Upregulated glial cell line-derived neurotrophic factor through cyclooxygenase-2 activation in the muscle is required for mechanical hyperalgesia after exercise in rats (original) (raw)
Abstract
Unaccustomed strenuous exercise that includes lengthening contraction (LC) often causes delayed onset muscle soreness (DOMS), characterised as muscular mechanical hyperalgesia. Previously we reported that a bradykinin-like substance released from the muscle during exercise plays a pivotal role in triggering the process of muscular mechanical hyperalgesia by upregulating nerve growth factor (NGF) in exercised muscle of rats. We show here that cyclooxygenase (COX)-2 and glial cell line-derived neurotrophic factor (GDNF) are also involved in DOMS. COX-2 inhibitors but not COX-1 inhibitors given orally before LC completely suppressed the development of DOMS, but when given 2 days after LC they failed to reverse the mechanical hyperalgesia. COX-2 mRNA and protein in exercised muscle increased six- to 13-fold in mRNA and 1.7–2-fold in protein 0–12 h after LC. COX-2 inhibitors did not suppress NGF upregulation after LC. Instead, we found GDNF mRNA was upregulated seven- to eight-fold in the exercised muscle 12 h–1 day after LC and blocked by pretreatment of COX-2 inhibitors. In situ hybridisation studies revealed that both COX-2 and GDNF mRNA signals increased at the periphery of skeletal muscle cells 12 h after LC. The accumulation of COX-2 mRNA signals was also observed in small blood vessels. Intramuscular injection of anti-GDNF antibody 2 days after LC partly reversed DOMS. Based on these findings, we conclude that GDNF upregulation through COX-2 activation is essential to mechanical hyperalgesia after exercise.
Key points
- Unaccustomed strenuous exercise that includes lengthening contraction often causes delayed onset muscle soreness (DOMS), characterised as muscular mechanical hyperalgesia.
- It has been reported that bradykinin triggers upregulation of nerve growth factor in exercised muscle, sensitizing nociceptors and resulting in DOMS, but additional mechanism(s) may be involved.
- We showed that pretreatment with cyclooxygenase (COX)-2 inhibitors completely suppressed the development of DOMS, but treatment 2 days after lengthening contraction failed to reverse existing mechanical hyperalgesia.
- We demonstrated that COX-2 induced upregulation of glial cell line-derived neurotrophic factor (GDNF) and that intramuscularly injected anti-GDNF antibody reduced muscle mechanical hyperalgesia after exercise.
- These results suggest that upregulation of GDNF through COX-2 activation is essential to mechanical hyperalgesia after exercise, and is another pathway alongside the bradykinin–nerve growth factor pathway that is involved in DOMS development.
Introduction
Delayed onset muscle soreness (DOMS) is described as an unpleasant sensation or pain after unaccustomed strenuous exercise (Armstrong, 1984). It is characterised as tenderness and movement-related pain, i.e. mechanical hyperalgesia. It typically appears after a period of no pain (about 1 day) and usually reaches a peak 1–2 days after exercise in humans and rats (Armstrong, 1984; Taguchi et al. 2005). DOMS can be induced by lengthening contraction (LC) but not by either shortening contraction (SC) or stretching.
DOMS itself is a common and rather unremarkable event in daily life. However, daily repetition of LC causes chronic muscular mechanical hyperalgesia in rats (Hayashi et al. 2011). In developed countries, increases in the proportion of aged persons and the obesity rate are becoming serious problems. Exercise is necessary to maintain the health of the elderly and to prevent obesity, but muscular mechanical hyperalgesia with unaccustomed exercise may hinder people who are not familiar with experiencing DOMS. Even for athletes DOMS is a problem when they start a new season or learn new techniques because it may alter contraction sequence and recruitment patterns of muscles (Cheung et al. 2003), causing changed movements as suggested by Hodges and Tucker (2011). Changed movements may pose unaccustomed stress on musculoskeletal structure and these may increase the risk of injury. Therefore, uncovering the mechanism of DOMS may lead to ways to prevent it, and thus help people to continue exercising without muscle hyperalgesia.
Various substances, including lactic acid, creatine kinase and oxygen radicals, are elevated after exercise (Smith, 1991; Cheung et al. 2003). Among them, several inflammatory cytokines, such as tumour necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β and some neurotrophins have been suggested to play roles in muscular mechanical hyperalgesia (Schafers et al. 2003; Hoheisel et al. 2005; Loram et al. 2007). Previously we reported that a bradykinin-like substance (BK, or precisely, Arg-BK in rats, Boix et al. 2002) released during exercise induced upregulation of nerve growth factor (NGF) in exercised muscle, and that NGF maintained muscular mechanical hyperalgesia (Murase et al. 2010).
Some studies have found that NSAIDs are able to suppress DOMS in human subjects treated before (prophylactic) and often after exercise (Tokmakidis et al. 2003; Rahnama et al. 2005), but studies on humans are often limited to the subjective evaluation of soreness level. It has been reported that stretching of myoblasts or myocytes induces cyclooxygenase (COX)-2 shortly after stretch and leads to their proliferation and cell growth (Vandenburgh et al. 1990; Otis et al. 2005).
Nociceptive neurons are commonly divided into two classes based on their neurotrophic factor dependence (Molliver et al. 1997). One group expresses the tyrosine kinase A (Trk A) receptor, and their survival depends on target tissue derived NGF. The other expresses the receptor tyrosine kinase Ret, the common signalling component for the receptor of glial cell line-derived neurotrophic factor (GDNF) (Buj-Bello et al. 1995; Molliver et al. 1997). Similar to NGF, GDNF is responsible for the growth and maintenance of a group of sensory neurones in fetal life (Buj-Bello et al. 1995), and it is involved in several pain states in adult animals. However, its effects on pain are inconsistent: it works as an analgesic in neuropathic pain (Boucher et al. 2000; Takasu et al. 2011), whereas it has a hyperalgesic effect in response to heat in an inflammation model (Malin & Davis, 2008). It is recently reported that intramuscular injection of GDNF decreased the mechanical threshold in rats (Mizumura et al. 2011; Hendrich et al. 2012). Therefore, we hypothesised GDNF works on nociceptive neurons, which express Ret and is involved in the muscular mechanical hyperalgesia in DOMS.
In the present experiment we examined the involvement of COX-2 (thus prostaglandins) and GDNF in mechanical hyperalgesia after exercise. We found that COX-2 was upregulated shortly after exercise, triggered upregulation of GDNF in the muscle with a delay of 12 h, and intramuscular injection of anti-GDNF antibody reversed existing mechanical hyperalgesia 2 days after LC. We also showed that BK is involved in upregulation of not only NGF (Murase et al. 2010) but also COX-2 and GDNF 12 h after LC.
Methods
Experimental animals
Male Sprague–Dawley rats (SLC Inc., Hamamatsu, Japan) weighing 250 g (7 weeks) at the beginning of the experiments were used in this study. The animals were kept two to three per cage under a 12 h light/dark cycle (light between 07.00 h and 19.00 h) in an air-conditioned room (22–24°C). They had food and water ad libitum throughout the experiment. All experimental procedures were conducted according to the Regulations for Animal Experiments in Nagoya University and Chubu University, and the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions in Japan.
Lengthening contraction, shortening contraction and stretching of the muscle
Lengthening and shortening contractions
LC was applied as reported previously (Taguchi et al. 2005). Briefly, on day 0 the animals were anaesthetised with sodium pentobarbital (50 mg kg−1, i.p.). LC was induced in the lower hind leg extensors [mainly the extensor digitorum longus (EDL) muscle] by electrical stimulation of the common peroneal nerve through a pair of needle electrodes inserted near the nerve. Electrical stimulation with the following parameters was applied for 1 s: current strength of three times the twitch threshold (<100 μA), and frequency of 50 Hz with pulse duration of 1 ms. The ankle joint was plantar flexed in synchrony with muscle contraction with use of a linearised servomotor (CPL28T08B-06C2T, Oriental Motor Co. Ltd., Tokyo, Japan) to stretch the lower hind leg extensors, and then returned to the starting position over 3 s. This cycle was repeated for a total of 500 times. The method of SC was similar to that for LC except for the absence of muscle stretching (i.e. the ankle joint was not fixed while the muscle was contracted). After the rats recovered from anaesthesia following exercise, they behaved, ate and drank normally.
Stretching the muscle
Without muscle contraction, the linearised servomotor could not stretch EDL muscle sufficiently because the entire hind leg was moved with the pulling of the ankle. To ensure intensive stretching similar to that during LC, direct stretching of the EDL muscle was performed as follows. A small incision (about 5 mm) was made on the dorsum of the right hindpaw, and the distal tendons of the EDL muscle were separated from the surrounding connective tissue. Each muscle was about 28 mm long from the proximal to distal tendon. The distal tendon was manually pulled about 2 mm (about 7% of the total muscle length) for 1 s while the knee was fixed by two acrylic bars on either side, and then returned to the starting position over 3 s. This cycle was repeated 500 times, the same as in LC. The tendons of the contralateral EDL muscle were also exposed without stretching, and served as a control.
Withdrawal threshold measurement
A Randall–Selitto analgesiometer (Ugo Basile, Comerio, Italy) equipped with a homemade probe of tip diameter 2.6 mm was used to measure the withdrawal threshold of the extensors of the lower hind leg (EDL muscle included). The use of probes with tip diameters of 2.6 mm and larger allows measurement of the muscle mechanical nociceptive threshold (Takahashi et al. 2005; Nasu et al. 2010). The animals were restrained around the trunk with a towel to calm them, and treated gently during the experiments. The probe was applied to the belly of the lower hind leg extensor muscles through shaved skin. The speed of force applied was set at 157 mN s−1 and there was a 2450 mN cut-off loading to avoid damaging the tissue. The intensity of force causing an escape reaction was defined as the withdrawal threshold. Each series of experiments was done at almost the same period of time during the day to avoid circadian fluctuations. Training sessions were carried out for at least four consecutive days. Measurements were performed 10 times at intervals of several minutes for the experiment of COX inhibitors or seven times for the test of anti-GDNF antibody, and the mean value of the latter five trials was taken as the threshold. Experimenters were blinded during measurements to which rats received which treatment, drug or vehicle.
Drugs
Cyclooxygenase-1 and -2 inhibitors
We used a highly selective COX-1 inhibitor, SC560 (10 mg kg−1; Sigma, St Louis, MO, USA). This dose of SC560 given orally inhibits COX-1-derived platelet thromboxane B2, gastric PGE2 and dermal PGE2 production (Smith et al. 1998). We also used another type of COX-1 inhibitor, ketorolac (10 mg kg−1; Sigma). This dose of ketorolac inhibits formalin-induced nociceptive behaviours (Padi et al. 2006). Celecoxib (2.5–10 mg kg−1, kindly synthesised by Nippon Chemiphar Co., Ltd, Tokyo, Japan) and zaltoprofen (2.5–10 mg kg−1, kind gift from Nippon Chemiphar Co., Ltd) were used as COX-2 inhibitors (Smith et al. 1998; Tegeder et al. 2001; Muratani et al. 2005). Celecoxib, SC560 and zaltoprofen were suspended in 1% methylcellulose. These drugs (0.5 ml) were administered orally either 1 h before exercise (pretreatment) or 1 h before the mechanical withdrawal threshold was measured on the second day after exercise. Ketorolac was dissolved in 0.5 ml of 0.01 m PBS and intraperitoneally injected either 30 min before exercise (pretreatment) or 30 min before measurement of the mechanical withdrawal threshold on the second day after exercise. The time elapsed between administration of a drug and measurement, and dosages were chosen based on previous reports (Tegeder et al. 2001; Muratani et al. 2005; Padi et al. 2006). An additional measurement was done 4.5 h before ketorolac injection on the second day after exercise. Groups that received vehicle (PBS or methylcellulose) were also included.
The percentage suppression of the decrease in muscle mechanical withdrawal threshold was calculated as follows. Differences in withdrawal threshold 2 days after exercise from the threshold on –1 day in the vehicle group (Δthreshold-veh) or drug group (Δthreshold-drug) were calculated, and differences (suppression) between Δthreshold-drug and Δthreshold-veh were normalised with Δthreshold-veh and expressed as percentages.
Bradykinin antagonists
HOE 140 (B2 antagonist, 0.1 mg kg−1, Sigma) was dissolved in saline and subcutaneously injected into the skin of the back 30 min before exercise. This antagonist is receptor specific (Hock et al. 1991), and the dosage used is sufficient to antagonise BK in a postoperative pain model (Muratani et al. 2005) and DOMS model (Murase et al. 2010).
Anti-GDNF antibody (5, 10 μg, R&D Systems, Minneapolis, MN, USA) was injected into the belly of the EDL muscle of the right hindlimb. The exact placement of the tip of the injection needle was assured by dorsiflexion of the middle three toes of the hindpaw in response to electrical stimulation through the electrode attached to the injection needle.
A volume of 5 μl of anti-GDNF antibody was injected under halothane anaesthesia (2%, Takeda Chemical Industries, Ltd., Osaka, Japan). An equivalent volume of normal mouse IgG (10 μg) in PBS was injected into the right EDL muscle in the control group. These substances were injected 2 days after exercise. The withdrawal threshold was measured twice on the second day after LC, before and 3 h after antibody/normal IgG injection.
RT-PCR
The EDL muscle was removed under anaesthesia immediately (0 h) and 3, 6, 12 h and 1, 2, 3 and 5 days after LC. To examine whether there was any interaction among BK, COX-2, NGF, GDNF, IL-6 and TNF-α, additional samples (_n_= 4, each) were taken after the administration of HOE 140 (0.1 mg kg−1, s.c.), celecoxib and zaltoprofen (10 mg kg−1, each). These drugs were administered before LC, and the EDL muscle was removed 12 h after LC, the time when both NGF and GDNF upregulation was reported to be at the highest without drug treatment (Murase et al, 2010). To compare effects of LC, SC and stretch on COX-2 and GDNF mRNA upregulation, EDL muscle samples were taken immediately and at 12 h after each treatment. Tissue samples from individual rats were weighed and about 30 mg of each sample was homogenised in lysis buffer and total RNA was extracted with RNeasy® Fibrous Tissue Mini Kit® (QIAGEN, Valencia, CA, USA). The amount of total RNA was evaluated by spectrophotometry (Genequantpro; Amersham Biosciences, Buckinghamshire, UK). One microgram samples of RNA preparations were transcribed to cDNA with M-MLV Reverse Transcriptase (Promega, Madison, WI, USA) at 42°C for 50 min. Five microlitre aliquots of the resultant solution of RT reaction were taken into each PCR tube and amplified with primers (Table 1). The data were measured at the exponential phases of the PCR reactions. PCR products were resolved by electrophoresis through a 1.5% agarose gel. The amplified DNA fragments were detected by ethidium bromide staining. The ethidium bromide stained bands were visualised by UV light and the optical density of the bands was analysed with Image J software (free software developed by the National Institutes of Health, USA). The values were normalised against glyceraldehyde-3-phosphate dehydrogenase for analysis.
Table 1.
Sequence of primers and cycle conditions for RT-PCR
| Gene product | Sequence of primers | Cycle conditions |
|---|---|---|
| Cyclooxygenase-2 | 5′-tgcagagttgaaagccctc-3′ | 30 s at 94°C, 30 s at 55°C, |
| 5′-cacaggaatcttcacaaatgg-3′ | 1 min at 72°C, 33 cycles | |
| Glial cell line-derived neurotrophic factor | 5′-cggacgggactctaagatga-3′ | 30 s at 94°C, 30 s at 55°C, |
| 5′-cgtcatcaaactggtcagga-3′ | 1 min at 72°C, 33 cycles | |
| Artemin | 5′-ccgacgagctgatacgtt-3′ | 1 min at 94°C, 1 min at 58°C, |
| 5′-ggtgctgttcacgtccat-3′ | 1 min at 72°C, 37 cycles | |
| Neurturin | 5′-cagctccctgctatctgtctg-3′ | 1 min at 94°C, 1 min at 61°C, |
| 5′-cagcccagggagaaagttctc-3′′ | 1 min at 72°C, 36 cycles | |
| Persephin | 5′-tgtcacaatggctgcaggaagactt-3′ | 1 min at 94°C, 1 min at 60°C, |
| 5′-agctcagccactggtagggtcagg-3′ | 1 min at 72°C, 36 cycles | |
| Nerve growth factor | 5′-ttcggacactctggatttagact-3′ | 30 s at 94°C, 30 s at 55°C, |
| 5′-gatttggggctcggcacttg-3′ | 1 min at 72°C, 33 cycles | |
| Interleukin-6 | 5′-atgttgttgacagccactgc-3′ | 30 s at 94°C, 30 s at 55°C, |
| 5′-acagtgcatcatcgctgttc-3′ | 1 min at 72°C, 35 cycles | |
| Tumour necrosis factor-α | 5′-atgtggaactggcagaggag-3′ | 30 s at 94°C, 30 s at 65°C, |
| 5′-ggccatggaactgatgagag-3′ | 1 min at 72°C, 35 cycles | |
| Glyceraldehyde-3-phosphate dehydrogenase | 5′-gtgaaggtcggtgtcaacggattt-3′ | 30 s at 94°C, 30 s at 55°C, |
| 5′-cacagtcttctgagtggcagtgat-3′ | 1 min at 72°C, 21 cycles |
Western blot analysis
LC-loaded EDL muscle tissue was homogenised in lysis buffer [10 mm Tris, pH 7.5, 10 mm NaCl, 0.5% Triton-X 100, 0.02% NaN3; before use, the buffer received a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA)], incubated for 30 min on ice and centrifuged for 15 min at 15,000 rpm (20,630 g). Twenty micrograms of protein per lane were separated discontinuously on sodium dodecyl sulphate polyacrylamide gels (10%) and were transferred to a polyvinyl difluoride membrane (Millipore, Eschborn, Germany). After blockade of non-specific binding sites with 5% skim milk in tris buffered saline with tween 20, membranes were incubated with anti-COX-2 antibodies (1:400; Cayman Chemical, Ann Arbor, MI, USA) at 4°C overnight, then followed by peroxidase-conjugated antirabbit IgG (1:2000; Amersham Pharmacia Biotech, Uppsala, Sweden) as a secondary antibody. Blots were developed using luminol-enhanced chemiluminescence (SuperSignal® West Pico Chemiluminescent Substrate; Pierce Biotechnology, Inc. Rockford, IL, USA). Digital images were captured using a chemiluminescent imaging system (Cool Scan Light Capture, ATTO, Tokyo, Japan). The band densities were determined with Image J. The COX values were normalised against glyceraldehyde-3-phosphate dehydrogenase for analysis.
In situ hybridisation histochemistry
To examine which cells produce COX-2 and GDNF, EDL muscle was removed immediately after LC for COX-2 and 12 h after LC for GDNF mRNA. These muscle samples were immediately frozen with liquid nitrogen and sectioned (16 μm thick) with a cryostat, thaw-mounted on to Vectabond (Vector Laboratories, Burlingame, CA, USA) coated slides and stored at –80°C until use. The procedure for in situ hybridisation histochemistry (ISHH) was basically the same as that used in a previous study (Kobayashi et al. 2008). Briefly, the rat COX-2 and GDNF cRNA probe corresponding to nucleotides of accession numbers AF233596, 1802–2456 and L15305, 33–378 were prepared, respectively. The sections were treated with 10 μg ml−1 proteinase K in 50 mm Tris–HCl and 5 mm EDTA for 5 min and acetylated with 0.25% acetic anhydride in 0.1 m triethanolamine, and rinsed with PB and dehydrated through an ascending ethanol series. A 35S-labelled RNA probe (5 × 106 cpm ml−1) in hybridisation buffer was then placed on these sections overnight at 55°C. Hybridised sections were rinsed in 5× standard saline citrate and 5 mm dithiothreitol for 30 min at 62°C, washed in high-stringency buffer for 30 min at 62°C, and treated with 2 μg ml−1 RNase A for 30 min at 37°C. Sections were rinsed, dehydrated in an ascending ethanol series, and air dried. For autoradiography, the sections were coated with NTB emulsion (Eastman Kodak, Rochester, NY, USA), diluted 1:1 with distilled water at 45°C, and exposed for 11 days (COX-2) and 25 days (GDNF) in light-tight boxes at 4°C. After development in D19 (Eastman Kodak) and fixation in 24% sodium thiosulfate, the sections were rinsed in distilled water, stained with haematoxylin–eosin, dehydrated in a graded ethanol series, cleared in xylene and coverslipped.
Statistical analysis
The data of the Randall–Selitto test are presented as means ±s.e.m. Two-way ANOVA with repeated measures was used (factors: drug and time), and post hoc comparisons were performed by Bonferroni's test when a significant interaction effect or drug effect was evident from the two-way ANOVA, compared with the threshold on –1 day or pretreatment. Change in percentage suppression was examined with one-way ANOVA followed by Bonferroni's multiple comparison test, compared with the control (vehicle) group. The change in mRNA and protein was examined with one-way ANOVA followed by Bonferroni's multiple comparison test. The effects of LC, SC and stretch on the expression of COX-2 mRNA were analysed with two-way ANOVA (factors: contraction pattern and time) followed by Bonferroni's test. The effect of HOE140 on COX-2 expression was examined with an unpaired t test. P < 0.05 was considered to indicate a significant change.
Results
Effects of cyclooygenase-2 and -1 inhibitors on delayed-onset muscle soreness
We studied the effects of COX2 inhibitors (celecoxib and zaltoprofen) on mechanical hyperalgesia after exercise. There were significant effects in drug, time and their interaction (_F_2,70= 7.895, P < 0.01; _F_5,70= 4.671, P < 0.001; _F_10,70= 3.346, P < 0.001). By post hoc analysis, we confirmed that exercise (LC) significantly decreased the mechanical withdrawal threshold 1–3 days after LC compared with –1 day in control animals that received vehicle (P < 0.05–0.001, Fig. 1_A_). Statistical analysis also showed differences between vehicle and celecoxib groups (P < 0.05), and vehicle and zaltoprofen groups (P < 0.01, post hoc analysis by Bonferroni's test for both). In both drug groups there was no significant change in withdrawal threshold after LC compared with –1 day (Bonferroni's multiple comparison test). This means development of mechanical hyperalgesia was completely blocked by application of these inhibitors before exercise. Smaller dosages (5 or 2.5 mg kg−1) of celecoxib failed to suppress the generation of mechanical hyperalgesia (Fig. 1_B_). Five mg kg−1 of zaltoprofen, administered before LC also suppressed generation of DOMS, but the smallest dosage (2.5 mg kg−1) had no effect (Fig. 1_B_). In contrast to pretreatment with COX-2 inhibitors, high doses of zaltoprofen (10 mg kg−1) and celecoxib (10 mg kg−1) applied on the second day, when the mechanical hyperalgesia is usually at its peak, failed to reverse mechanical hyperalgesia (Fig. 1_B_, 2 days after LC).
Figure 1. Two selective cyclooxygenase (COX)-2 inhibitors, celecoxib and zaltoprofen, administered before exercise blocked development of muscle mechanical hyperalgesia.
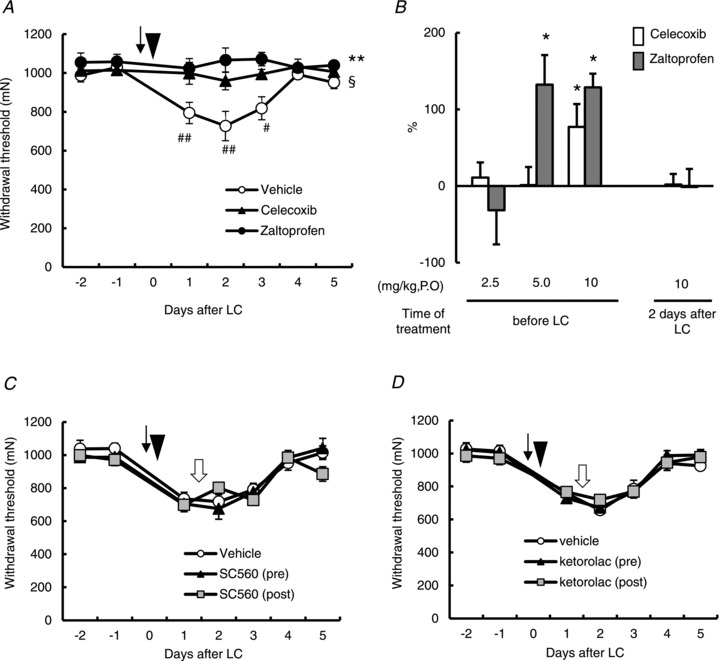
A, change in the mechanical withdrawal threshold of exercised muscle. COX-2 inhibitors, celecoxib (10 mg kg−1, p.o., triangle) and zaltoprofen (10 mg kg−1, p.o., solid circle) were administered 1 h before lengthening contraction (LC). The arrow and arrowhead indicate the time points for administration and LC loading (on day 0), respectively. Means ±s.e.m. (_n_= 5 for vehicle group and n_= 6 for other groups). §_P < 0.05 vehicle vs. celecoxib group and **P < 0.01 vehicle vs. zaltoprofen group. ## P < 0.01, #P < 0.05 compared with –1 day. B, percentage suppression of mechanical hyperalgesia (decrease of the withdrawal threshold), see Materials and methods for calculation; 100% suppression corresponds to no mechanical hyperalgesia. Celecoxib, open column; zaltoprofen, grey column. _n_= 6, each. *P < 0.05 compared with the vehicle group. C and D, effects of COX-1 inhibitors, ketorolac (10 mg kg−1, i.p., C) and SC560 (10 mg kg−1, p.o., D). Black arrow, white arrow and arrowhead indicate the time points for antagonist administration (1 h before LC, 2 days after LC) and LC loading (on day 0), respectively.
A COX-1 inhibitor, SC560 (10 mg kg−1, p.o.) administered before and after LC failed to block the development of mechanical hyperalgesia and to reverse the existing mechanical hyperalgesia, respectively (Fig. 1_C_, significant time effect, _F_5,125= 39.788, P < 0.001, but no significant treatment effect, _F_2,125= 0.531, _P_ > 0.05). Another COX-1 inhibitor, ketorolac (10 mg kg−1, i.p.), also failed to block the development of DOMS or to reverse the existing mechanical hyperalgesia (Fig. 1_D_, significant time effect, _F_5,80= 55.835, P < 0.001, but no significant treatment effect _F_2,80= 0.118, _P_ > 0.05).
It is reported that several cytokines and neurotrophins increase after exercise and might be involved in muscular mechanical hyperalgesia. We examined whether COX-2 inhibitors affected mRNA expression of NGF, IL-6 and TNF-α, which increased 12 h after LC (Murase et al. 2010). COX-2 inhibitors had no effect on upregulation of these mRNAs 12 h after LC (Fig. 2).
Figure 2. Expression of NGF, IL-6 and TNF-α 12 h after LC was not modified by pretreatment with COX-2 inhibitors.
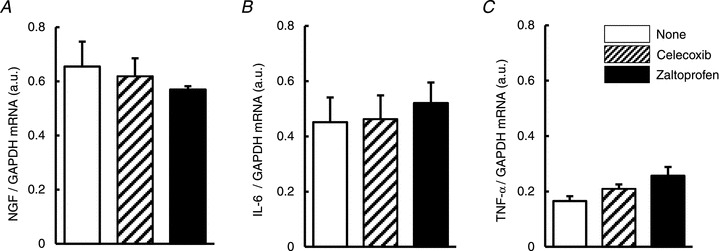
Expression of NGF (A), IL-6 (B) and TNF-α (C) mRNA at 12 h after LC were measured with or without treatment with COX-2 inhibitors before LC and normalised with GAPDH mRNA. Open column: no treatment, hatched column: celecoxib (10 mg kg−1) treatment, solid column: zaltoprofen (10 mg kg−1) treatment. _n_= 4, each. IL, interleukin; LC, lengthening contraction; NGF, nerve growth factor; TNF, tumour necrosis factor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The time course of cyclooygenase-2 and glial cell line-derived neurotrophic factor upregulation
In the previous section, we observed that suppression of COX-2 activity by administration of celecoxib or zaltoprofen before exercise blocked development of mechanical hyperalgesia after exercise. Because COX-2 expression in the control muscle is usually low, COX-2 must have been induced during or shortly after the exercise. Therefore, we next examined the change of COX-2 expression in the muscle after exercise. COX-2 mRNA in the exercised EDL muscle significantly increased immediately after LC (0 h in Fig. 3_A_, P < 0.001 compared with the control, one-way ANOVA followed by Bonferroni's multiple comparison test) and remained increased up to 12 h after LC (P < 0.05–0.01). It must be noted that the highest expression was observed immediately after LC, and COX-2 mRNA was not increased 1–3 days after LC when the muscle was hyperalgesic. No increase in COX-2 mRNA was observed in the EDL muscle contralateral to the exercised muscle.
Figure 3. Upregulation of COX-2 after LC in the muscle.
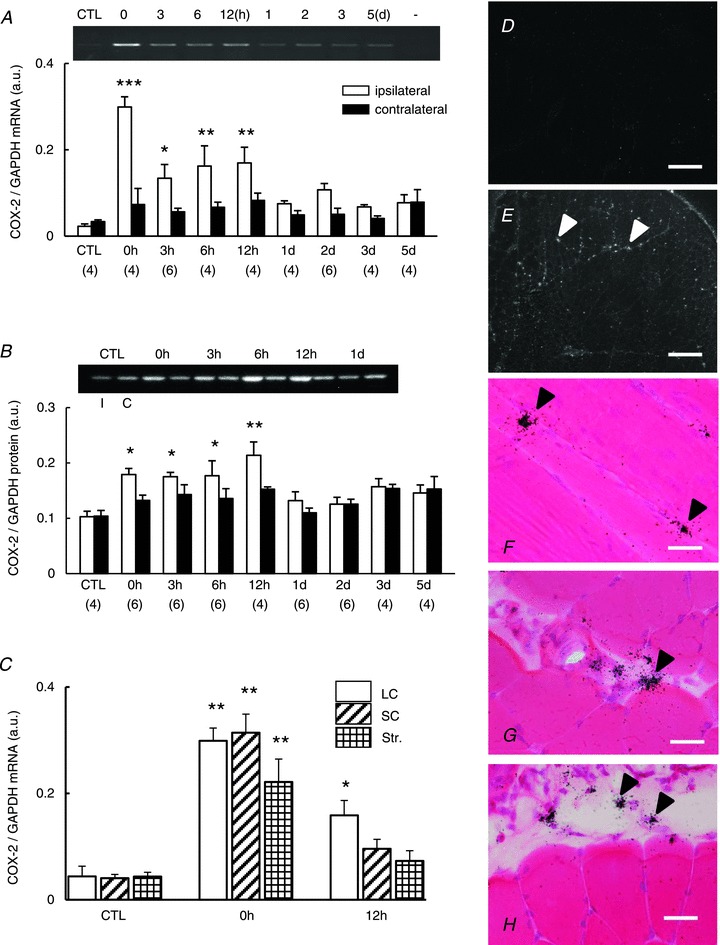
A, COX-2 mRNA level in exercised extensor digitorum longus muscle (open column) and the contralateral side (solid column). Representative RT-PCR profile of COX-2 mRNA is at the top. (−) shows the PCR result of sample without RT. B, COX-2 protein measured by Western blotting increased from 0 to 12 h after LC. Representative Western blotting profile of COX-2 protein is at the top. I, ipsilateral side; C, contralateral side. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with CTL both in A and B. Number of samples examined is in parentheses. C, changes of COX-2 mRNA expression after SC (obliquely hatched column), Str (cross-hatched column) and LC loading (LC: white column). *P < 0.05 and **P < 0.01 compared with the control groups (_n_= 5, each). _n_= 4, each for other groups. D–H, photomicrographs of ISHH. Transverse section except F (longitudinal section). D and E, Expression of COX-2 mRNA (white arrowheads in E) in extensor digitorum longus muscle immediately after LC, using dark-field photomicrographs of ISHH, was increased in the ipsilateral side (E) compared with the contralateral side (D). Scale bar, 100 μm in D and E. F–H, bright-field photomicrograph shows ISHH signals for COX-2 mRNA in the ipsilateral muscle and fascia. F, signals (black arrowheads) near the muscle cell and/or satellite cell nuclei. G, signals (black arrowheads) in the smooth muscle of the blood vessel. H, signals at the epimysium (black arrowheads). Scale bar, 10 μm in F_–_H. COX-2, cyclooxygenase-2; CTL, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ISHH, in situ hybridisation histochemistry; LC, lengthening contraction; SC, shortening contraction; Str, stretch.
COX-2 protein increased in the exercised EDL muscle in a time course quite similar to that of COX-2 mRNA; namely, it started to increase immediately after LC (0 h in Fig. 3_B_, P < 0.05 one-way ANOVA followed by Bonferroni's multiple comparison test). It remained at this higher level up to 12 h after LC. No increase in COX-2 protein was observed in the EDL muscle contralateral to the exercised muscle. We also examined whether upregulation of COX-2 was specific to LC. For this we measured COX-2 mRNA after LC, SC and stretching. There were significant effects in pattern of exercise and time (two-way ANOVA, _F_2,32= 3.912, P < 0.05; _F_2,32= 71.138, P < 0.001, respectively) but no significant interaction of these factors (_F_4,32= 1.814, _P_= 0.151). Independent of contraction pattern COX-2 mRNA was significantly increased immediately after contraction (P < 0.001 compared with the control group, Bonferroni's post hoc test) (Fig. 3_C_, 0 h). However, 12 h later, a significant COX-2 upregulation was observed only in the LC group (P < 0.05, Bonferroni's post hoc test) (Fig. 3_C_, 12 h), suggesting that COX-2 upregulation at 12 h is specifically related with DOMS.
To determine which cells produced COX-2, we performed in situ hybridisation for COX-2 mRNA in EDL muscle tissue immediately after LC. The signals of COX-2 mRNA in the ipsilateral muscle increased compared with the contralateral side (Fig. 3_D_ and E). Strong signals were observed especially around skeletal muscle cell nuclei and/or satellite cells (Fig. 3_F_). Signals were also observed in smooth muscle cells in the blood vessel (Fig. 3_G_) and fibroblasts or other connective tissue cells in the epimysium (Fig. 3_H_), and increased expression after LC was detected in vascular smooth muscle cells.
Next we examined possible involvement of GDNF expression. GDNF mRNA in exercised EDL muscle significantly increased 12 h–1 day after LC (Fig. 4_A_), later than the increase of COX-2 mRNA expression (Fig. 3_A_). The signals of GDNF mRNA visualised with in situ hybridisation in the ipsilateral EDL muscle 12 h after LC increased compared with the contralateral side (Fig. 4_B_ and C). Many signals were observed especially at the periphery and around nuclei of skeletal muscle cells and/or satellite cells (Fig. 4_D_). Expression of other GDNF family (artemin, neurturin and persephin) mRNA was also examined, but these mRNAs did not change after LC (Fig. 4_F_–H). GDNF mRNA upregulation was observed after 12 h only in the LC group, and neither in SC nor in stretch group (Fig. 4_I_).
Figure 4. Upregulation of GDNF in the muscle and its suppression by cyclooxygenase-2 inhibitors and B2 receptor antagonist.
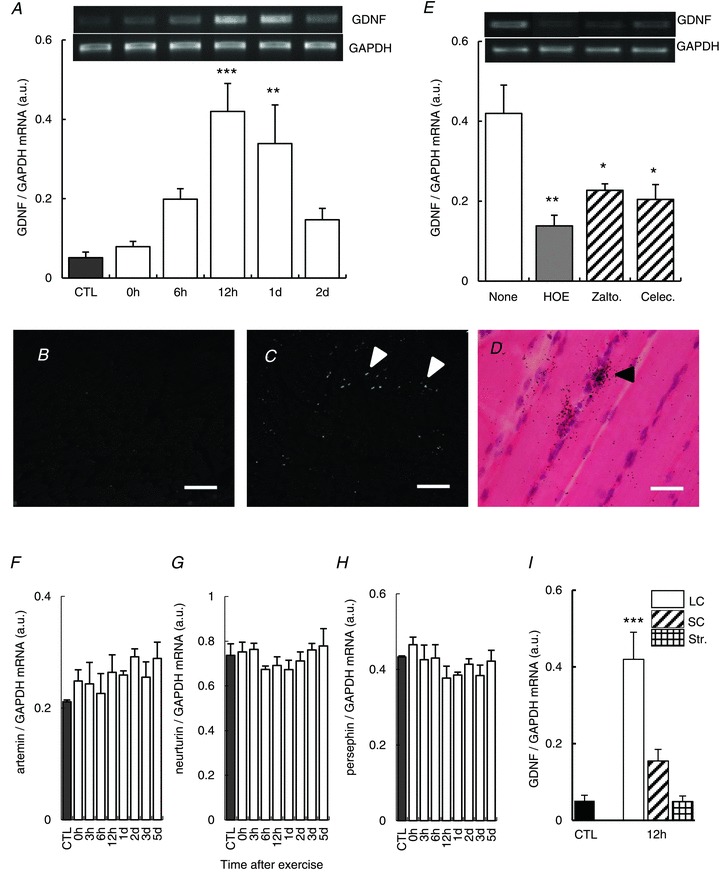
A, change of GDNF mRNA level in exercised EDL muscle. **P < 0.01 and ***P < 0.001 compared with CTL group (black column). _n_= 4. B and C, expression of GDNF mRNA (white arrowheads) in EDL muscle 12 h after LC, shown using dark-field photomicrographs of ISHH (oblique sections), was increased on the ipsilateral side (C) compared with the contralateral side (B). Scale bar, 100 μm in B and C. D, bright-field photomicrograph (longitudinal section) in larger magnification shows ISHH signals for GDNF mRNA (black arrowhead) in the ipsilateral muscle. Scale bar, 10 μm. E, effects of cyclooxygenase-2 inhibitors, zaltoprofen and celecoxib (10 mg kg−1, p.o. each, administered 1 h before LC) on GDNF mRNA 12 h after LC. Bradykinin B2 receptor antagonist, HOE 140 (0.1 mg kg−1, s.c., administered 30 min before LC) also suppressed upregulation of GDNF mRNA. *P < 0.05 and **P < 0.01 compared with the no drug group (none, white column). _n_= 4 each. F–H, expression of mRNAs of other members of GDNF family, artemin (F), neurturin (G) and persephin (H) in the EDL muscle did not change after LC. _n_= 4, each. I, neither SC (obliquely hatched column, _n_= 4) nor Str (cross-hatched column, _n_= 4) increased GDNF mRNA 12 h after treatment unlike the LC group (LC: white column, _n_= 4). ***P < 0.001 compared with the control group. CTL, control; EDL, extensor digitorum longus; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GDNF, glial cell line-derived neurotrophic factor; ISHH, in situ hybridisation histochemistry; LC, lengthening contraction; SC, shortening contraction; Str, stretch.
Relationships among cyclooygenase-2, glial cell line-derived neurotrophic factor and bradykinin
To understand the causal relationship between COX-2 and GDNF, we examined upregulation of GDNF under pretreatment with COX-2 inhibitors, zaltoprofen and celecoxib. Upregulation of GDNF mRNA 12 h after LC was suppressed by these COX-2 inhibitors administered 1 h before LC (Fig. 4_E_).
We previously reported that a B2 BK receptor antagonist, HOE140, administered 30 min before LC completely blocked the development of DOMS by suppressing NGF upregulation in exercised muscle (Murase et al. 2010). We therefore examined whether HOE140 affects GDNF and COX-2 upregulation. It was found that HOE140 (0.1 mg kg−1) administered before exercise also suppressed GDNF mRNA upregulation (Fig. 4_E_). Increases in COX-2 mRNA and protein 12 h after LC were not observed after HOE140 treatment, but rather suppressed (Fig. 5_A_ and B). However, the notable increase of COX-2 expression immediately after LC was also observed (Fig. 5_A_ and B).
Figure 5. Effects of B2 receptor antagonist on COX-2 mRNA expression after exercise.
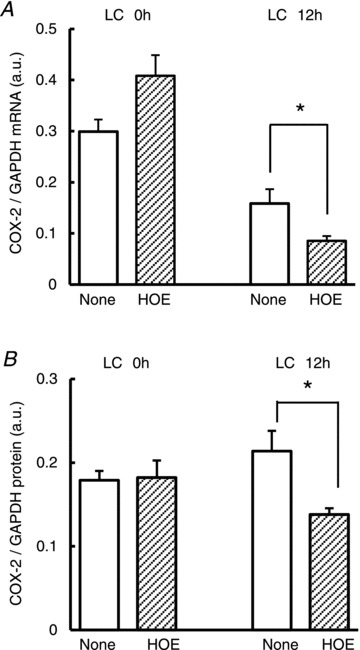
Upregulated COX-2 mRNA (A) and protein (B) 12 h after LC was suppressed by B2 receptor antagonist, HOE 140 (0.1 mg kg−1, s.c., administered 30 min before LC), but that immediately after LC (LC 0 h) was not. *P < 0.05 compared with the None group (without antagonist), unpaired t test. _n_= 4, each. COX-2, cyclooxygenase-2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LC, lengthening contraction.
Figure 6. Effects of intramuscular injection of anti-GDNF antibody.
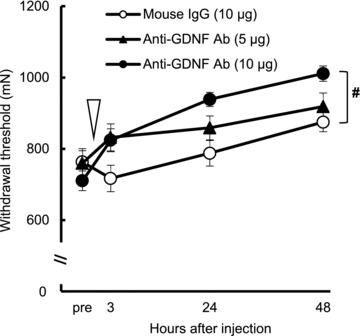
Anti-GDNF antibody (10 μg) injected into the extensor digitorum longus muscle 2 days after lengthening contraction partially reversed the mechanical hyperalgesia (significantly different from the control group (IgG group), #P < 0.05). Time point of treatment is indicated with an open arrowhead. _n_= 6 for the control and anti-GDNF Ab (5 μg) group and _n_= 8 for anti-GDNF Ab (10 μg) group. Ab, antibody; GDNF, glial cell line-derived neurotrophic factor.
Involvement of glial cell line-derived neurotrophic factor in the muscle mechanical hyperalgesia after lengthening contraction
To confirm the involvement of GDNF in muscular mechanical hyperalgesia after LC, we injected anti-GDNF antibody (5 and 10 μg) intramuscularly on the second day. Before injection the mechanical withdrawal threshold was 911 ± 32, 1018 ± 42 and 1019 ± 43 mN in normal mouse IgG and anti-GDNF antibody (5 μg and 10 μg) groups, respectively (no significant difference). After injection, there were statistically significant effects in time, treatment and their interaction (_F_3,51= 27.146, P < 0.001; _F_2,51= 4.438, _P_ < 0.05; _F_6,51= 3.678, _P_ < 0.01, respectively). Anti-GDNF antibody 10 μg intramuscular injection induced a significant recovery in the withdrawal threshold (_P_ < 0.05) compared with the control group. In another experiment we examined the effect of anti-GDNF antibody 10 μg on the mechanical withdrawal thresholds in normal rats, and found no effect (before 1100 ± 20 mN and 1 day after injection 1101 ± 81 mN, _n_= 6, _P_ > 0.05). We also found that the antibody (10 μg) administered intraperitoneally 2 days after LC did not attenuate muscle mechanical hyperalgesia [no significant difference in the withdrawal threshold before (742 ± 34 mN) and after (777 ± 39 mN) injection, _n_= 5, P > 0.05], suggesting that GDNF at this dosage does not work systemically.
Discussion
Upregulation of cyclooxygenase-2 triggers processes leading to delayed-onset muscle soreness
The present experiment demonstrated that COX-2 inhibitors, but not COX-1 inhibitors, applied before exercise suppressed the generation of mechanical hyperalgesia after LC. The absence of effects of COX-1 inhibitors was not due to their low dosage as formalin-evoked nociceptive behaviour was suppressed with this same dosage (Tegeder et al. 2001; Padi et al. 2006). Neither COX-1 nor COX-2 inhibitors failed to reverse already generated mechanical hyperalgesia when applied 2 days after LC. These observations are in agreement with previous reports that prophylactic administration of NSAIDs is effective in reducing DOMS (Tokmakidis et al. 2003; Rahnama et al. 2005). Previously, we reported that NGF, IL-6 and TNF-α mRNA increased in the exercised muscle 12 h after LC, and that B2 receptor antagonist inhibited both the NGF and IL-6 increase (Murase et al. 2010). In contrast, COX-2 inhibitors did not inhibit these upregulations at all. This result suggests that COX-2 works in a different pathway from the one triggered by BK. Both COX-2 mRNA and protein increased 0 to 12 h after LC. COX-2 synthesises prostaglandins from arachidonic acid and prostaglandin increases almost immediately after COX-2 upregulation (Inoue et al. 2006). These data suggest that COX-2 (prostaglandins) works during and shortly after exercise as a trigger of the development of DOMS. This study provides a scientific basis for inhibitory effects of pre-administration of COX-2 inhibitors on DOMS.
As is well known, COX-2 is produced by mast cells and macrophages during inflammation (Tilley et al. 2001). However in our model, there is no inflammatory cell infiltration associated with necrotic muscle cells, which did not exist, either, in our model (Fujii et al, 2008). The result of ISHH demonstrated that COX-2-producing cells are skeletal muscle cells or satellite cells, and vascular smooth muscle cells and cells in the epimysium (fibroblasts and/or other connective tissue cells). The ability of myoblasts and myocytes to upregulate COX-2 after mechanical events has been reported (Vandenburgh et al. 1990; Otis et al. 2005). Thus it is clear that muscle/satellite cells can produce COX-2. Although we did not quantitatively compare COX-2 upregulation in muscle/satellite cells and vascular smooth muscle cells, the importance of COX-2 upregulation in muscle/satellite cells in DOMS is clear because GDNF upregulation triggered by COX-2 upregulation (thus prostaglandin upregulation) discussed below was observed only in muscle/satellite cells.
An immediate increase of COX-2 mRNA was induced by not only LC but also SC and stretch. However, neither stretching nor SC itself induced mechanical hyperalgesia (Murase et al. 2010). The upregulation of COX-2 at 12 h after stretching or SC was absent, unlike after LC. These results suggest that the upregulation of COX-2 at a later time point is important for the development of DOMS. Alternatively, perhaps additional mechanisms associated with LC are needed to trigger the pathway inducing DOMS. BK is just one possibility, as a B2 BK antagonist, HOE 140, blocked the later (12 h after LC) upregulation of COX-2, although it failed to block the immediate upregulation after LC. This suggests that B2 receptor activation is needed for later upregulation of COX-2.
Involvement of glial cell line-derived neurotrophic factor in delayed-onset muscle soreness
The second important observation of this study is that COX-2 induced GDNF upregulation. Upregulated GDNF was involved in the maintenance of DOMS as anti-GDNF antibody injected 2 days after LC partially but significantly reversed already generated mechanical hyperalgesia. This observation is in good agreement with the finding that the increase of GDNF mRNA after exercise showed a suitable time course (12 h–1 day after LC) for the appearance of mechanical hyperalgesia after LC. In addition SC and stretch failed to induce upregulation of GDNF. Intramuscular injection of GDNF induces muscle mechanical hyperalgesia (Mizumura et al. 2011; Hendrich et al. 2012) and decreases the cutaneous mechanical threshold in GDNF-overexpression mice (Albers et al. 2006). The significant decrease of withdrawal threshold was started 1 h after and maintained up to 1 day after intramuscular injection of GDNF (0.03 μm) (Mizumura et al. 2011). It is also reported that other members of the GDNF family, artemin and neurturin, regulate inflammatory pain or neuropathic pain (Malin et al. 2006; Yoshida et al. 2011), but in the present DOMS model, upregulation of no other GDNF family members (artemin, neurturin and persephin) was observed after LC. Thus, only GDNF is considered to be responsible for DOMS.
In situ hybridisation showed that skeletal muscle cells and/or satellite cells produced both GDNF and COX-2. It has been reported that subcultured skeletal muscle cells (C2C12 cells) can produce GDNF to help motor neurones survive (Vianney & Spitsbergen, 2011). GDNF sends signals via the tyrosine kinase RET and specific coreceptor GFRα1 (Carmillo et al. 2005). In rat DRG neurones, RET and GFRα1 are expressed in a subpopulation of both small and large diameter afferents projecting through the sciatic nerve (about 60% and 40%, respectively) (Bennett et al. 2000). Another report that showed cutaneous C fibres were sensitised to mechanical stimulation by GDNF 15 min after injection (Bogen et al. 2008), but it provided no information about whether this sensitising effect lasted longer. We recently showed that GDNF sensitised the mechanical response of Aδ fibres but not of C fibres (Mizumura et al. 2011). The mechanical threshold of Aδ fibres significantly decreased from 1 h after intramuscular injection of GDNF, and this time course fitted well with the latency of mechanical hyperalgesia observed in behavioural tests (Mizumura et al. 2011). Thus augmentation of mechanical sensitivity of Aδ fibres by GDNF plays an important role in DOMS.
The interaction of cyclooxygenase-2–glial cell line-derived neurotrophic factor and bradykinin–nerve growth factor pathway
We showed in a previous report (Murase et al. 2010) and in this experiment that there are two pathways leading to DOMS: B2 receptor activation to NGF and COX-2 upregulation to GDNF. When we consider the role of these two pathways we must take into consideration that pretreatment of either BK B2 receptor antagonist or COX-2 inhibitor completely blocked the development of DOMS. A simple way to view this is that DOMS stands on two feet, and if one foot is blocked DOMS cannot occur. Alternatively, there may be interactions between the two pathways. The reason that B2 receptor antagonist completely blocked the development of DOMS may be that BK B2 antagonist inhibited both NGF and GDNF upregulation. The latter is induced by inhibition of later (12 h after LC) upregulation of COX-2. Induction of COX-2 by BK B2 receptor upregulation is known to occur in DRG neurones (Inoue et al. 2006), airway epithelial cells and arterial smooth muscle cells (Chen et al. 2004). Thus, COX-2 is one site where interaction occurs between the two pathways.
In conclusion, LC-induced and BK-facilitated COX-2 expression in the muscle triggers the process leading to mechanical hyperalgesia via GDNF upregulation.
Acknowledgments
The authors E. Terazawa and K. Hirate passed away from illness during the course of this experiment. The corresponding author expresses her condolences and dedicates this paper to their important contributions.
Glossary
BK
bradykinin
COX
cyclooxygenase
DOMS
delayed onset muscle soreness
EDL
extensor digitorum longus
GDNF
glial cell line-derived neurotrophic factor
IL
interleukin
LC
lengthening contraction
NGF
nerve growth factor
SC
shortening contraction
TNF
tumour necrosis factor
Additional information
Competing interests
None.
Author contributions
S.M. performed all experimental work, and analysed and interpreted the data. E.T., H.O. and F.Q. performed part of behavioural studies. H.Y., H.K. and K.N. designed and performed ISHH. K.M. conceived the study. S.M., K.H., T.T. and K.M. designed the experiments and drafted the report. All remaining authors approved submission of the final version.
Funding
This work was supported in part by Grants-in-Aid for Scientific Research on Priority areas (no. 21026015) and (B) (no. 23390154).
References
- Albers KM, Woodbury CJ, Ritter AM, Davis BM, Koerber HR. Glial cell-line-derived neurotrophic factor expression in skin alters the mechanical sensitivity of cutaneous nociceptors. J Neurosci. 2006;26:2981–2990. doi: 10.1523/JNEUROSCI.4863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RB. Mechanisms of exercise-induced delayed onset muscular soreness: a brief review. Med Sci Sports Exerc. 1984;16:529–538. [PubMed] [Google Scholar]
- Bennett DL, Boucher TJ, Armanini MP, Poulsen KT, Michael GJ, Priestley JV, Phillips HS, McMahon SB, Shelton DL. The glial cell line-derived neurotrophic factor family receptor components are differentially regulated within sensory neurones after nerve injury. J Neurosci. 2000;20:427–437. doi: 10.1523/JNEUROSCI.20-01-00427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen O, Joseph EK, Chen X, Levine JD. GDNF hyperalgesia is mediated by PLCgamma, MAPK/ERK, PI3K, CDK5 and Src family kinase signalling and dependent on the IB4-binding protein versican. Eur J Neurosci. 2008;28:12–19. doi: 10.1111/j.1460-9568.2008.06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boix F, Rosenborg L, Hilgenfeldt U, Knardahl S. Contraction-related factors affect the concentration of a kallidin-like peptide in rat muscle tissue. J Physiol. 2002;544:127–136. doi: 10.1113/jphysiol.2002.025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290:124–127. doi: 10.1126/science.290.5489.124. [DOI] [PubMed] [Google Scholar]
- Buj-Bello A, Buchman VL, Horton A, Rosenthal A, Davies AM. GDNF is an age-specific survival factor for sensory and autonomic neurons. Neuron. 1995;15:821–828. doi: 10.1016/0896-6273(95)90173-6. [DOI] [PubMed] [Google Scholar]
- Carmillo P, Dago L, Day ES, Worley DS, Rossomando A, Walus L, Orozco O, Buckley C, Miller S, Tse A, Cate RL, Rosenblad C, Sah DW, Gronborg M, Whitty A. Glial cell line-derived neurotrophic factor (GDNF) receptor alpha-1 (GFR alpha 1) is highly selective for GDNF versus artemin. Biochemistry. 2005;44:2545–2554. doi: 10.1021/bi049247p. [DOI] [PubMed] [Google Scholar]
- Chen BC, Yu CC, Lei HC, Chang MS, Hsu MJ, Huang CL, Chen MC, Sheu JR, Chen TF, Chen TL, Inoue H, Lin CH. Bradykinin B2 receptor mediates NF-kappaB activation and cyclooxygenase-2 expression via the Ras/Raf-1/ERK pathway in human airway epithelial cells. J Immunol. 2004;173:5219–5228. doi: 10.4049/jimmunol.173.8.5219. [DOI] [PubMed] [Google Scholar]
- Cheung K, Hume P, Maxwell L. Delayed onset muscle soreness: treatment strategies and performance factors. Sports Med. 2003;33:145–164. doi: 10.2165/00007256-200333020-00005. [DOI] [PubMed] [Google Scholar]
- Fujii Y, Ozaki N, Taguchi T, Mizumura K, Furukawa K, Sugiura Y. TRP channels and ASICs mediate mechanical hyperalgesia in models of inflammatory muscle pain and delayed onset muscle soreness. Pain. 2008;140:292–304. doi: 10.1016/j.pain.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ozaki N, Kawakita K, Itoh K, Mizumura K, Furukawa K, Yasui M, Hori K, Yi SQ, Yamaguchi T, Sugiura Y. Involvement of NGF in the rat model of persistent muscle pain associated with taut band. J Pain. 2011;12:1059–68. doi: 10.1016/j.jpain.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Hendrich J, Alvarez P, Chen X, Levine JD. GDNF induces mechanical hyperalgesia in muscle by reducing I(BK) in isolectin B4-positive nociceptors. Neuroscience. 2012;219:204–213. doi: 10.1016/j.neuroscience.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock FJ, Wirth K, Albus U, Linz W, Gerhards HJ, Wiemer G, Henke S, Breipohl G, Konig W, Knolle J, et al. Hoe 140 a new potent and long acting bradykinin-antagonist: in vitro studies. Br J Pharmacol. 1991;102:769–773. doi: 10.1111/j.1476-5381.1991.tb12248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges PW, Tucker K. Moving differently in pain: a new theory to explain the adaptation to pain. Pain. 2011;152:S90–98. doi: 10.1016/j.pain.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Hoheisel U, Unger T, Mense S. Excitatory and modulatory effects of inflammatory cytokines and neurotrophins on mechanosensitive group IV muscle afferents in the rat. Pain. 2005;114:168–176. doi: 10.1016/j.pain.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Inoue A, Iwasa M, Nishikura Y, Ogawa S, Nakasuka A, Nakata Y. The long-term exposure of rat cultured dorsal root ganglion cells to bradykinin induced the release of prostaglandin E2 by the activation of cyclooxygenase-2. Neurosci Lett. 2006;401:242–247. doi: 10.1016/j.neulet.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signalling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram LC, Fuller A, Fick LG, Cartmell T, Poole S, Mitchell D. Cytokine profiles during carrageenan-induced inflammatory hyperalgesia in rat muscle and hind paw. J Pain. 2007;8:127–136. doi: 10.1016/j.jpain.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Malin SA, Davis BM. Postnatal roles of glial cell line-derived neurotrophic factor family members in nociceptors plasticity. Sheng Li Xue Bao. 2008;60:571–578. [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Molliver DC, Koerber HR, Cornuet P, Frye R, Albers KM, Davis BM. Glial cell line-derived neurotrophic factor family members sensitize nociceptors in vitro and produce thermal hyperalgesia in vivo. J Neurosci. 2006;26:8588–8599. doi: 10.1523/JNEUROSCI.1726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumura K, Murase S, Taguchi T. Effects of glial cell line-derived neurotrophic factor (GDNF) on the mechanical response of rat muscle thin-fibre receptors in vitro. SfN's 41st Annual Meeting. 2011:GG15. [Google Scholar]
- Molliver DC, Wright DE, Leitner ML, Parsadanian AS, Doster K, Wen D, Yan Q, Snider WD. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997;19:849–861. doi: 10.1016/s0896-6273(00)80966-6. [DOI] [PubMed] [Google Scholar]
- Murase S, Terazawa E, Queme F, Ota H, Matsuda T, Hirate K, Kozaki Y, Katanosaka K, Taguchi T, Urai H, Mizumura K. Bradykinin and nerve growth factor play pivotal roles in muscular mechanical hyperalgesia after exercise (delayed-onset muscle soreness) J Neurosci. 2010;30:3752–3761. doi: 10.1523/JNEUROSCI.3803-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani T, Doi Y, Nishimura W, Nishizawa M, Minami T, Ito S. Preemptive analgesia by zaltoprofen that inhibits bradykinin action and cyclooxygenase in a post-operative pain model. Neurosci Res. 2005;51:427–433. doi: 10.1016/j.neures.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Nasu T, Taguchi T, Mizumura K. Persistent deep mechanical hyperalgesia induced by repeated cold stress in rats. Eur J Pain. 2010;14:236–244. doi: 10.1016/j.ejpain.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Otis JS, Burkholder TJ, Pavlath GK. Stretch-induced myoblast proliferation is dependent on the COX2 pathway. Exp Cell Res. 2005;310:417–425. doi: 10.1016/j.yexcr.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Padi SS, Naidu PS, Kulkarni SK. Involvement of peripheral prostaglandins in formalin-induced nociceptive behaviours in the orofacial area of rats. Inflammopharmacology. 2006;14:57–61. doi: 10.1007/s10787-006-1495-7. [DOI] [PubMed] [Google Scholar]
- Rahnama N, Rahmani-Nia F, Ebrahim K. The isolated and combined effects of selected physical activity and ibuprofen on delayed-onset muscle soreness. J Sports Sci. 2005;23:843–850. doi: 10.1080/02640410400021989. [DOI] [PubMed] [Google Scholar]
- Schafers M, Sorkin LS, Sommer C. Intramuscular injection of tumor necrosis factor-alpha induces muscle hyperalgesia in rats. Pain. 2003;104:579–588. doi: 10.1016/S0304-3959(03)00115-5. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci U S A. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LL. Acute inflammation: the underlying mechanism in delayed onset muscle soreness. Med Sci Sports Exerc. 1991;23:542–551. [PubMed] [Google Scholar]
- Taguchi T, Matsuda T, Tamura R, Sato J, Mizumura K. Muscular mechanical hyperalgesia revealed by behavioural pain test and c-Fos expression in the spinal dorsal horn after eccentric contraction in rats. J Physiol. 2005;564:259–268. doi: 10.1113/jphysiol.2004.079483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Taguchi T, Itoh K, Okada K, Kawakita K, Mizumura K. Influence of surface anaesthesia on the pressure pain threshold measured with different-sized probes. Somatosens Mot Res. 2005;22:299–305. doi: 10.1080/08990220500420475. [DOI] [PubMed] [Google Scholar]
- Takasu K, Sakai A, Hanawa H, Shimada T, Suzuki H. Overexpression of GDNF in the uninjured DRG exerts analgesic effects on neuropathic pain following segmental spinal nerve ligation in mice. J Pain. 2011;12:1130–1139. doi: 10.1016/j.jpain.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Niederberger E, Vetter G, Brautigam L, Geisslinger G. Effects of selective COX-1 and -2 inhibition on formalin-evoked nociceptive behaviour and prostaglandin E(2) release in the spinal cord. J Neurochem. 2001;79:777–786. doi: 10.1046/j.1471-4159.2001.00613.x. [DOI] [PubMed] [Google Scholar]
- Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokmakidis SP, Kokkinidis EA, Smilios I, Douda H. The effects of ibuprofen on delayed muscle soreness and muscular performance after eccentric exercise. J Strength Cond Res. 2003;17:53–59. doi: 10.1519/1533-4287(2003)017<0053:teoiod>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Vandenburgh HH, Hatfaludy S, Sohar I, Shansky J. Stretch-induced prostaglandins and protein turnover in cultured skeletal muscle. Am J Physiol Cell Physiol. 1990;259:C232–240. doi: 10.1152/ajpcell.1990.259.2.C232. [DOI] [PubMed] [Google Scholar]
- Vianney JM, Spitsbergen JM. Cholinergic neurons regulate secretion of glial cell line-derived neurotrophic factor by skeletal muscle cells in culture. Brain Res. 2011;1390:1–9. doi: 10.1016/j.brainres.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Kobayashi K, Yu L, Wang S, Na R, Yamamoto S, Noguchi K, Dai Y. Inhibition of TRPA1 channel activity in sensory neurons by the glial cell line-derived neurotrophic factor family member, artemin. Mol Pain. 2011;7:41. doi: 10.1186/1744-8069-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]