing the antigen expression pattern in viable U373 cells, 7 days after co-culture with various T cell products in 1:5 T cell to tumor cell ratio (20,000 T cells and 100,000 tumor cells). Table depicts the breakdown of antigen positivity (IL-13Rα2 only, HER2 only, IL-13Rα2 and HER2 positive and IL-13Rα2 and HER2 negative) as a percentage of surviving tumor cells. A dot plot of IL-13Rα2 and HER2 co-expression on tumor cells is displayed in Supplementary Figure S4. Data shown are representative of an experiment repeated three times.
Bispecific T cell products have enhanced activation and tumor cell killing in ex vivo immunoassays
We hypothesized that the bispecific T cell products would have enhanced activation and cytolytic activity by virtue of being able to recognize two tumor-associated antigens simultaneously. In cocultures, CAR T cells secreted Th1 cytokines (IFN-γ and IL-2) upon encountering HER2 and IL-13Rα2-positive U373-GBM cells. Both IFN-γ and IL-2 secretion were significantly higher with biCAR T cells in comparison with unispecific T cells or the pooled product (Figure 5a,b). Though the pooled product consistently secreted higher amounts of cytokines than the unispecific T cells, these differences did not reach statistical significance. No cytokine release was detected with NT T cells.
Figure 5.

Enhanced activation of the biCAR T cells in vitro, upon simultaneously encountering two tumor-restricted antigens. In cocultures, HER2 and IL-13Rα2 specific CAR T cells secreted Th1 cytokines (IFN-γ and IL-2) upon encountering HER2 and IL-13Rα2 expressing U373 cells (25,000 T cells: 100,000 tumor cells), as detected by ELISA. biCAR T cells secreted significantly higher amounts of (a) IFN-γ in comparison with HER2 CAR T cells (P = 0.03), IL-13Rα2 CAR T cells (P = 0.04) and the pool of equal number of unispecific T cells (50% HER2 CAR T cells and 50% IL-13Rα2 CAR T cells; P = 0.046) and (b) IL-2 in comparison with HER2 CAR T cells (P = 0.04), IL-13Rα2 CAR T cells (P = 0.02) and their pooled-product (P = 0.05). Both IFN-γ and IL-2 secretion with the pooled product were consistently higher than unispecific T cells but the difference was not statistically significant (P = 0.09–0.13). No cytokine release was detected with NT T cells from the same donor. (c) While unispecific HER2 CAR and IL-13Rα2 CAR T cells showed comparable degrees of proliferative capacity, as evidenced by 3H-thymidine uptake, biCAR T cells showed significantly higher 3H-thymidine uptake in comparison to HER2 CAR T cells (P = 0.01), IL-13Rα2 CAR T cells (P = 0.03) and their pooled product (P = 0.04) upon encountering HER2 and IL-13Rα2 plate bound proteins simultaneously. CAR T cells stimulated with OKT3 were used as positive controls and NT T cells as negative control. (d) biCAR T cells exhibited higher cytolytic function in vitro, in CCK-8 cytotoxicity assays. CAR T cells recognized and killed HER2 and IL-13Rα2 expressing U373 cells at different effector to target ratios (4:1, 2:1, 1:1, and 1:2) after 8 hour incubation. Improved tumor cell killing shown by the pooled product (50% HER2 CAR and 50% IL-13Rα2 CAR T cells) was lost at low T cell to tumor ratios. Conversely, biCAR T cells consistently showed most cytolytic activity and maintained the effect even at low T cell to tumor cell ratios. NT T cells did not induce any significant tumor cell lysis. Data are M ± SD from one of three experiments done in triplicates. Validation of the methodology used in (c) is described in Supplementary Figure 6a.
The distinct activation of the T cells and their proliferation in response to each individual target and not to target null entities is of utmost importance. We therefore used and optimized an ex vivo assay to distinctly test the specificity of various effector products in a titrable manner and to ensure an antigen density/response correlation. Triplicate wells of nontissue culture-treated plates were coated with HER2-Fc chimeric protein or IL-1Rα2-Fc chimeric protein. Control wells were not coated or coated with OKT3 (negative and positive controls, respectively) or encountered a mock target. The assay was optimized to use concentrations of chimeric proteins in the correlative range of the dose/response. This optimization is shown in Supplementary Figure S6a. In Supplementary Figure S6a, an experiment was performed to demonstrate the distinct reactivity of each effector T cell product to the respective antigens evidenced by IL-2 release. Further, in a cytotoxicity assay, we ensure the reactivity of biCAR T cells to antigen positive (U373-GBM) but not to antigen null (Raji) tumor cells (Supplementary Figure S6b).
On the basis of this specificity platform, in proliferation assays, we show that unispecific HER2 and IL-13Rα2 CAR T cells exhibited comparable degrees of proliferation evidenced by 3H-thymidine uptake (Figure 5c) upon exposure to recombinant human HER2-Fc- and IL-13Rα2-Fc-coated polystyrene culture plates. biCAR T cells showed significantly more proliferation compared with either unispecific T cells and their pooled product (P = 0.01, 0.03, and 0.04, respectively). Again, the difference in proliferation between the pooled product and unispecific T cells was not statistically significant.
Standard CCK-8 assays were used to quantitate cytolytic activity against U373-GBM cells. Unispecific T cells recognizing either HER2 or IL-13Rα2 killed tumor efficiently at standard T cell: tumor cell ratios (Supplementary Figure S7), but less efficiently at lower such ratios (Figure 5d). The pooled product of HER2 and IL-13Rα2-specific T cells showed improved killing, but only at high T cell to tumor cell ratios (Figure 5d). biCAR T cells showed the highest lytic activity and maintained this high activity at low T cell to tumor cell ratios in most experiments. NT T cells from the same donor showed no lytic activity against GBM cells, eliminating the possibility of an allogeneic response. Differences between the degrees of killing were quite consistent but were not statistically significant.
Collectively, these ex vivo results indicate that a pool of unispecific T cells exhibit enhanced activity above unispecific T cell products and that biCAR T cells show enhanced activation and antitumor activity over a pooled product of unispecific T cells.
Primary bispecific T cell products from GBM patients exhibit enhanced functionality against autologous glioma cells
In order to validate and substantiate our results in primary human samples, we procured tumor cells and peripheral blood mononuclear cells sets from two GBM patients (UPN1 and UPN10) and generated autologous T cell effectors (HER2 and IL-13Rα2-specific CAR T cells, their pooled product, HER2 and IL-13Rα2-bispecific biCAR T cells, and NT control effectors). CAR transduction rates were similar for all experimental conditions (Supplementary Figure S8). One exception was the IL-13Rα2 CAR transduction in UPN-10 that was lower than its unispecific counterpart. Figure 6a,b represents the analysis of a 4 to 1 tumor cell to T cell effector ratio coculture assays to test for T cell activation (IFN-γ as a signal 1 product and IL-2 as a signal 2 product) in the supernatant using ELISA. The results were similar to those seen upon encounter of U373-GBM cells with T cell effectors generated from healthy volunteer donors. biCAR T cells released significantly higher IFN-γ and IL-2 than HER2 or IL-13Rα2 CAR T cells or their pooled product. Again, the latter had slightly higher functionality than HER2 or IL-13Rα2 CAR T cells but this was largely not statistically significant. Similarly, we performed cytotoxicity assays from those GBM patients using autologous effectors (Figure 6c). These were also similar to the healthy effector versus U373-GBM cell line findings shown in Figure 5d. Further, in addition to the low effector to tumor cell ratios, we performed cytotoxicity assays using the standard 40:1, 20:1, 10:1, 5:1, and 2.5:1 ratios (Supplementary Figure S9), which largely confirmed a tendency toward better functionality in bispecific products, with no statistical significance.
Figure 6.

Primary bispecific T-cell products from glioblastoma (GBM) patients exhibit enhanced functionality against autologous glioma cells. Effector cells were generated from two GBM patients (UPN-1 and UPN-10; transduction rates are shown in Supplementary Figure S8) and were tested against autologous primary glioma cells. Supernatants from 25,000 T cells: 100,000 tumor cell co-cultures were harvested and analyzed for (a) IFN-γ and (b) IL-2, for both patients. Further, cytotoxicity assays were performed; displayed in (c). Collectively, these ex vivo results using patient material in an autologous set up confirmed, as seen with healthy donors tested against GBM-U373, that a pool of unispecific T cells exhibited some enhanced (yet not statistically significant) activity above unispecific T cell products and that biCAR T cells show enhanced activation and antitumor activity over unispecific T cells and the pooled product thereof. Data are M ± SD from one of at least two experiments done in triplicates.
Collectively, these ex vivo results using patient material in an autologous set up confirmed, as seen with healthy donors tested against GBM-U373, that a pool of unispecific T cells exhibited some enhanced (yet not statistically significant) functionality above unispecific T cell products and that biCAR T cells show significantly enhanced activation and antitumor activity over unispecific T cells and the pooled product thereof.
Accentuated specific signaling in T cells coexpressing two distinct CAR molecules
We reasoned that the enhanced in vitro activity of biCAR T cells could result from increased ζ-chain signaling by virtue of simultaneous engagement of HER2 and IL-13Rα2 and examined the intracellular expression of21 pZAP-70 in T cells upon antigen encounter.22
On flow cytometric analysis, both HER2-specific and IL-13Rα2-specific T cells exhibited increased pZAP-70 expression above the unstimulated and NT controls following simultaneous exposure to HER2-Fc and IL-13Rα2-Fc proteins. biCAR T cells showed consistent and statistically significant higher degree of pZap-70 compared with both unispecific T cell products (Figure 7a and Table 2 shows the statistical analysis of these results).
Figure 7.

Accentuated phosphorylation of Zap70 in biCAR T cells. CAR T cells were exposed to a nontissue culture plate coated with either HER2-Fc or IL-13Rα2-Fc or both chimeric proteins for 2 hours at 37 °C. Activated CAR T cells were permeated with monoclonal antibody to pZap70 and analyzed using flow cytometry. biCAR T cells showed (a) a consistently higher statistically significant degree of phosphorylated Zap70 (pZap70) in comparison with both the unispecific CAR T cell products on exposure to both chimeric proteins; (b) while we observed comparable degrees of pZap70 on exposure to either HER2-Fc or IL-13Rα2-Fc individually, the pZap70 signaling was consistently enhanced upon encountering both HER2-Fc and IL-13Rα2-Fc simultaneously. The comparison of the latter mean fluorescence intensity was not statistically significant though. Displayed histograms are representatives of three experiments. The statistical analysis of fluorescence is displayed in Table 2.
Table 2. Flowcytometric analysis of specific signaling through Zap70.
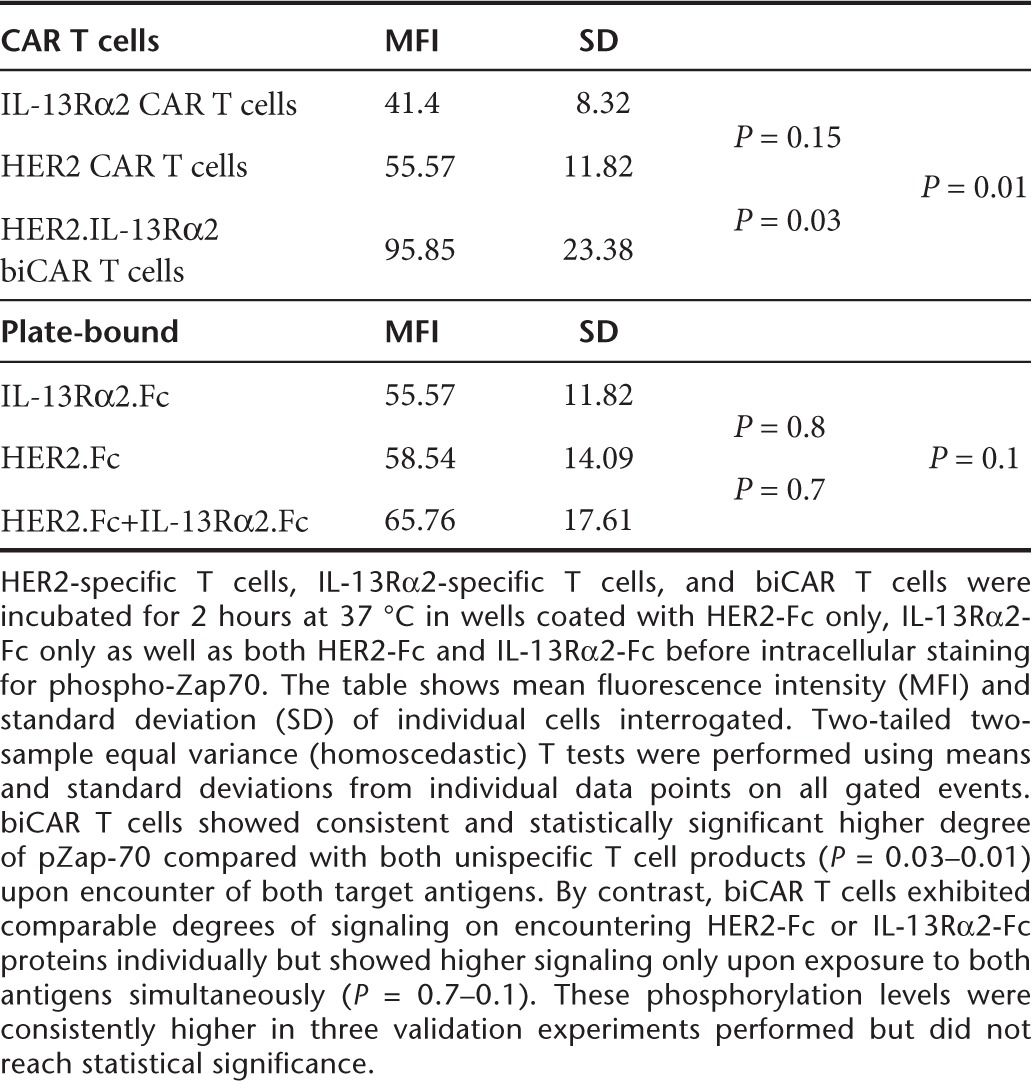
To test if enhanced activation reflected a T cell product with a lower activation threshold, biCAR T cells were exposed to either HER2-Fc or IL-13Rα2-Fc protein alone or to both simultaneously. biCAR T cells exhibited comparable degrees of signaling on encountering HER2-Fc or IL-13Rα2-Fc proteins individually but showed higher signaling only upon exposure to both antigens simultaneously (Figure 7b and Table 2 shows the statistical analysis of these results). While these phosphorylation levels were consistently higher in three validation experiments performed, they did not reach statistical significance.
Hence, biCAR T cells have accentuated ζ-chain signaling upon simultaneous encounter of two distinct antigens and this effect is specific to antigen encounter, indicating their potential for enhanced yet specific effector functions.
Adoptively transferred biCAR T cells improve tumor control in an orthotopic xenograft model of human GBM
To determine the efficacy of bispecific T cell products in vivo, we used an orthotopic xenograft model of human GBM. Tumor size was monitored by serial bioluminescence imaging (Figure 8a,b). Tumors grew exponentially in untreated animals and those treated with NT T cells. HER2-specific T cells induced tumor regression similar to that induced by IL-13Rα2-specific T cells, but the durability of this response was significantly increased using the bispecific T cell products. There was a statistically significant difference between all CAR-expressing effectors and both control groups (tumor only and NT T cell treated) past day 8, between bispecific and unispecific effectors past day 49 and between biCAR and pooled T cells past day 74. Kaplan–Meier survival analysis (Figure 8c) showed that mice in both control groups (untreated and NT T cell treated) had a median survival of 35 days. Tumor control by unispecific HER2 and IL-13Rα2 CAR T cells improved the median survival to 79 and 84 days, respectively. Mice treated with pooled product had a median survival of 120 days and biCAR T cells further improved tumor control with 80% of mice surviving >160 days (pooled product versus unispecific T cells, P = 0.04; biCAR versus unispecific T cells, P = 0.0007; biCAR versus pooled product, P = 0.002 using Log-rank test).
Figure 8.

Adoptively transferred biCAR T cells show improved tumor control in vivo. Tumors were established by stereotactic injection of 2.5 × 105 eGFP. Firefly luciferase expressing U373 cells into the right frontal cortex of SCID mice. On day 6 after tumor cell injection, mice received an intra-tumoral injection of 2 × 106 unispecific HER2 CAR T cells (n = 5), IL-13Rα2 CAR T cells (n = 5), pooled product of equal number of T cells containing 50% HER2-specific and 50% IL-13Rα2-specific CAR T cells (n = 5), biCAR T cells (n = 5) or NT T cells (n = 5) from the same donor. A subset of animals were not treated (n = 5). (a) Tumors grew exponentially in untreated mice and those treated with NT T cells as shown in one representative animal from each group. Unispecific HER2 CAR and IL-13Rα2 CAR T cells induced transient tumor regression that was comparable. Both bispecific T cell products, the pool of unispecific HER2 CAR and IL-13Rα2 CAR T cells as well as biCAR T cells, induced tumor regression that was sustained for significantly longer duration. Mice treated with the pooled T cell product started relapsing after the mean of day 60; (b) quantitative bioluminescence imaging showing the group median, photons/cm2/seconds/area imaged; (c) Survival analysis on a Kaplan–Meier plotted at 160 days after the tumor was established. Mice treated with unispecific HER2 CAR or IL-13Rα2 CAR T cells had median survival of 79 and 84 days respectively in comparison to median survival of 35 days in untreated mice and those treated with NT T cells (P = 0.03). Median survival improved to 120 days in mice treated with the pooled product (P = 0.04). biCAR T cells further improved tumor control with 80% of the mice surviving >160 days (biCAR versus unispecific T cells, P = 0.0007; biCAR versus pooled product, P = 0.002).
Discussion
In this study, we investigated the single cell antigen expression pattern in primary GBM and showed that near complete targeting of the tumor cells can be achieved by using a bispecific combinational approach. Further, our results indicate that simultaneous targeting of the two glioma-restricted antigens, HER2 and IL-13Rα2, using CAR T cell products could offset antigen escape and achieve better tumor control, conferring survival advantage to the treated animals.
On exploring the antigen expression landscape in primary GBM, we encountered substantial heterogeneity between patients as observed by others.13,14 This makes the selection of any single antigen incapable of capturing most patients as candidates for targeted therapies. While a well-chosen single CAR T-cell product can target the majority of the tumor cell bulk in most patients, most nontargeted cells express one of two other antigens tested. This, in addition to the limited T cell persistence in vivo,23,24,25 could explain why targeting a single antigen using CAR T cells allows an initial robust antitumor response that is followed by relapse26,27,28,29 due to outgrowth of antigen null tumor cells. While GBM is particularly heterogeneous, these findings could certainly apply to other tumors since with few exceptions, there exists no universal tumor antigen for targeted therapies.
We reasoned that targeting multiple distinct tumor-restricted antigens simultaneously could reduce the possibility of tumor escape, since multipotent T cells will maintain effector function in the event that one of the targets is downregulated or mutated or if therapy selects tumor antigen-loss variants. We constructed a binominal mathematical model, comparing the odds of targeting two versus three tumor antigens to define how broad of an approach is necessary to achieve near-complete (>95%) tumor cell capture30 and found that while dual targeting was clearly superior to single targeting, targeting of three antigens (or more) did not predict a significantly superior advantage in the patient cohort we studied. We therefore investigated if a bispecific T cell product could overcome this drawback of adoptive T cell transfer.
We generated T cell lines coexpressing HER2 and IL-13Rα2 CARs (biCAR T cells), since these two antigens had the highest coprevalence in tumor samples from our patient cohort, and compared their functionality to unispecific T cell products (HER2-specific T cells and IL-13Rα2-specific T cells) and their pooled product. The tumor antigen pair could vary if a larger cohort is considered. Indeed, EphA2, as recently demonstrated by our group, could well be very valid in the context of targeting multiple antigens, and paired with either HER2 or IL-13Rα2 or both, could represent a more valid option in particular patient's scenarios.31 Our results indicate that bispecific T cell products, probably by virtue of being able to recognize both target antigens, efficiently offset antigen escape variants and induced uniform depletion of tumor cell populations expressing HER2 and IL-13Rα2. Approximately 0.18% of the input tumor cells survived biCAR T cells targeting and were null for both HER2 and IL-13Rα2. This very small tumor cell population could theoretically continue to pose an antigen escape variant. Nevertheless, the intent of this work is to test the proof-of-concept that the multiple targeting gets sufficiently close to a certain efficacy; it gets the antigen escape variant arbitrarily close to zero (0.18% in this case). Indeed, further expansion of this concept by targeting a multiplex of antigens could substantiate these findings in a larger cohort of patients enough to both achieve complete tumor remission and yield a broad spectrum universal CAR T cell product.
Bystander tumor cell killing could play a role in this improvement, yet was not formally tested here. In most of the experiment described, we deliberately downtitrated the effector: tumor ratio to minimize the effect of mass/bystander killing induced by T cell effectors and to more closely simulate the tumor: T cell dominance that occurs in most clinical scenarios of testing. In vitro, pooled product exhibited marginally more cytolytic ability, secretory functions, and proliferative functions that were consistent albeit not statistically significant. biCAR T cells, however, exhibit significantly enhanced effector functions including cytolysis, cytokine release, and potential to proliferate after antigen encounter. Therefore, the magnitude of response was not solely due to collective recognition of both tumor antigens.
It has been shown that the magnitude of T cell response is influenced by the balance between the density of target antigens expressed on the tumor and the density of CAR molecules expressed on T cells.32 Accordingly, biCAR T cells would have functional advantage by virtue of their ability to “see” a more antigen dense tumor cell and their collective expression of a higher density of CARs on the cell surface. Such an advantage would be particularly favorable where the expression of individual tumor antigens is modest.28 This was demonstrated by the ability of biCAR T cells to maintain their cytolytic activity even at relatively low T cell to tumor cell ratios. In fact, though both bispecific T cell products showed enhanced effector functions in vitro and achieved better tumor control in vivo, biCAR T cells were the most potent, indicating they may be superior to bispecific pooled product in clinical studies. While a spontaneous model would be an ideal setup for testing in vivo efficacy, xenogeneic models have been particularly useful in the context of preclinical testing of human T cell products developed for cancer and have served as a justification for most early-phase clinical trials.4,5,6 In addition, the ability to amplify the antitumor response in situ could compensate for small cell doses, therefore avoiding the potential for adverse reactions such as the cytokine storms associated with very high T cell doses.33,34,35 Collectively, we observed multiple indicators of functional superiority of biCAR T cells to a bispecific pooled product. This could be explained by the enhanced downstream signaling from ζ-chains upon encountering both target antigens simultaneously, as evidenced by a significantly higher degree of ZAP-70 phosphorylation upon simultaneous antigen encounter.21,22 Also, particularly important is that activation of biCAR T cells was comparable with unispecific T cells on exposure to single target antigen but was maximal only when both targets were encountered simultaneously. This finding can potentially be utilized to increase the specificity of effector cells for malignant versus normal target cells as only coexpressers would trigger a sufficient T cell activation. Finally, a theoretical advantage of targeting multiple antigens simultaneously is the ability to customize therapies for patients based on their tumor antigen make-up; personalizing targeted therapies and/or targeting tumor cells and elements of the tumor microenvironment.
We demonstrate that biCAR T cells can be efficiently generated by sequential retroviral transduction without compromising T cell activation, proliferation potential, or antitumor activity. Generating a clinical grade biCAR T cell product by sequential retroviral transduction would nevertheless substantially increase the cost and understandably be faced by multiple regulatory hurdles. To overcome this, one strategy is to use internal ribosomal entry site or 2A-containing retroviral bicistronic vectors, allowing for simultaneous expression of two (or more) CAR molecules from the same RNA transcript.36,37 Alternatively, nonviral carriers such as sleeping beauty or PiggyBac transposon systems can be used to genetically modify T cells to coexpress multiple CARs with distinct specificities; we have optimized the latter system for scale up.37,38
Broader targeting and potentially enhanced activation of T cells introduces the additional risk of adverse events related to lower specificity and off-target effects. Suicide genes that allow for conditional elimination of transgene expressing T cells enabling quick elimination of adoptively transferred T cells in case of adverse events.39,40,41 Our group recently showed the remarkable efficacy of such an approach using a small molecule-dimerizable iCasp9 cell-suicide system.42 Alternatively, others have used a functionally bipartite CAR molecule that conditionally triggers downstream signaling only on simultaneous engagement of two distinct tumor-restricted antigens.43,44
In summary, we demonstrated the pattern of heterogeneity in GBM justifies cotargeting multiple antigens. We show that simultaneous targeting of two glioma-restricted antigens using bispecific T cell products does indeed result in improved tumor control by minimizing the possibility of tumor selection on the basis of target antigen downregulation or alteration.
Materials and Methods
Blood donors, primary tumor cells, and cell lines. Blood samples and primary tumor cells were obtained from healthy donors and patients with GBM respectively, on a protocol approved by the institutional review board of Baylor College of Medicine and The Methodist Hospital. Written, informed consent was obtained from all patients and healthy donors. The U373-GBM cell line was purchased from the American Type Culture Collection (ATCC; Manassas, VA) and grown in Dulbecco's Modified Eagle's Medium (Invitrogen, Carlsbad, CA) with 10% FCS, 2 mmol/l GlutaMAX-I, 1.5 g/l sodium bicarbonate, 0.1 mmol/l nonessential aminoacids, and 1.0 mmol/l sodium pyruvate. The Characterized Cell Line Core Facility at MD Anderson Cancer Center, Houston, TX, performed cell line validation by short-tandem repeat method. The authentication was done in February of 2011 and we have an authenticated cell bank. Once cells are thawed, we do not culture them longer than 6 months to meet the time requirement. T cells were maintained in T cell media (250 ml RPMI-1640, 200 ml CLICKS with 10% FCS containing 2 mmol/l GlutaMAX-I; Invitrogen). Tumor samples were processed aseptically and primary cell cultures were initiated using DMEM with 15% heat-inactivated fetal calf serum, 2 mmol/l GlutaMAX-I, 1% Insulin-Transferrin-Selenium-X supplement, and 1% Penicillin-Streptomycin mixture (Invitrogen). Cells were used within 7 days of plating or established as primary cell lines.
Immunofluorescent staining and imaging. Primary GBM samples embedded in paraffin were used for immunofluorescent staining. Sections were deparaffinized in xylene and rehydrated in descending concentrations of ethanol. Antigen retrieval was completed by immersing slides in 1X DAKO citrate buffer under pressure for 45 minutes. Blocking with 0.05% Tween in PBS plus 5% donkey serum was done in humidified conditions for 1 hour at room temperature, followed by overnight incubation at 4 °C with primary antibodies; mouse CB11 anti-HER2 in 1:25 (BioGenex, Fremont, CA), rabbit anti-EphA2 in 1:300 (Invitrogen) and goat anti-IL-13ra2 in 1:500 dilution (R&D Systems, Minneapolis, MN). Slides were incubated for 1 hour at room temperature in secondary antibodies diluted at 1:200 (anti-rabbit FITC, anti-mouse Texas-Red, and anti-goat Cy5; Invitrogen), counterstained with nuclear stain DAPI, and examined under a fluorescent microscope.
Sample slides processed via immunocytochemistry were viewed using an Olympus Fluoview FV1000 (Olympus America, NY) upright confocal laser scanning microscope, using a 60X PlanApo oil immersion objective (numerical aperture 1.4). The instrument is equipped with diode laser with 405-nm emission line, argon laser with 458, 488, and 515 nm emission lines, and a HeNe with 559 and 635 nm emission lines. The excitation/emission wavelengths were 405/460 nm (430–470 nm band-pass emission filter, DAPI; nucleus), 488/519 nm (505–525 nm band-pass emission filter, Alexa Fluor 488; HER2), 543/618 nm (560–660 nm band-pass emission filter, Alexa Fluor 594; EphA2), and 643/690 nm (655–755 nm band-pass emission filter, Alexa Fluor 647; IL-13α2). Low laser power intensity was used to avoid photobleaching of the dyes. The photomultiplier gain was set to 1 to give an average luminal fluorescence intensity of 100–150 (full scale, 255) in order to mitigate detection of tissue auto fluorescence, and the photomultiplier voltage was adjusted to minimize pixel saturation. Although some preparation-to-preparation variations in the samples were noted, overall photomultiplier settings were maintained across samples to obtain similar fluorescence intensities. Negative control sections from normal brain received identical preparations for immunohistochemical staining including dilutions of primary and secondary antibodies. No specific staining was visible on control preparations. The resolution was same in the various preparations at 0.276 × 0.276 μm per pixel in the image plane (512 × 512 pixels) and differed in the z-direction at about 1.5–2.5 μm in axial dimension (6–12 planes). A 2X line average Kalman filter was used to improve the signal-to-noise ratio. Images were sequentially obtained at each of the four wavelengths with each laser separately on, with the same starting plane and ending plane along the z axis. Sequential excitation was implemented, to remove “cross-talk” between the four color channels. Each section was examined individually and then used to generate maximum intensity projections. The four projections were merged in a single overlay image to give simultaneous visualization of the four antigens. The only tool used to improve image quality was contrast enhancement, in order to produce print quality images for publication.
Generation of retroviral constructs. The construction of the HER2-specific CAR (pSFG.FRP5.CD28.ζ) has been previously described.26,27,28 The construction of the IL-13Rα2-specific CAR is described in detail elsewhere (Krebs et al. submitted for publication). Briefly, an IL-13 mutein with enhanced affinity to IL-13Rα2 was subcloned into a SFG retroviral vector containing an expression cassette encoding a CH2CH3 domain, a CD28 transmembrane domain, and a CD28.ζ signaling domain.45,46 For in vivo studies, a retroviral vector encoding the fusion protein eGFP-Firefly Luciferase (eGFP.FFLuc) was used to generate firefly luciferase expressing GBM cells.26,28
Retrovirus production and transduction of T cells. To produce retroviral supernatant, 293T cells were cotransfected with the FRP5.CD28.ζ or IL-13Rα2.CD28.ζ retroviral vector containing plasmid, Peg-Pam-e plasmid encoding the sequence for MoMLV gag-pol, and plasmid pMEVSVg containing the sequence for VSV-G, using GeneJuice transfection reagent (EMD Biosciences, San Diego, CA). Supernatants containing the retrovirus were collected 48 and 72 hours later.
OKT3/CD28-activated T cells were transduced with retroviral vectors as described previously.27 Peripheral blood mononuclear cells were isolated by Lymphoprep (Greiner Bio-One, NC) gradient centrifugation. 5 × 105 peripheral blood mononuclear cells per well in a 24-well plate were activated with OKT3 (Ortho Biotech, Raritan, NJ) and CD28 monoclonal antibodies (BD Biosciences, San Jose, CA) at a final concentration of 1 μg/ml. On day 2, recombinant human IL-2 (Chiron, Emmeryville, CA) was added at 100 units/ml, and cells were harvested for retroviral transduction on day 3. A nontissue culture treated 24-well plate was precoated with recombinant fibronectin fragment (Retronectin; Takara Shuzo, Otsu, Japan). Wells were washed with phosphate-buffered saline (PBS; Sigmaaldrich, St Louis, MO) and incubated 30 minutes with retrovirus. Subsequently, 3 × 105 T cells/well were transduced with retrovirus in the presence of IL-2 (100 units/ml). After 48–72 hours, cells were removed and expanded in the presence of IL-2 (100 units/ml) for 10–15 days before use.
T cells coexpressing HER2.CD28 ζ and IL-13Rα2.CD28 ζ (biCAR T cells) were generated by tandem retroviral transductions to express the second CAR molecule on a unispecific T cell platform such that the same unispecific T cell product was used in comparative experiments to normalize for heterogeneity in transduction efficiencies.
Flow cytometry. Analysis was done with a FACS calibur instrument (Becton Dickinson, Franklin Lakes, NJ). CellQuest software (BD Biosciences) or Kaluza software (Beckman Coulter, Brea, CA) was used for all flow cytometric analyses of >10,000 events; negative controls included isotype antibodies. Cells were washed with PBS containing 2% FBS and 0.1% sodium azide (FACS buffer; Sigmaaldrich) before adding the the antibody. After 30–60 minutes incubation at 4 °C in dark, cells were washed with FACs buffer and fixed in 0.5% paraformaldehyde for analysis.
Surface staining of tumor cells was done using mouse anti-HER2.APC (BD Biosciences), mouse anti-IL-13Rα2.PE (Abcam, Cambridge, MA), and goat anti-EphA2 with rabbit anti-goat IgG.FITC (Southern Biotech, Birmingham, AL) antibodies. Cell surface expression of HER2-CAR was detected using goat anti-mouse Fab fragment specific antibody conjugated with DyLight (Jackson ImmunoResearch, West Grove, PA) and that of IL-13Rα2-CAR was detected using goat antihuman Fc fragment specific antibody conjugated with FITC (Millipore, Billerica, MA).
To measure downstream signaling, a coated plate method was used to activate T cells. Recombinant human HER2-Fc and IL-13Rα2-Fc (R&D systems) diluted in sterile water at concentrations of 2 and 5 µl/ml, respectively were added to a nontissue culture-treated 24-well plate (BD Falcon, Franklin Lakes, NJ) and kept overnight at 4 °C. HER2-specific T cells, IL-13Rα2-specific T cells and biCAR T cells were incubated for 2 hours at 37 °C in wells coated with HER2-Fc only, IL-13Rα2-Fc only as well as both HER2-Fc and IL-13Rα2-Fc before intracellular staining for phospho-Zap70. CAR T cells not exposed to Fc-protein and NT T cells served as negative controls. CAR T cells stimulated with OKT3 were used as positive controls. Cells were taken off the 24-well plate and incubated in 4% PFA for 10 minutes followed by 1% Saponin for 30 minutes on ice with washes in FACS buffer in between. Cells were then incubated with 5 µl phospho-Zap70 antibody (BD Biosceinces) for 30 minutes, followed by 30 minutes with rat anti-mouse IgG1.PE secondary antibody (BD Biosceinces), washed in FACS buffer, and fixed in 0.5% paraformaldehyde for analysis.
Analysis of cytokine production and T cell expansion. Equal numbers of HER2-specific, IL-13Rα2-specific T cells, their pooled product (50% HER2-specific and 50% IL-13Rα2-specific), and biCAR T cells were cocultured with U373 cells in 1:4 (25,000 T cells: 100,000 tumor cell) ratios. After 24 hours incubation, conditioned-culture medium were collected and the presence of IFN-γ and IL-2 were determined by ELISA as per manufacturer's instructions (R&D Systems).
T cell proliferation was measured by 3H-Thymidine uptake assay. T cell products (HER2-specific, IL-13Rα2-specific, pool of unispecific T cells, biCAR T cells and NT T cells) were incubated at 37 °C in 96-well nontissue culture-treated plate coated with recombinant human HER2-Fc (0.1 µg/ml) and IL-13Rα2-Fc (0.8 µg/ml) T cell stimulation with OKT3 was used as positive control. On day 5, 25 µl of 40 µCi/ml 3H-Thymidine (1 µCi/well) was added to T cells and incubated overnight. On day 6, cells were harvested, placed in fiber discs in 1.5-ml Eppendorf tubes with 1-ml liquiscint, and counted with a Beta-Scintillation counter. Data were expressed as the stimulation index found by dividing the CPM in wells with stimulant by the CPM in the sample without stimulant.
Cytotoxicity assays. Cytolytic activity of T cells was assessed using the Cell Counting Kit-8 (CCK-8; Dojindo, Gaithersburg, MD), a sensitive colorimetric assay that allows the determination of the number of viable cells in cell proliferation and cytotoxicity experiments. CCK-8 assay utilizes a highly water-soluble tetrazolium-salt (WST-8) that is reduced by the dehydrogenases in the viable cells to water soluble orange colored formazan-dye. U373-GBM cells were cocultured in vitro, with unispecific HER2, IL-13Rα2 CAR-T cells, their pooled product (50% HER2-specific and 50% IL-13Rα2-specific T cells) or biCAR T cells in low T cell to tumor cell ratios to simulate the in vivo scenario and to minimize the bystander killing of tumor cells. 10,000 U373 cells/well were incubated for 12 hours at 37 °C in a 96-well nontissue culture-treated plate. T cell products were added at 4:1, 2:1, 1:1, and 1:2 T cell to tumor cell ratios, after washing the wells with complete medium to remove any nonadherent tumor cells and then incubated for 8 hours at 37 °C. After adding 10 µl of CCK-8 solution, the plate was incubated at 37 °C and absorbance was measured at 450 nm using a microplate reader at 60 and 90 minutes. T cells incubated with complete medium alone were used to determine spontaneous lysis.
Orthotopic xenogeneic severe combined immunodeficiency mouse model of GBM. All animal experiments were conducted on a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Recipient NOD-severe combined immunodeficiency mice were purchased from Taconic, Hudson, USA (C.B-Igh-1 b/IcrTac-Prkdc scid; FOX CHASE CB-17 SCID ICR). Male 9–12-week-old mice were anesthetized with rapid sequence inhalation using Isofluorane (Abbot Laboratories, England, UK) followed by intraperitoneal injection of 225–240 mg/kg Avertin solution and maintained on Isofluorane by inhalation throughout the procedure. Mouse's head was shaved, immobilized in a Cunningham Mouse/Neonatal Rat Adaptor (Stoelting, Wood Dale, IL) stereotactic apparatus fitted into an E15600 Lab Standard Stereotaxic Instrument (Stoelting), then scrubbed with 1% povidone-iodine. A 10-mm skin incision was made along the midline. The tip of a 31G half-inch needle mounted on a Hamilton syringe (Hamilton, Reno, NV) served as the reference point. A 1-mm burr-hole was drilled into the skull, 1-mm anterior to and 2 mm to the right of bregma.
The generation of the U373 cell lines expressing an enhanced GFP firefly luciferase fusion protein (U373.eGFP.FFluc) has been previously described.26 Firefly-luciferase expression was confirmed in vitro prior to usage in a luminometer. Firefly-luciferase-expressing U373 cells (1 × 105 in 2 µl) were injected 3 mm deep to bregma, corresponding to the center of right caudate nucleus over 3 minutes for six groups of five mice each. Needle was left in place for 1 minute, avoiding tumor cell extrusion, and withdrawn slowly over 3 minutes. All animals had progressively growing xenografts, evidenced by progressive and exponential increments in bioluminescence signal. Each group was randomly assigned a condition of no treatment, NT T cells, HER2-specific T cells, IL-13Rα2-specific T cells, biCAR T cells, or the pool of unispecific T cells (50% HER-specific and 50% IL-13Rα2-specific) and received an intratumoral injection of 2 × 106 T cells on day 6 following tumor injection. T cell injections were performed following the same protocol as tumor injection. Incision was closed with two to three interrupted 7.0 Ethicon sutures (Ethicon, Somerville, NJ). Subcutaneous injection of 0.03–0.1 mg/kg buprenorphine (Buprenex RBH, England) was given for pain control.
Bioluminescence imaging. Isofluorane anesthetized animals were imaged using the IVIS system (Xenogen, Alameda, CA) 10 minutes after intraperitoneal injection of 150 mg/kg D-luciferin (Xenogen). Photons emitted from luciferase-expressing cells within the animal body and transmitted through the tissue were quantified using “Living Image” software program (Xenogen). A pseudocolor image representing light intensity (blue least intense and red most intense) was generated and superimposed over the grayscale reference image. Animals were imaged every other day for 1 week after injections, biweekly the next 2 weeks, and weekly thereafter until 76 days while regularly examining for neurological deficits, weight loss, or signs of stress and euthanizing according to preset criteria by the Baylor College of Medicine's Center for Comparative Medicine guidelines.
Statistical analysis. Proportions of cells identifying with one or more of HER2, EphA2, or IL-13Rα2 antigens using flow cytometry and immunofluorescence were analyzed for each patient. Differences in proportions between cells with two antigens and cells with one antigen, and between cells with three antigens and cells with two antigens were estimated with 95% binomial CI. The exact binomial method tested whether the difference was greater than a nominal 5%.
For the analysis of phosphor-flow studies, two-tailed two-sample equal variance (homoscedastic) _t_-tests were performed using means and standard deviations from individual data points on all gated events.
For the bioluminescence experiments, intensity signals were log-transformed and summarized using mean ± SD at baseline and multiple subsequent time points for each mouse group. Two separate pilot experiments were performed to assess the trends for bioluminescence among various groups along time. Intensity signals from periodic imaging were log-transformed and summarized using mean ± SD at baseline and multiple subsequent time points for each mouse group. On the basis of preliminary experiment, repeat simulations were used to determine the number of mice per group in the experiment presented with P values as indicated and in a predetermined end point of 160 days. Signal intensity changes from baseline at each time point were calculated and compared using paired both _t_-test and Wilcoxon signed-ranks test. Log-rank test was used to compare the survival distribution between treatment groups.
SUPPLEMENTARY MATERIAL Figure S1. Gating strategy for tumor cell analysis performed in Figures 1 and 2 and Supplemental Figure 3.Figure S2. Coimmunofluorescence for HER2, IL-13Rα2, and EphA2 demonstrating the heterogenous expression of antigens within the same tumor by changing the field examined.Figure S3. Representative co-immunofluorescence captures for HER2, IL13Rα2 and EphA2 from six serially diagnosed GBM patients and normal brain tissue.Figure S4. Flowcytometric analysis of IL-13Rα2 and HER2 expression in the glioma cell line U373-GBM upon encounter of HER2 CAR T cells, IL-13Rα2 CAR T cells, their pooled product and biCAR T cells at 1, 3, 7, and 14 days of coculture.Figure S5. Surface expression of HER2 and IL-13Rα2 on biCAR T cells (a) and their CD4/CD8 phenotype (b).Figure S6. Optimization of experimental set up for functional testing of biCAR T cellsFigure S7. A standard 4-hour 51Cr release cytotoxicity assay of HER2 CAR T cells, IL-13Rα2 CAR T cells, their pooled product (HIL) and biCAR T cells against the HER2, IL-13Rα2-positive glioma line U373-GBM.Figure S8. Transduction efficiencies unispecifc CAR T cells and of biCAR T cells generated from two GBM patients, UPN-1, and UPN-10.Figure S9. An extended standard 4 hour 51Cr release cytotoxicity assay of primay GBM patient effectors, namely: HER2 CAR T cells, IL-13Rα2 CAR T cells, their pooled product (HIL), and biCAR T cells against autologous glioma cells.
Acknowledgments
We thank Malcolm K. Brenner for helpful discussions and advice, and for critical review of the manuscript. M.H is a Hyundai scholar and was supported by Hyundai Hope on Wheels and Alex Lemonade Stand Foundation. K.K.H.C is a Melnick scholar and was supported by NIH grants 5T32HL092332 and 5T32GM007330 and H.E.H by a Dan L Duncan Chair. This work was also supported by the Alliance for Cancer Gene Therapy (ACGT), the Dana Foundation, the Clayton Foundation for Research, and the James S McDonnell Foundation. We also appreciate the support of the Cell and Vector Production at Baylor College of Medicine shared NIH resource in P30CA125123. The Characterized Cell Line Core facility at MD Anderson Cancer Center is funded by NCI grant CA16672. The Center for Cell and Gene Therapy has research collaboration with Celgene and bluebird bio. K.K.H.C, V.S.B, S.K, S.G,and N.A have patent application in the field of T-cell and gene-modified T-cell Therapy for cancer.
Supplementary Material
Supplementary Information
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phase I Study of Cellular Immunotherapy for Recurrent/Refractory Malignant Glioma Using Intratumoral Infusions of GRm13Z40-2, An Allogeneic CD8+ Cytolitic T-Cell Line Genetically Modified to Express the IL 13-Zetakine and HyTK and to be Resistant to Glucocorticoids, in Combination With Interleukin-2. 1-11-2013.
- Study of Administration of CMV-specific Cytotoxic T Lymphocytes Expressing CAR Targeting HER2 in Patients With GBM (HERT-GBM). 1-6-2011.
- White Blood Cells With Anti-EGFR-III for Malignant Gliomas. 1-11-0013.
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S, et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res. 2007;13 2 Pt 1:566–575. doi: 10.1158/1078-0432.CCR-06-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Diehn M, Watson N, Bollen AW, Aldape KD, Nicholas MK, et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci USA. 2005;102:5814–5819. doi: 10.1073/pnas.0402870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. 2008;88:245–250. doi: 10.1007/s11060-008-9566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64:4980–4986. doi: 10.1158/0008-5472.CAN-03-3504. [DOI] [PubMed] [Google Scholar]
- Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67:7983–7986. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–551. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- Wykosky J, Gibo DM, Stanton C, Debinski W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin Cancer Res. 2008;14:199–208. doi: 10.1158/1078-0432.CCR-07-1990. [DOI] [PubMed] [Google Scholar]
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell. 1992;71:649–662. doi: 10.1016/0092-8674(92)90598-7. [DOI] [PubMed] [Google Scholar]
- Isakov N, Wange RL, Burgess WH, Watts JD, Aebersold R, Samelson LE. ZAP-70 binding specificity to T cell receptor tyrosine-based activation motifs: the tandem SH2 domains of ZAP-70 bind distinct tyrosine-based activation motifs with varying affinity. J Exp Med. 1995;181:375–380. doi: 10.1084/jem.181.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Ratnayake M, Savoldo B, Perlaky L, Dotti G, Wels WS, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–5964. doi: 10.1158/0008-5472.CAN-06-4309. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther. 2009;17:1779–1787. doi: 10.1038/mt.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainusso N, Brawley VS, Ghazi A, Hicks MJ, Gottschalk S, Rosen JM, et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012;19:212–217. doi: 10.1038/cgt.2011.83. [DOI] [PubMed] [Google Scholar]
- Guess HA, Thomas JE. A rapidly converging algorithm for exact binomial confidence intervals about the relative risk in follow-up studies with stratified incidence-density data. Epidemiology. 1990;1:75–77. doi: 10.1097/00001648-199001000-00016. [DOI] [PubMed] [Google Scholar]
- Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–637. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. 2000;7:35–42. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslop HE, Rooney CM. Adoptive cellular immunotherapy for EBV lymphoproliferative disease. Immunol Rev. 1997;157:217–222. doi: 10.1111/j.1600-065x.1997.tb00984.x. [DOI] [PubMed] [Google Scholar]
- Hoyos V, Savoldo B, Dotti G. Genetic modification of human T lymphocytes for the treatment of hematologic malignancies. Haematologica. 2012;97:1622–1631. doi: 10.3324/haematol.2012.064303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther. 2011;19:2133–2143. doi: 10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuri PV, Wilson MH, Maiti SN, Mi T, Singh H, Olivares S, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 2010;21:427–437. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. 2007;110:2793–2802. doi: 10.1182/blood-2007-02-072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straathof KC, Spencer DM, Sutton RE, Rooney CM. Suicide genes as safety switches in T lymphocytes. Cytotherapy. 2003;5:227–230. doi: 10.1080/14653240310001497. [DOI] [PubMed] [Google Scholar]
- Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhankumar AB, Mintz A, Debinski W. Interleukin 13 mutants of enhanced avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia. 2004;6:15–22. doi: 10.1016/s1476-5586(04)80049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. 2012;18:5949–s5960. doi: 10.1158/1078-0432.CCR-12-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information