Implementing genomic medicine in the clinic: the future is here (original) (raw)
Abstract
Although the potential for genomics to contribute to clinical care has long been anticipated, the pace of defining the risks and benefits of incorporating genomic findings into medical practice has been relatively slow. Several institutions have recently begun genomic medicine programs, encountering many of the same obstacles and developing the same solutions, often independently. Recognizing that successful early experiences can inform subsequent efforts, the National Human Genome Research Institute brought together a number of these groups to describe their ongoing projects and challenges, identify common infrastructure and research needs, and outline an implementation framework for investigating and introducing similar programs elsewhere. Chief among the challenges were limited evidence and consensus on which genomic variants were medically relevant; lack of reimbursement for genomically driven interventions; and burden to patients and clinicians of assaying, reporting, intervening, and following up genomic findings. Key infrastructure needs included an openly accessible knowledge base capturing sequence variants and their phenotypic associations and a framework for defining and cataloging clinically actionable variants. Multiple institutions are actively engaged in using genomic information in clinical care. Much of this work is being done in isolation and would benefit from more structured collaboration and sharing of best practices.
Genet Med 2013:15(4):258–267
Keywords: medical genomics, practice standards
Introduction
The potential for genomics to contribute to clinical care has long been recognized, and many optimistic scenarios for clinical use of information about a patient's genome have been proposed.1,2,3 The pace of realizing this potential has appeared slow to some,4,5 although clinical adoption of scientific discoveries has been estimated to take up to 17 years6 and a recent genetic example7 required 18 years after the initial 1991 report.8 Indeed, relatively robust genotype–phenotype associations for common, complex diseases only began to become available around 2005.9 Yet several academic medical centers and integrated health systems have already begun programs for implementing genomic medicine, which we define here as using an individual patient's genotypic information in his or her clinical care. This definition encompasses both Mendelian and multigenic complex diseases, and at present probably focuses most on single Mendelian variants of large effect, although emphasis is expected to shift soon to assaying and using multiple variants simultaneously in clinical care. This is not only because of an expanding knowledge base but also because the logistic complexities and costs of assessing genomic variation on a large scale are approaching those of one-at-a-time testing of individual variants, permitting a more holistic approach to incorporating genomic findings into clinical care.
These initial and separate forays have encountered similar obstacles and developed many of the same nascent solutions, often quite independently. In expectation that sharing of lessons learned in these efforts can facilitate broader and more effective implementation of genomic medicine, the National Human Genome Research Institute in collaboration with several leaders in this area (R.L.C., G.S.G., D.M.R., M.S.W., and R.Wilson) brought together about 20 groups working to implement genomic medicine for a Genomic Medicine Colloquium in June 2011. This article summarizes the projects described by these groups, describes challenges they have encountered in implementation, identifies common infrastructure and research needs, and outlines an implementation framework for introducing similar genomic medicine programs more broadly.
Brief Landscape of Genomic Medicine Projects
Participating sites reported a broad range of genomic medicine activities, both in pilot and full implementation forms (Table 1). These included genotyping (used here and throughout to refer to the detection of genetic variants by single-nucleotide polymorphism assay, sequencing, or other technologies) of somatic mutations in malignant tumors to guide treatment decisions. The expanding scope and growing acceptance of such techniques places tumor genotyping at the forefront of genotype-directed care. All the participating sites (and most cancer centers nationwide) conduct tumor genotyping of cancers such as melanoma and those of the breast, colon, and lung for the targeting of therapy. Several also use genotyping to select patients for interventional protocols, but the main focus here was on the use of genomics outside of cancer diagnosis and treatment.
Table 1. Examples of genomic medicine projects currently in implementation at participating sites.

Several centers have begun targeted screening for highly penetrant germline mutations for Lynch syndrome and in BRCA1/210,11,12 to identify genetically at-risk individuals. With appropriate immunohistochemical or other evidence suggestive of germline mutations, patients can be actively sought and counseled regarding the importance of genomic testing for their own treatment and for potential preventive care of their relatives. Direct outreach to patients at the Cleveland Clinic, for example, increased the uptake of germline testing in patients with colorectal cancer by nearly sixfold—from 14% with information provided directly to surgeons to 80% with a genetic counselor seeing patients at follow-up examinations (C.E., unpublished data). Highly penetrant variants such as these provide robust, evidence-based paradigms to help move a clinical site toward implementing broader genomic medicine approaches. More important, perhaps, successful implementation of screening programs at early adopter sites provides valuable lessons and refined approaches that may be adopted elsewhere.13
Self-reported family history information can also be used in risk assessment for individual patients with documented clinical validity and utility,14,15 despite its potential weaknesses such as incomplete or inaccurate reporting.16 Family history is generally not difficult to collect, especially with electronic data collection tools such as the Surgeon General's My Family Health Portrait,17 although integrating the information into an independent electronic medical record (EMR) is not trivial. Clinicians are generally familiar with family history and appreciate its importance, and evidence-based guidelines are available for several conditions.12,14 A Duke University pilot project, for example, is collecting three-generation family history information on 48 diseases using a Web-based computerized tool.18 Patient-entered information is integrated into the medical record and generates a pedigree, tabular family history, and reports for the patient and clinician, as well as decision support information for four conditions (breast, ovarian, and colorectal cancer and venous thrombosis). Outcomes being assessed include patient and clinician satisfaction and ease of use; changes in patient behaviors such as diet and smoking; changes in physician behaviors such as screening and referral; and estimates of sensitivity, specificity, net reclassification, and cost.18 Similarly, a Cleveland Clinic prototype collects three-generation family histories for the assessment of all inherited cancer syndromes.19 Intermountain Healthcare has also deployed a patient-entered family tool (“Our Family Health”) in their electronic patient portal.20
Pharmacogenomics provides additional opportunities in genomic medicine, particularly if appropriate decision support tools present relevant data to clinicians only when needed. Patients genetically unable to activate prodrugs such as clopidogrel, tamoxifen, and codeine, for example, do not need their genetic data presented to their clinicians unless and until these drugs are about to be prescribed. Although “reactive” genotyping for pharmacogenomic variants can be ordered at many sites, turnaround is often slow and uptake low. To address this, the Vanderbilt University Medical Center and St. Jude Children's Research Hospital have both implemented programs to conduct preemptive genotyping in patients very likely to receive medications with relevant genetically based dosing algorithms. At Vanderbilt, for example, selected patients, including those undergoing coronary angiography for acute coronary syndromes receive genotyping on a 184-variant platform that includes CYP2C19 alleles before being prescribed clopidogrel,21 based on a review of available evidence of poorer outcomes in persons with variant CYP2C19 alleles.22,23,24 At St. Jude Children's Research Hospital, array-based testing for 225 genes is performed, and results for those genes with the strongest clinical evidence are placed in the EMR.25 Clinical pharmacogenomic testing is expanding to several sites affiliated with the Pharmacogenomics Research Network (PGRN)26 with a goal of implementing proactive genotyping of a large number of variants, such as those identified by PharmGKB27 or the US Food and Drug Administration (FDA).28
Preemptive testing, embedding this information into the EMR with appropriate decision support, and then seamlessly presenting it at the point of care when needed, rather than requiring tests to be ordered each time a relevant medication is prescribed, could help to minimize delay, cost, lack of follow-through, and duplicate testing. This is a testable hypothesis and one that is likely to be explored in programs such as the Electronic Medical Records and Genomics (eMERGE) Network.29 Preemptive pharmacogenomic information could be even more useful were informed patients able to access it and work proactively with their clinicians in making treatment decisions. Putting patients in control of their genomic information is another potential strategy for genomic medicine implementation that needs evaluation. Such implementation strategies could be evaluated by measuring improvement, or lack thereof, in efficiency, cost, and health outcomes.
Whole-exome (protein-coding exons plus other highly conserved regions) and whole-genome sequencing are beginning to be used for identifying genetic causes of rare, unknown conditions, particularly those presenting in childhood or in strongly affected pedigrees for which novel mutations are more easily detected.30 The recent cases of a young boy with severe, atypical, refractory inflammatory bowel disease31 and of three families with severe arterial calcifications32 demonstrate the potential for this approach to detect novel disease-causing mutations and point the way toward appropriate therapies. The Medical College of Wisconsin and Children's Hospital of Wisconsin have implemented a genome sequencing program to which clinicians can nominate patients who remain undiagnosed after appropriate clinical evaluation and testing. Nominations are reviewed by a multidisciplinary patient selection committee chaired by the hospital's chief medical officer. Patients and their families undergo 6–8 h of assessment and counseling before enrollment. Sequencing is provided by a commercial laboratory certified by the College of American Pathologists (CAP) Laboratory Accreditation Program and the Clinical Laboratory Improvement Amendments (CLIA) Program. The detected variants are analyzed using an in-house, validated software package, focusing first on specific candidate genes nominated by the referring clinicians to limit the chances of incidental, potentially unrelated findings. Analysis is then broadened if initially nominated genes are uninformative. Potentially relevant variants are confirmed by an independent method, and those results are placed in the medical record. The Baylor College of Medicine has implemented a Whole Genome Laboratory33 with “in-house” sequencing and analysis of variants in a CLIA/CAP environment with review and sign-out by a board-certified clinical molecular geneticist. A number of other centers (Geisinger Clinic, Partners Healthcare, Washington University) and commercial laboratories such as Ambry Genetics,34 GeneDx,35 and SeqWright36 are also now offering whole-genome sequencing that can be used in clinical care.
These four briefly described applications—tumor-based screening, family history–directed decision support, pharmacogenomics, and diagnostic genome sequencing—demonstrate that genomic medicine is no longer on the threshold; it has arrived. Although much of this work is being pursued as part of research programs, some has been adopted institutionally and offered in a CLIA-certified laboratory environment as part of regular clinical care.13 Evaluating the impact of such programs and expanding their reach to diverse settings and populations is a high priority within the National Human Genome Research Institute's research agenda and mission. Information from such programs is helping to provide a needed evidence base to support more widespread implementation and reimbursement. Consideration of the barriers encountered and strategies used to surmount them by “early adopter” genomic medicine sites may help speed their evaluation and facilitate their expansion to the mainstream of clinical care.
Challenges in Implementation
Numerous challenges and barriers have been encountered in launching genomic medicine projects and many common solutions have emerged (Table 2). The greatest challenge may be the lack of appreciation by clinicians, health-care institutions, and payers of the potential for genomics to improve patient care, as compelling evidence of clinical validity and utility is limited at present.37 Even for genomic applications with proven validity and utility, such as family history, there is lack of adoption and implementation, suggesting the lack of evidence is far from being the only barrier.
Table 2. Challenges in implementation and potential solutions.
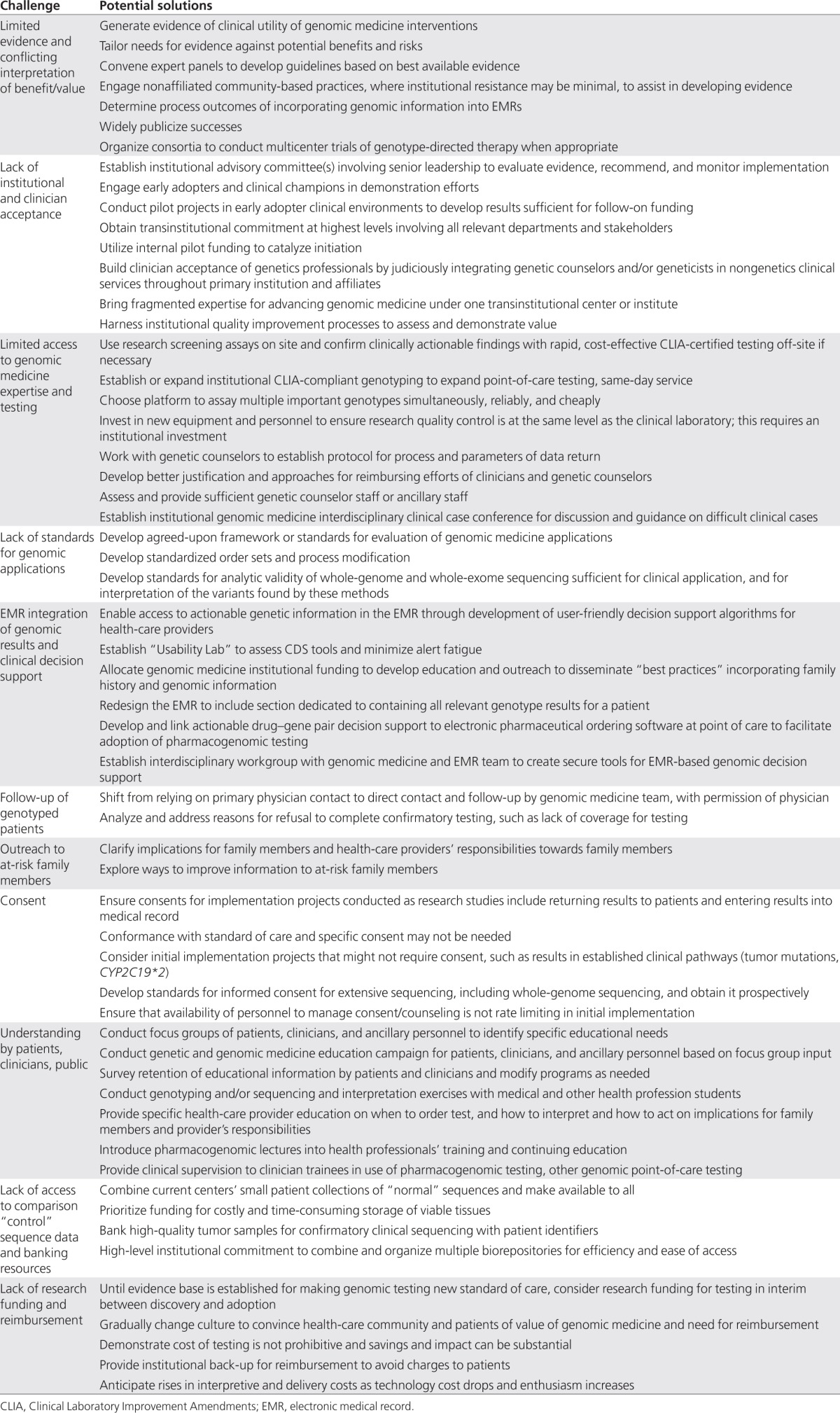
Cost concerns and institutional inertia typically demand convincing arguments and hard data before clinical practice is changed, yet the genome-wide nature of results to be expected by incorporating dense genotyping technologies into clinical care will soon force clinicians and institutions to deal with genomic information for which evidence may be quite limited. Evidentiary thresholds may thus need to be aligned with the intended use of the information, as the impact of genotype-driven care on morbidity and mortality cannot be tested for every variant.
Institutions have frequently relied on expert panels and local committees to evaluate available evidence and recommend particular new initiatives in genomic medicine, such as testing for specific pharmacogenomic variants or estimating risk to carriers. Such panels have tended to work in isolation from institution to institution, surveying the same evidence and often coming to similar conclusions. Far better would be to harness the collective knowledge of these groups in a more systematic way, distributing among them the genomic medicine issues for which evidence is compiled and evaluated, such as antidepressant therapy, coronary disease risk assessment, or risk in relatives of patients with colorectal cancer. These groups could then use a mutually accepted protocol for evaluation, with conclusions of each process shared among all. This is similar to the approach taken by the Centers for Disease Control and Prevention's Evaluation of Genomic Applications in Practice and Prevention (EGAPP),38 the Clinical Pharmacogenomics Implementation Consortium,39 and by some early adopter institutions in developing committees of their own. Given the rapid spread of diagnostic sequencing and direct-to-consumer testing, it may be appropriate to begin to identify an intermediate category of “actionable” variants, for which evidence is insufficient to establish unequivocal clinical utility but is sufficient to determine how already available information could be used. Consideration should be given to tailoring of required levels of evidence to the risk/benefit ratio of the specific clinical setting at issue; development of consensus on evidence needed in different scenarios would be a valuable contribution. Recommendations for drastic or irreversible interventions such as genotype-driven mastectomy or salpingo–oophorectomy, for instance, should require far stricter evidence than should low-risk interventions such as modifying choice between two proven, roughly equivalent medications such as codeine and hydromorphone.40
For more modest interventions, the risks are more likely to involve factors such as burden to clinicians and patients in obtaining, interpreting, and managing the result, including learning of incidental findings not sought by clinician or patient; cost of testing; and psychological impact of the information on patients or their families. The first two of these can be addressed in part by improving the accessibility and lowering the costs of obtaining genomic information suitable for clinical use, which is occurring rapidly with expanding certification of genomic technologies and laboratories under CLIA and CAP. Testing fees can be substantial, especially when individual genetic variants are assayed sequentially, and indeed are soon likely to rival the cost of obtaining an entire genome sequence.41 For a genetically heterogeneous condition, such as Charcot-Marie-Tooth disease, that is potentially due to variation at one or more of over 40 genetic loci, for example, whole-exome sequencing can already be performed for less than half the price of the current clinically available “multigene panel.”42 Third-party reimbursement of these costs is thus crucial for ensuring uptake by institutions and patients.
Integrating effective clinical decision support tools into the EMR, to query a patient's genomic data as care is being delivered, will be needed to present relevant variants and recommendations only when they are indicated. Such real-time, genomically driven clinical decision support can help avoid “alert fatigue” and relieve the clinician of trying to sift through the massive amount of genomic information without impeding clinical workflow. Sharing the effort of following up actionable genetic variants and at-risk family members with a dedicated genomic medicine team, with the primary physician's agreement and in ways that do not “compete” for the patient, can also minimize burden on patients and clinicians and increase efficiency. Looking to the future, the potential to update EMRs automatically as new discoveries are made and their clinical utility validated provides exciting possibilities for rapid implementation of research advances. Growing knowledge and understanding, however, mean that variants that are today of uncertain importance may tomorrow be found to be of great clinical relevance and vice versa. The responsibility of the clinician to recontact the patient with this new information is uncertain and could pose a major burden as well as medico-legal risks.43
Psychological impact on patients and their families has traditionally been addressed by the informed consent process and genetic counseling, but expanding genomic medicine efforts will likely soon outstrip the available supply of genetic counselors.44 Innovative models utilizing ancillary personnel or social media tools to provide counseling and education, along with broad-ranging educational campaigns to increase familiarity with genomic medicine among practitioners, patients, and payers, will have the greatest impact if they can be linked to early successes and readily observable local efforts.
Reports of successes such as reduced hospitalization rates following genotype-driven adjustments in warfarin dosing,45 e.g., or symptomatic improvement or reduced adverse effects after genotype-driven selection of antidepressants,46 can help stimulate discussion of the potential for genomic medicine to improve outcomes and shift an institution's culture to embrace genotype-guided care,30 although even these examples need a more complete evidence base and remain controversial.47,48,49,50
Infrastructure and Research Needs
Genomic medicine programs share needs for basic informational and policy infrastructure as well as evidence and outcomes produced through research (Table 3). Key infrastructure needs include a comprehensive knowledge base that captures sequence variants and their phenotypic associations and is openly accessible to clinical groups attempting to interpret sequence data. Such a knowledge base would have enormous value for research as well, particularly to the degree that it entails a careful curation and consensus process to reduce error and promote consistency. Here again, many genomic medicine groups appear to be doing this in isolation, capturing and reviewing such information internally but finding themselves constrained in sharing it outside their institutions. The policy aspects of establishing such a knowledge base, ensuring the accuracy of associations to be reported within it, and providing wide access to it are far from trivial and involve patient privacy and confidentiality of health information. Linking to appropriate supporting evidence and providing frequent updates are also daunting tasks, given the rapid pace of discovery, but are tasks that could readily be distributed among collaborating groups.
Table 3. Infrastructure and research needs.
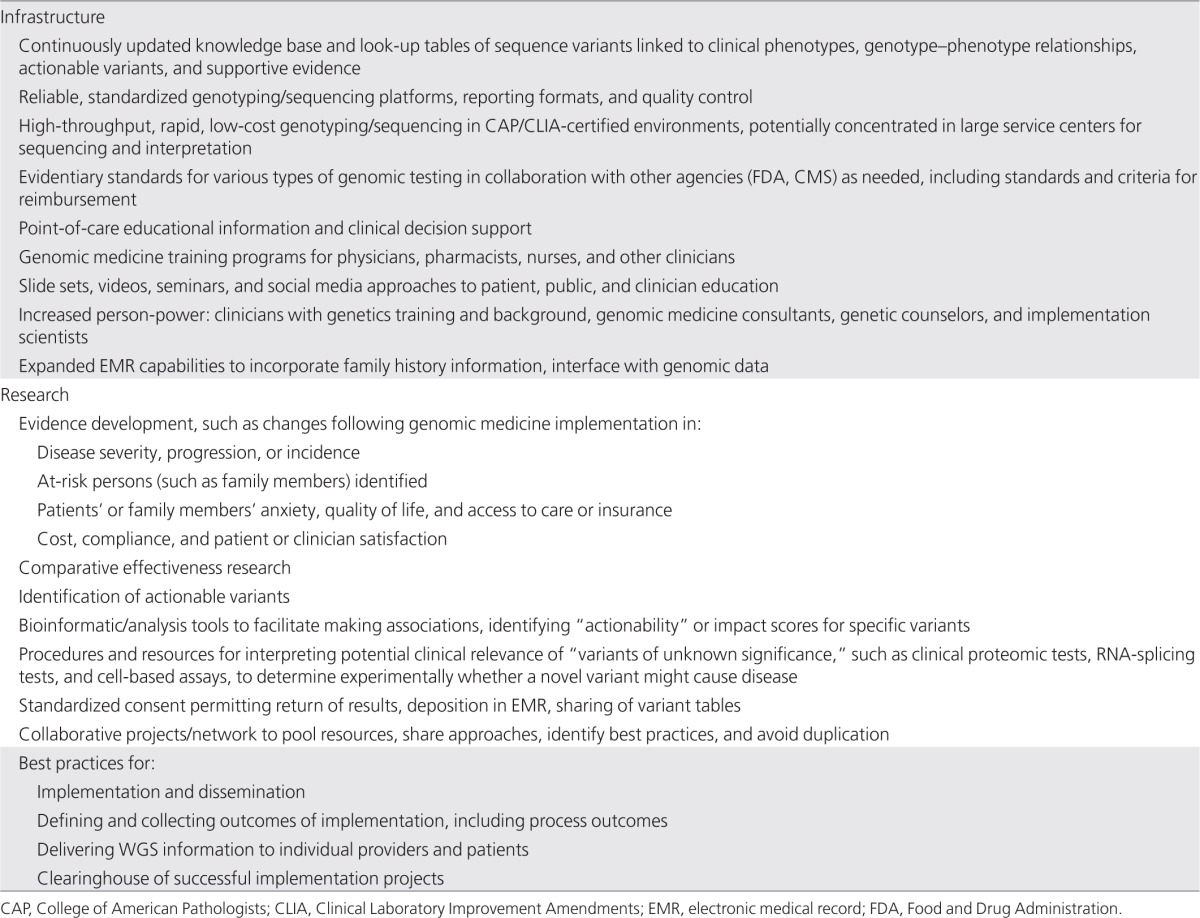
Perhaps an even greater informational need addressable by data sharing among genomic medicine groups and other genome sequencing projects is the ability to determine whether a newly discovered variant has been reported before. This is currently done by surveying available large-scale databases, such as the 1000 Genomes Project,51 the Exome Variant Server,52 and local institutional catalogs, on the presumption that a patient with a very rare or undiagnosed condition likely has a private or even unique variant.53 Interpretation can be difficult because these reference databases generally lack phenotypic information. Far better than individual small cohorts of sequenced patients would be to aggregate cohorts and make their data widely available to qualified clinicians and researchers. Sequence variants in these databases should also be linked to phenotypic information, when possible, rather than forcing the assumption that all available sequenced individuals are free of the disease under consideration. The recently initiated Centers for Mendelian Genomics54,55 will identify variants responsible for a great number of rare disorders and make this information widely available, which should help in this regard. The nascent ClinVar database of the National Center for Biotechnology Information56 and the International Standards for Cytogenomic Arrays database available in dbGaP and dbVar57 are two open-access databases that can provide valuable phenotypic information associated with rare sequence variants and structural variation.
Placing all this information in a single queryable source, regardless of where the actual data reside, would be a substantial advance in interpreting the three million or more differences from the human reference sequence identifiable per individual.58 Genomic sequencing, in contrast to genotyping microarray technology, reveals rare or even unique alleles lacking scientific literature support for physiological effects. Computational approaches to predicting function may need to be incorporated into clinical decision support methods in trying to determine the implications of such variants for clinical care. Such analyses would be greatly facilitated by standard formats for reporting the data produced by individual sequencing groups rather than expecting individual clinicians and clinical sites to handle and interpret multiple different formats. The Centers for Disease Control and Prevention are currently working to develop performance specifications for next-generation sequencing.59 Access to accurate, low-cost genome sequencing in CAP/CLIA-certified environments will remain a major limiting factor in utilizing sequencing in clinical care across diverse settings. Provision of genome sequencing as a commodity service in national or regional hubs, including interpretation of potential clinical relevance of identified variants, could be a major advance.
Appropriate professional organizations that can develop genomic medicine practice guidelines, such as the American College of Medical Genetics and Genomics, the College of American Pathologists, and the Association of Molecular Pathologists, as well as regulatory agencies such as the FDA and Centers for Medicare and Medicaid Services, should be involved in developing evidentiary standards tailored to specific interventions under consideration and criteria for reimbursement. Targeted education for active practitioners should also be tailored to specific settings and delivered as succinctly as possible at the point of care for maximum uptake and value, whereas broader educational efforts can capture clinicians earlier in their training.60 Fellowships in pharmacogenomics and genomic medicine will be needed to develop specialists (perhaps “clinical genomicists,” akin to radiologists or pathologists) capable of interpreting patients' genomic information and advising clinicians on appropriate actions to be taken for a given set of variants in a given clinical setting. Given the broad range of possible genomic medicine applications, the training of physicians and scientists enrolled in certified programs of the American Board of Medical Genetics and Genomics and the American Board of Pathology in clinical genetics, molecular genetics, biochemical genetics, and cytogenetics is particularly relevant. Consideration should be given to the potential for expanding the curricula of these programs to include more genomic medicine content or providing fellowships within these specialties that focus on genomic medicine.
Other educational modes such as seminars, slide sets, webinars, and videos need to be developed and shared across institutions doing similar types of work. Expanded EMR capabilities to permit querying of sequence or other dense genomic data, and incorporation of family history, are readily addressable. EMRs also have the ability to deliver context-specific educational information at the point of care during an eminently teachable moment, with a clinical conundrum at hand, as well as more sophisticated clinical decision support to aid clinicians in the appropriate use of genomic information.61 Challenges in implementing point-of-care tools need to be recognized, however, including the potential for overwhelming clinicians with alerts leading to alert fatigue.62 Here again, consideration should be given to the evidence needed for implementing such tools in genomic medicine and for ensuring the consistent adoption of point-of-care interventions for high priority health-care targets with the best risk/benefit ratio and strongest evidence, whether genomic or nongenomic.
Research is acutely needed to generate, collect, and make widely available the evidence needed for determining which variants are actionable, in whom, and in what clinical situations. Intermediate steps are needed that lie between genomic discovery research and routine clinical implementation to generate the evidence base to justify implementation. Consensus should be sought on the levels of evidence needed to justify the introduction of genomic medicine interventions, taking into account the degree of complexity or cost (such as family history vs. whole-genome sequencing) and risk/benefit (Lynch syndrome screening vs. universal colonscopy) involved, and the comparisons and outcome measures needed to answer these questions. Generating clear evidence of benefit for genomic interventions will remain challenging, however, in part because the rarity of many genomic markers of risk means large samples must be studied to accrue benefit/risk data in the small subsets of patients affected. Even with large samples, there can be controversy about the interpretation of the evidence, as shown by the data on the relationship between CYP2C19 variants and failure of antiplatelet efficacy of clopidogrel.63 In one meta-analysis that included only patients with acute coronary syndromes, there was strong evidence for an effect of variant alleles on outcomes,23 whereas another analysis that included trials with different indications (and in which the efficacy of clopidogrel itself appears to be less) reported a much smaller effect of genetic variation on outcome.24 Thus, the evidence supporting an effect of pharmacogenomic variants on variable drug outcomes may also depend on indication, substantially increasing the complexity of generating and evaluating evidence.
Research efforts should also aim to establish and validate criteria for actionability. They should facilitate association analysis and assessment of actionability and influence on outcomes from available data. Given that it is impossible, however, to perform prospective studies of the impact of all potentially important variants on clinical outcomes, comprehensive strategies will be needed to generate the necessary data. Real-world studies quantifying the impact of implementing genomic medicine programs on patient outcomes, cost and acceptability of care, and clinician and patient satisfaction are needed to promote adoption and dissemination of genomic medicine. Such studies fall under the rubric of “implementation research,” defined as the study of methods to promote the systematic uptake of proven interventions into routine practice.64 Comparative effectiveness research assessing the added value of genomic information, such as genomic variants added to family history added to clinical risk factors, and consensus on the outcomes and evidence needed to answer such questions, would help to define priorities for collecting and using such information in a cost-effective way.
Informed consents for genomic research should be broadened to include return of results, where desired and appropriate, so that lack of such consent (or indeed, consent forms declaring “no results will ever be returned”) does not become an obstacle, if not an excuse, for failing to report actionable findings. The consent process should also explicitly address deposition of data in the EMR or research databases and sharing of variant information among researchers and clinicians.
Assembling early adopters of genomic medicine into a confederation or community should facilitate addressing the many infrastructure and research needs outlined here. Collaborative projects such as those of the PGRN65 can facilitate dissemination and testing of genomic medicine approaches in diverse settings to maximize their generalizability and usefulness. Such projects can help develop best practices for complex tasks such as implementation and dissemination, defining and collecting outcomes of implementation, and delivering genomic information to clinicians and patients. A clearinghouse of successful implementation projects, with detailed protocols addressing steps needed for patients, clinicians, laboratories, departments, and institutions, would go a long way toward disseminating this work beyond early adopter sites. Indeed, a critical question to ask is whether and how approaches developed at highly specialized and resourced tertiary care centers can be adopted in less resource-intensive settings.
Implementation Roadmap
Incorporating genomic results into clinical care is as much a cultural and political exercise within a given institution as a scientific one, although a firm scientific grounding is essential to convincing the various stakeholders to accept an implementation project, particularly the first one (Figure 1). Such grounding can be obtained from a comprehensive review of the available scientific literature, recommendations of expert groups, and examination of ongoing successful projects at other institutions. Available policy or institutional guidelines on implementing changes to clinical care must also be considered. Critical in choosing an initial implementation project is the pragmatic issue of identifying a group of ready adopters or clinical champions, such as a subspecialty group or local practice, willing to try a new approach. Indeed, selection of a pilot project among several possibilities may hinge upon the availability of an enthusiastic group of clinicians.
Figure 1.

Implementation roadmap: one approach to initial implementation at a single institution with subsequent evaluation and ultimately dissemination.
Identifying and engaging the myriad stakeholders within an institution, including those needed to conduct genetic testing, interpret it, integrate it with the EMR, provide results and recommendations to the clinician, and pay for it, require that senior leaders who are familiar with the institution and its culture be on the genomic medicine team. It also often requires patience, persistence, and sufficient standing within the organization to convince skeptics and overcome institutional inertia. Some programs have found it simpler to begin at smaller affiliated hospitals or practices rather than at dominant academic medical centers.
Engagement and support from institutional leadership is obviously crucial, and most successful programs have involved them directly in decision making, as chairs of committees selecting interventions to implement, for example, or evaluating their impact. Funding may be provided through local institutional support, in anticipation perhaps of eventual reimbursement by major payers, or through foundation or research funding, including National Institutes of Health–supported projects such as the Clinical Translational Science Awards and the PGRN. Institutional stakeholders are key in developing practical and efficient clinical workflows for obtaining testing and providing results. Development and testing of streamlined educational tools, which may be as simple as a pop-up window with appropriate links for the clinician and letters or brochures for patients, will be essential both for initiating a program and encouraging its acceptance. Once launched, careful monitoring of initial cases for adherence to established workflows and unanticipated barriers permits tailoring of procedures as needed. Outcome data related to ease of use, adherence, patient and clinician satisfaction and behaviors, cost, and, where possible, measures of morbidity and mortality can be used to modify the program and inform design and implementation of future efforts. Evaluation of the effectiveness of an implementation project should focus on outcomes of the implementation itself rather than the outcomes of treatment.66 Several conceptual models for such evaluations are available for consideration.67,68
Conclusion
Multiple institutions are actively engaged in using patients' genomic information in their clinical care, either as pilot demonstration and research projects or as part of clinical care as a reimbursable service. The Genomic Medicine Colloquium revealed that much of this work is being done in isolation and would benefit from more structured collaboration and sharing of approaches across groups and institutions. Implementation of genomics in medicine would also benefit from establishing common infrastructure, such as a catalog of variants observed across large numbers of sequenced individuals to date, an updatable database of actionable variants, evidentiary standards tailored to benefits and risks, common clinical decision support tools including point-of-care educational materials, and collaborative projects to pool resources and identify best practices. Systematic and timely incorporation of advances in genomic research related to somatic and germline DNA, as well as consideration of RNA, protein, and epigenomic applications, will also be needed. The National Human Genome Research Institute, in collaboration with several other National Institutes of Health institutes and in accord with its recent strategic plan,69 is currently exploring approaches for facilitating the research needed to implement genomic medicine on an ever-widening scale. Such efforts should also take full advantage of the extant momentum, information, and experience of “early adopters” across the United States, as well as the tools of implementation science to address the cultural barriers described above.
The substantial National Institutes of Health investment in basic genomic research has yielded and will continue to yield new insights into the role of genetic variants in health and disease. It is now time to ensure those results are translated and used to the maximum benefit of patients by periodically convening relevant stakeholders and building on the vanguard of nationwide genomic medicine implementation projects. Bringing these groups together in this way is anticipated to increase their diffusion, efficiency, and, ultimately, their value to patients and health care.
Disclosure
C.E. is an unpaid member of the External Scientific Advisory Board of MyMercury.com and of the Genomic Medicine Advisory Board of Complete Genomics, Inc. The other authors declare no conflict of interest.
Acknowledgments
This article summarizes the deliberations of a colloquium convened by the National Human Genome Research Institute on 29 June 2011 to examine opportunities and barriers for implementing genomic medicine in clinical care. The authors gratefully acknowledge the contributions of Gary Gibbons to the workshop.
References
- Dulbecco R. A turning point in cancer research: sequencing the human genome. Science. 1986;231:1055–1056. doi: 10.1126/science.3945817. [DOI] [PubMed] [Google Scholar]
- Collins FS. Shattuck lecture–medical and societal consequences of the Human Genome Project. N Engl J Med. 1999;341:28–37. doi: 10.1056/NEJM199907013410106. [DOI] [PubMed] [Google Scholar]
- Guttmacher AE, Collins FS. Genomic medicine–a primer. N Engl J Med. 2002;347:1512–1520. doi: 10.1056/NEJMra012240. [DOI] [PubMed] [Google Scholar]
- Evans JP, Meslin EM, Marteau TM, Caulfield T. Genomics. Deflating the genomic bubble. Science. 2011;331:861–862. doi: 10.1126/science.1198039. [DOI] [PubMed] [Google Scholar]
- Varmus H. Ten years on–the human genome and medicine. N Engl J Med. 2010;362:2028–2029. doi: 10.1056/NEJMe0911933. [DOI] [PubMed] [Google Scholar]
- Committee on Quality of Health Care in America, Institute of Medicine Crossing the Quality Chasm: A New Health System for the 21st Century. National Academy Press: Washington, DC; 2001 [Google Scholar]
- England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology. Practice Parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation Neurology 200972185–192. [DOI] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Ann Intern Med. 2011;155:69–79. doi: 10.7326/0003-4819-155-2-201107190-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates R, Williams M, Melillo S, Gudgeon J. Genetic testing for lynch syndrome in individuals newly diagnosed with colorectal cancer to reduce morbidity and mortality from colorectal cancer in their relatives. PLoS Curr. 2011;3:RRN1246. doi: 10.1371/currents.RRN1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Preventive Services Task Force Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med. 2005;143:355–361. doi: 10.7326/0003-4819-143-5-200509060-00011. [DOI] [PubMed] [Google Scholar]
- Gudgeon JM, Williams JL, Burt RW, Samowitz WS, Snow GL, Williams MS. Lynch syndrome screening implementation: business analysis by a healthcare system. Am J Manag Care. 2011;17:e288–e300. [PubMed] [Google Scholar]
- Qureshi N, Armstrong S, Dhiman P, et al. for the ADDFAM (Added Value of Family History in CVD Risk Assessment) Study Group. Effect of adding systematic family history enquiry to cardiovascular disease risk assessment in primary care: A Matched-Pair, Cluster Randomized Trial Ann Intern Med 2012156253–262. [DOI] [PubMed] [Google Scholar]
- Rubinstein WS, Acheson LS, O'Neill SM, et al. Family Healthware Impact Trial (FHITr) Group. Clinical utility of family history for cancer screening and referral in primary care: a report from the Family Healthware Impact Trial Genet Med 201113956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury MJ, Feero WG, Valdez R. Family history and personal genomics as tools for improving health in an era of evidence-based medicine. Am J Prev Med. 2010;39:184–188. doi: 10.1016/j.amepre.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Surgeon General's My Family Health Portrait. .https://familyhistory.hhs.gov/fhh-web/home.action.
- Orlando LA, Hauser ER, Christianson C, et al. Protocol for implementation of family health history collection and decision support into primary care using a computerized family health history system. BMC Health Serv Res. 2011;11:264. doi: 10.1186/1472-6963-11-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr M, Leach B, Men X, et al. Patient-entered, web-based, EMR-compatible, comprehensive cancer family history collection tool: clinical workflow, demographics, and patient satisfaction from pilot studyAmerican Society of Human Genetics 2010 Annual Meeting: Abstract 1992. Washington, D.C., 2–6 November 2010.
- Hulse NC, Ranade-Kharkar P, Post H, Wood GM, Williams MS, Haug PJ. Development and early usage patterns of a consumer-facing family health history tool. AMIA Annu Symp Proc. 2011;2011:578–587. [PMC free article] [PubMed] [Google Scholar]
- Pulley JM, Denny JC, Peterson JF, et al. Operational implementation of prospective genotyping for personalized medicine: the design of the Vanderbilt PREDICT project. Clin Pharmacol Ther. 2012;92:87–95. doi: 10.1038/clpt.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet. 2009;373:309–317. doi: 10.1016/S0140-6736(08)61845-0. [DOI] [PubMed] [Google Scholar]
- Mega JL, Simon T, Collet JP, et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA. 2010;304:1821–1830. doi: 10.1001/jama.2010.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Perel P, Shah T, Hingorani AD, Casas JP. CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: a systematic review and meta-analysis. JAMA. 2011;306:2704–2714. doi: 10.1001/jama.2011.1880. [DOI] [PubMed] [Google Scholar]
- St. Jude Children's Research Hospital PG4KDS program .http://www.stjude.org/pg4kds.
- Pharmacogenomics Research Network (PGRN) .http://www.nigms.nih.gov/Research/FeaturedPrograms/PGRN/.
- Pharmacogenomics Knowledge Base (PharmGKB) .http://www.pharmgkb.org/search/guidelineList.action.
- US Food and Drug Administration Table of Pharmacogenomic Biomarkers in Drug Labels .http://www.fda.gov/drugs/scienceresearch/researchareas/ pharmacogenetics/ucm083378.htm.
- McCarty CA, Chisholm RL, Chute CG, et al. eMERGE Team. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies BMC Med Genomics 2011413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- St Hilaire C, Ziegler SG, Markello TC, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor College of Medicine, Whole Genome Laboratory .http://www.bcm.edu/geneticlabs/index.cfm?PMID=21319.
- Ambry Genetics .http://www.ambrygen.com/clinical-diagnostic-exome.
- GeneDx .http://www.genedx.com/site/xomedx.
- SeqWright .http://www.seqwright.com/Exome.
- Scheuner MT, Sieverding P, Shekelle PG. Delivery of genomic medicine for common chronic adult diseases: a systematic review. JAMA. 2008;299:1320–1334. doi: 10.1001/jama.299.11.1320. [DOI] [PubMed] [Google Scholar]
- Teutsch SM, Bradley LA, Palomaki GE, et al. EGAPP Working Group. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group Genet Med 2009113–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011;89:464–467. doi: 10.1038/clpt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype Clin Pharmacol Ther 201291321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Pertea M. Do-it-yourself genetic testing. Genome Biol. 2010;11:404. doi: 10.1186/gb-2010-11-10-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyeritz RE. The coming explosion in genetic testing–is there a duty to recontact. N Engl J Med. 2011;365:1367–1369. doi: 10.1056/NEJMp1107564. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute, Research-tested Intervention Programs .http://rtips.cancer.gov/rtips/programDetails.do?programId=277363.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011;364:1144–1153. doi: 10.1056/NEJMra1010600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli S, Drago A, Fabbri C, Gibiino S, Calati R, Serretti A. Pharmacogenetics of antidepressant response. J Psychiatry Neurosci. 2011;36:87–113. doi: 10.1503/jpn.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoef TI, Schalekamp T, Redekop WK, de Boer A, Maitland-van der Zee AH. Clinical and economic consequences of pharmacogenetic-guided dosing of warfarin. Expert Rev Pharmacoecon Outcomes Res. 2010;10:375–378. doi: 10.1586/erp.10.42. [DOI] [PubMed] [Google Scholar]
- Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks and costs of warfarin pharmacogenomic testing. Pharmacoeconomics. 2010;28:61–74. doi: 10.2165/11318240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group Recommendations from the EGAPP Working Group: testing for cytochrome P450 polymorphisms in adults with nonpsychotic depression treated with selective serotonin reuptake inhibitors. Genet Med. 2007;9:819–825. doi: 10.1097/gim.0b013e31815bf9a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II) Circulation. 2012;125:1997–2005. doi: 10.1161/CIRCULATIONAHA.111.070920. [DOI] [PubMed] [Google Scholar]
- 1000 Genomes Project .http://www.1000genomes.org/.
- Exome Variant Server .http://evs.gs.washington.edu/EVS/.
- Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelian Disorders Sequencing Centers .http://www.genome.gov/27546192.
- Bamshad MJ, Shendure JA, Valle D, et al. Centers for Mendelian Genomics. The Centers for Mendelian Genomics: a new large-scale initiative to identify the genes underlying rare Mendelian conditions Am J Med Genet A 2012158A1523–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information .http://www.ncbi.nlm.nih.gov/clinvar/.
- International Standards for Cytogenomic Arrays (ISCA) database .http://www.iscaconsortium.org.
- Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, Next Generation Sequencing: Standardization of Clinical Testing (Nex-StoCT) Working Group .http://www.cdc.gov/osels/lspppo/Genetic_Testing_Quality_Practices/ Nex-StoCT.html.
- Dhar SU, Alford RL, Nelson EA, Potocki L. Enhancing exposure to genetics and genomics through an innovative medical school curriculum. Genet Med. 2012;14:163–167. doi: 10.1038/gim.0b013e31822dd7d4. [DOI] [PubMed] [Google Scholar]
- Hoffman MA, Williams MS. Electronic medical records and personalized medicine. Hum Genet. 2011;130:33–39. doi: 10.1007/s00439-011-0992-y. [DOI] [PubMed] [Google Scholar]
- Shah NR, Seger AC, Seger DL, et al. Improving acceptance of computerized prescribing alerts in ambulatory care. J Am Med Inform Assoc. 2006;13:5–11. doi: 10.1197/jamia.M1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Roden DM, Lesko LJ, Ashley E, Klein TE, Shuldiner AR. Clopidogrel: a case for indication-specific pharmacogenetics. Clin Pharmacol Ther. 2012;91:774–776. doi: 10.1038/clpt.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles MP, Foy R, Sales A, Wensing M, Mittman B. Implementation Science six years on–our evolving scope and common reasons for rejection without review. Implement Sci. 2012;7:71. doi: 10.1186/1748-5908-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharmacogenomics Research Network Research Groups .http://pgrn.org/display/pgrnwebsite/Research+Groups+and+Network+Resources.
- Proctor E, Silmere H, Raghavan R, et al. Outcomes for implementation research: conceptual distinctions, measurement challenges, and research agenda. Adm Policy Ment Health. 2011;38:65–76. doi: 10.1007/s10488-010-0319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor EK, Landsverk J, Aarons G, Chambers D, Glisson C, Mittman B. Implementation research in mental health services: an emerging science with conceptual, methodological, and training challenges. Adm Policy Ment Health. 2009;36:24–34. doi: 10.1007/s10488-008-0197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglio B, Glasgow RE.Evaluation approaches for dissemination and implementation researchIn: Brownson RC, Colditz GA, Proctor EK (eds.), Dissemination and Implementation Research in Health: Translating Science to Practice. Oxford University Press, New York, NY; 2012334–337. [Google Scholar]
- Green ED, Guyer MS.National Human Genome Research Institute. Charting a course for genomic medicine from base pairs to bedside Nature 2011470204–213. [DOI] [PubMed] [Google Scholar]