Molecular response to the pathogen Phytophthora sojae among ten soybean near isogenic lines revealed by comparative transcriptomics (original) (raw)
Abstract
Background
Phytophthora root and stem rot (PRR) of soybean, caused by Phytophthora sojae, is controlled by Rps genes. However, little is known regarding the _Rps_-induced molecular responses to P. sojae and how they actually overlap. We thus sequenced, analyzed, and compared the transcriptomes of 10 near isogenic lines (NILs), each with a unique Rps gene/allele, and the susceptible parent Williams, pre- and post-inoculation with the pathogen.
Results
A total of 4,330 differentially expressed genes (DEGs) were identified in Williams versus 2,014 to 5,499 DEGs in individual NILs upon inoculation with the pathogen. Comparisons of the DEGs between the NILs and Williams identified incompatible interaction genes (IIGs) and compatible interaction genes (CIGs). Hierarchical cluster and heatmap analyses consistently grouped the NILs into three clusters: Cluster I (Rps1-a), Cluster II (Rps1-b, 1-c and 1-k) and Cluster III (Rps3-a, 3-b, 3-c, 4, 5, and 6), suggesting an overlap in _Rps_-induced defense signaling among certain NILs. Gene ontology (GO) analysis revealed associations between members of the WRKY family and incompatible reactions and between a number of phytohormone signaling pathways and incompatible/compatible interactions. These associations appear to be distinguished according to the NIL clusters.
Conclusions
This study characterized genes and multiple branches of putative regulatory networks associated with resistance to P. sojae in ten soybean NILs, and depicted functional “fingerprints” of individual _Rps_-mediated resistance responses through comparative transcriptomic analysis. Of particular interest are dramatic variations of detected DEGs, putatively involved in ethylene (ET)-, jasmonic acid (JA)-, (reactive oxygen species) ROS-, and (MAP-kinase) MAPK- signaling, among these soybean NILs, implicating their important roles of these signaling in differentiating molecular defense responses. We hypothesize that different timing and robustness in defense signaling to the same pathogen may be largely responsible for such variations.
Keywords: Comparative transcriptomics, Resistance to Phytophthora sojae, Soybean
Background
Phytophthora root and stem rot (PRR) is one of the most devastating diseases of soybean (Glycine max), causing nearly $200 million in annual yield losses in the U.S. alone [1]. PRR is caused by the soil-borne, hemibiotrophic pathogen Phytophthora sojae, and is most effectively controlled by Rps (Resistance to P. sojae) genes. The resistance conferred by an Rps gene is race specific, and the interaction between an Rps gene and the corresponding avirulence (Avr) gene in P. sojae, follows the gene-for-gene model [2]. Currently, 18 Rps genes/alleles from soybean and 12 Avr genes from P. sojae have been identified [3-5].
Like most resistance (R) genes, the Rps gene family encodes, or is predicted to encode nucleotide binding leucine rich repeat (NB-LRR)-type proteins [6-8], which are able to recognize the Avr effector proteins of pathogens directly or indirectly to induce the appropriate defense response [9,10]. The first evidence of a direct interaction between an R and Avr protein was reported in the flax-Melampsora lini pathosystem [11]. Indirect interactions, however, are more common. In these cases, the R protein appears to require the existence of a ‘guard protein’ or ‘decoy’ in order to recognize an Avr protein [12,13]. A classical example of this type of interaction is the RPM1-interacting protein 4 (RIN4), which is ‘guarded’ by RPM1 (RESISTANCE TO PSEUDOMONAS SYRINGAE PV MAC_ULICOLA 1)_ and RPS2 (RESISTANCE TO P. SYRINGAE 2) proteins in _Arabidopsis_[14,15].
The recognition of pathogen effectors triggers an innate immunity response that is mediated by two distinct types of NB-LRR proteins, the toll-interleukin-1 receptor (TIR)-NB-LRR proteins and the coiled-coil (CC)-NB-LRR proteins. The former requires EDS1 (ENHANCED DISEASE SUSCEPTIBILITY 1), a central regulator of effector-triggered immunity (ETI), which functions together with PAD4 (PHYTOALEXIN DEFICIENT 4). In contrast, the latter NB-LRR proteins are independent of EDS1 but require NDR1 (NON-RACE-SPECIFIC DISEASE RESISTANCE 1) [16-21]. The interaction among these intracellular proteins results in a regulation network of phytohormone signaling, which is mainly mediated by salicylic acid (SA) for biotrophic and hemibiotrophic pathogens, and jasmonic acid (JA) and ethylene (ET) for necrotrophic pathogens [8,22]. In addition to SA, JA and ET, other phytohormones such as brassinosteroids (BR), abscisic acid (ABA), auxin, gibberellins (GA) and cytokinin (CK) also contribute to plant immune responses, with complex crosstalk between one another [23].
It is suggested that the resistance to P. sojae conferred by Rps genes is mediated by the SA signaling pathway, with the induction of SA-mediated systemic acquired resistance (SAR) occurring several days post infection (dpi) via expression of the gene _NPR1_[24-26]. During this process, two putative regulators of the chromosome condensation 1 (RCC1) gene family are down-regulated during the incompatible interaction [27]. Besides SA, exogenous treatment of 1-aminocyclopropane-1-carboxylic acid (ACC, a precursor of ET) has been shown to enhance resistance while applications of GA and ABA induce susceptibility, highlighting the complexity of phytohormone signaling pathways in response to attack by _P. sojae_[26,28,29].
A microarray study of transcriptomes from one susceptible and two partially resistant soybean genotypes indicated that 97-99% of all detectable genes experienced transcriptional modulation five dpi in response to infection by P. sojae_[30]._ However, the majority of these differences were less than two fold. Another microarray study of transcriptomic changes in soybean revealed a peak in most defense related genes at 24 hours post inoculation (hpi) with _P. sojae_[24]. Recently, Zhang et al conducted a proteomics study in which 46 differentially expressed proteins were identified in soybean after infection with _P. sojae_[31]. Among these, 26 were affected during the incompatible interaction, while the other 20 were altered during the compatible interaction. These studies have contributed to our understanding of the interaction between soybean and P. sojae. Nevertheless, what mechanisms underlie compatible and incompatible interactions, how molecular responses mediated by a variety of Rps genes/alleles resemble or differ from one another, as well as the nature of these similarity or difference, remain largely unknown.
Access to the soybean genome sequence of Williams 82 (Rps1-k) [32], and the advent of high-throughput RNA sequencing (RNA-Seq) by next-generation sequencing technology, have allowed researchers to take a novel look at the molecular interaction between soybean and P. sojae. Kim et al reported the first application of RNA-Seq for profiling gene expression in soybean in response to pathogen attack [33]. In this study, the transcriptomes of two near isogenic lines (NILs), one resistant and one susceptible to bacterial leaf pustule, were analyzed 0, 6, and 12 hpi and a total of 2,761 differentially expressed genes (DEGs), including a set of defense response genes, were identified.
NILs are pure breeding lines that are developed by backcrossing a donor line with the recurrent parent for at least five generations to achieve introgression of a desired trait. As such, they share more than 98% genetic identity with the recurrent parent, except for the region where the desired gene is located. In order to determine the molecular mechanisms responsible for _Rps_-mediated defense and to understand molecular basis for diverse responses to the pathogen, we analyzed the transcriptomes of 10 NILs, each with a unique Rps gene/allele, along with the susceptible recurrent parent Williams, pre- and post-inoculation with a race 1 isolate of P. sojae (avirulent towards NILs; virulent towards Williams).
Results
Symptom development in 10 NILs and the recurrent susceptible parent inoculated with P. sojae
The 10 soybean NILs carrying Rps1-a, Rps1-b, Rps1-c, Rps1-k, Rps3-a, Rps3-b, Rps3-c, Rps4, Rps5, or Rps6, within the Williams background, provided a unique resource for investigating the common and specific defense responses mediated by individual Rps genes against P. sojae (Additional file 1). Phenotypic reactions of the 10 NILs and Williams to P. sojae 7 days post-inoculation (dpi) with the pathogen were evaluated under greenhouse conditions. In each of three experimental replications, approximately half the number of seedlings from each line was inoculated with race 1 of P. sojae. The remaining seedlings were “mock” inoculated in the same manner without the pathogen. In each experimental replicate, Williams was susceptible (expanding lesion/plant death) to P. sojae, while all NILs were resistant (Additional file 2). NILs containing Rps1-a, Rps1-b, Rps1-c, Rps1-k and Rps4 displayed 100% survival when challenged with the pathogen. In contrast, NILs containing Rps3-a, Rps3-b, Rps3-c, Rps5 and Rps6 showed a slight variation in the proportion of surviving seedlings across the three replicates, which may be attributed to minor differences in environmental conditions. Despite this variation, the percentage of surviving seedlings in each replicate was equal to or greater than 75%, which is generally used as a criterion for defining a pure line resistance [34]. All mock-inoculated seedlings were asymptomatic of PRR (Additional file 2).
Transcriptional changes in 10 NILs and the recurrent susceptible parent in response to P. sojae
RNAs, representing the entire transcriptomes of P. sojae inoculated and mock-inoculated seedlings from each of the 10 NILs and Williams 24 hpi with the pathogen, were sequenced and 14.5 to 50.1 million raw reads were generated for individual samples (Table 1). Trimming adaptor sequences and elimination of low quality reads and reads shorter than 101 bp, resulted in 13.5 to 39.5 million clean reads for each sample. Among these, 71.5% to 87.8% per sample were uniquely mapped to the soybean reference genome v1.0 (Table 1).
Table 1.
Statistics of pair-end reads in RNA sequencing experiment
| Soybean line | Raw reads | Clean reads a | Uniquely mapped reads b | % of uniquely mapped reads c |
|---|---|---|---|---|
| Inoculated Group | ||||
| Williams (rps) | 50,126,404 | 39,475,154 | 28,222,946 | 71.5 |
| Union (Rps1-a) | 25,614,448 | 20,169,804 | 14,969,954 | 74.2 |
| L77-1863 (Rps1-b) | 28,308,350 | 22,460,578 | 16,591,206 | 73.9 |
| L75-3735 (Rps1-c) | 19,696,090 | 15,483,052 | 11,084,896 | 71.6 |
| Williams82 (Rps1-k) | 14,508,392 | 13,527,604 | 11,527,676 | 85.2 |
| L83-570 (Rps3-a) | 21,982,116 | 17,531,234 | 12,954,567 | 73.9 |
| L91-8347 (Rps3-b) | 32,063,808 | 25,210,242 | 18,704,692 | 74.2 |
| L92-7857 (Rps3-c) | 29,003,280 | 22,775,218 | 16,776,075 | 73.7 |
| L85-2352 (Rps4) | 27,891,876 | 21,857,446 | 15,860,686 | 72.6 |
| L85-3059 (Rps5) | 35,204,260 | 30,651,204 | 24,816,154 | 81.0 |
| L89-1581 (Rps6) | 26,761,058 | 23,224,142 | 19,322,975 | 83.2 |
| Mock-inoculated Group | ||||
| Williams (rps) | 21,949,578 | 20,385,960 | 17,841,238 | 87.5 |
| Union (Rps1-a) | 25,680,502 | 23,860,934 | 20,936,764 | 87.7 |
| L77-1863 (Rps1-b) | 20,303,214 | 18,830,218 | 16,453,627 | 87.4 |
| L75-3735 (Rps1-c) | 26,452,482 | 22,841,526 | 19,049,873 | 83.4 |
| Williams82 (Rps1-k) | 19,289,852 | 16,845,090 | 14,241,574 | 84.5 |
| L83-570 (Rps3-a) | 33,576,796 | 29,006,366 | 24,493,712 | 84.4 |
| L91-8347 (Rps3-b) | 25,609,764 | 23,775,078 | 20,880,124 | 87.8 |
| L92-7857 (Rps3-c) | 24,806,160 | 22,983,728 | 20,078,725 | 87.4 |
| L85-2352 (Rps4) | 24,677,426 | 22,899,582 | 20,085,744 | 87.7 |
| L85-3059 (Rps5) | 26,874,804 | 23,304,436 | 19,637,730 | 84.3 |
| L89-1581 (Rps6) | 27,363,314 | 25,362,532 | 22,151,262 | 87.3 |
Based on the 46,367 high-confidence gene models annotated in the soybean reference genome [32], the relative change in the abundance of reads mapped to the same genes for inoculated and mock-treated samples, and the criteria for defining DEGs pre- and post-inoculation, a total of 9,847 non-redundant gene models were identified as DEGs in at least one of the 11 soybean lines. The numbers of DEGs identified between Williams and the 10 NILs or among the 10 NILs vary greatly. For example, 2,014 to 2,366 DEGs were found in individual NILs containing Rps1-a, Rps1-b, Rps1-c, or Rps1-k, while 3,038 to 5,499 DEGs were detected in individual NILs containing Rps3-a, Rps3-b, Rps3-c, Rps4, Rps5 or Rps6 (Table 2). Among the 10 NILs, 1,274 to 2,823 DEGs were up-regulated, while 643 to 2,744 DEGs were down-regulated in response to inoculation with the pathogen (Table 2). In comparison, 2,460 up-regulated and 1,870 down-regulated DEGs were identified in Williams upon inoculation with P. sojae.
Table 2.
Differentially expressed genes (DEGs) in each soybean line
| Soybean line | Up-regulated DEGs | Down-regulated DEGs | Total |
|---|---|---|---|
| Williams (rps) | 2460 | 1870 | 4330 |
| Union (Rps1-a) | 1432 | 643 | 2075 |
| L77-1863 (Rps1-b) | 1536 | 831 | 2366 |
| L75-3735 (Rps1-c) | 1532 | 698 | 2230 |
| Williams82 (Rps1-k) | 1274 | 740 | 2014 |
| L83-570 (Rps3-a) | 2362 | 1827 | 4189 |
| L91-8347 (Rps3-b) | 2823 | 2166 | 4989 |
| L92-7857 (Rps3-c) | 2749 | 2247 | 4996 |
| L85-2352 (Rps4) | 2470 | 2143 | 4613 |
| L85-3059 (Rps5) | 2755 | 2744 | 5499 |
| L89-1581 (Rps6) | 2033 | 1005 | 3038 |
To validate DEGs profiled by RNA-Seq analysis, six detected DEGs per soybean line (Additional file 3) were randomly chosen for quantitative RT-PCR (qRT-PCR) analysis with the same RNA samples as used for RNA-Seq. Subsequently, the patterns of differential expression of these genes detected by RNA-Seq and qRT-PCR were compared. As shown in Additional file 4, significant correlations between the patterns of DEGs detected by the two approaches were observed for each set of genes chosen from 11 individual lines (Pearson’s correlation coefficient (r), ranging from 0.725 to 0.994), indicating the reliability of DEGs identified by RNA-Seq (Additional file 4).
Characterization of genes associated with incompatible, compatible, and basal interactions
In an attempt to decipher the molecular basis of resistance and susceptibility to P. sojae, we applied a comparative transcriptomics approach to identify DEGs specifically associated with a host response. Three major groups of genes were classified: 1) Incompatible interaction genes (IIGs), are DEGs identified in NILs and associated with resistance; 2) Compatible interaction genes (CIGs) are DEGs identified in Williams and specifically associated with the susceptible response; and 3) Basal interaction genes (BIGs), which are DEGs shared by all NILs and Williams (Figure 1).
Figure 1.
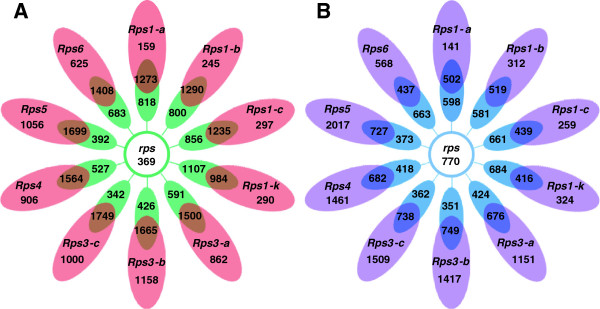
Comparison of differentially expressed genes (DEGs) between Williams (susceptible to Phytophthora sojae ) and 10 near isogenic lines (NILs) each containing a single Rps (Resistance to P . sojae ) gene. A: up-regulated DEGs. B: down-regulated DEGs. Red and purple represent the number of DEGs specific to individual Rps genes and collectively referred to as incompatible interaction genes. Brown and dark purple represent the number of DEGs shared by an individual NIL and Williams. Green and light blue represent the number of DEGs specifically expressed in ‘Williams’ when compared to a specific NIL. The central green and light blue circles represent the common proportion of Williams specific DEGs that are collectively referred to as compatible interaction genes.
A total of 5,806 non-redundant IIGs, 1139 CIGs, and 835 BIGs were identified (Figure 1). The number of up-regulated and down-regulated IIGs range from 159 to 1,158 and from 141 to 2,017, respectively, among the 10 NILs. Of the 1,139 CIGs, 369 were up-regulated and 770 were down-regulated; and of the 835 BIGs, 696 were up-regulated and 139 were down-regulated (Figure 1).
Clusters of _Rps_-mediated IIGs
To understand how transcriptomic changes mediated by different Rps genes/alleles overlap, we performed hierarchical cluster analysis using pvclust, an R package for assessing the uncertainty in hierarchical clustering [35], with log2 fold change (FC) of the 5,806 IIGs identified in at least one of the ten NILs. The NILs were grouped into three clusters, Clusters I, II, and III (Figure 2). Cluster I (C-I) consisted of the NIL containing Rps1-a only. Cluster II (C-II) was composed of those NILs containing Rps1-b, Rps1-c and Rps1-k, while Cluster III (C-III) included those NILs containing Rps3-a, Rps3-b, Rps3-c, Rps4, Rps5, and Rps6. This clustering of IIGs was further supported by the hierarchical cluster structure of the ten NILs revealed by heatmap analysis [36] (Additional file 5). In addition, the numbers of IIGs identified for each individual NIL, were unevenly distributed among different clusters (Figures 1 and 2). For example, 300, 1,193-3,073, and 556-614 IIGs were found in C-I, C-II, and C-III, respectively. The numbers of IIGs in NILs containing different alleles at a same gene locus also varied (Figures 1 and 2).
Figure 2.

Hierarchical cluster analysis (pvclust) of incompatible interaction genes identified for 10 soybean near isogenic lines, each containing a single Rps gene. The numbers behind each node represent AU (Approximately Unbiased) / BP (Bootstrap Probability) for an estimation of the confidence of each node.
The IIGs among the three clusters were analyzed by both broad range comparison, where all the genes within a cluster were counted, and narrow range comparison, where only those genes shared by the NILs within a cluster were counted. Broad range comparison showed that 75 up-regulated and 68 down-regulated IIGs were shared by the three clusters, whereas 62, 214, and 4,488 IIGs were unique to NILs in Clusters I, II, and III, respectively (Figure 3A,B). In contrast, narrow range comparison indicated that only 16 up-regulated and 7 down-regulated IIGs were shared among the three clusters (Figure 3C,D). The annotation of these 23 IIGs is listed in Table 3.
Figure 3.

Comparison of incompatible interaction genes between three clusters, C-I, C-II, and C-III. A: Broad range comparison for up-regulated genes. B: Broad range comparison for down-regulated genes. C: Narrow range comparison for up-regulated genes. D: Narrow range comparison for down-regulated genes.
Table 3.
Incompatible interaction genes (IIGs) shared by all the near isogenic lines and their annotation
| Gene | Arabidopsis homologous gene | Gene symbol | Gene annotation | GO group |
|---|---|---|---|---|
| Up regulated | ||||
| Glyma01g32130 | AT2G40435 | - | Transcription factor, bHLH | Response to biotic or abiotic stress |
| Glyma02g16710 | AT1G03220 | - | Eukaryotic aspartyl protease family protein | Metabolism |
| Glyma03g01010 | AT4G26010 | - | Peroxidase superfamily protein | Response to biotic or abiotic stress |
| Glyma05g34870 | AT1G14870 | PCR2 | Plant cadmium resistance 2 | - |
| Glyma06g09220 | AT5G25880 | NADP-ME3 | NADP-malic enzyme 3 | Response to biotic or abiotic stress |
| Glyma06g13090 | AT5G24110 | WRKY30 | WRKY DNA-binding protein 30 | Response to biotic or abiotic stress |
| Glyma09g15090 | AT4G21380 | RK3 | Receptor kinase 3 | Metabolism |
| Glyma11g07430 | AT5G03260 | LAC11 | Laccase 11 | Metabolism |
| Glyma13g17800 | AT4G04450 | WRKY42 | WRKY family transcription factor | Biological regulation |
| Glyma13g30770 | AT1G28480 | GRX480 | Thioredoxin superfamily protein | Response to biotic or abiotic stress |
| Glyma13g36070 | AT3G20660 | 4-Oct | Organic cation/carnitine transporter4 | Biological regulation |
| Glyma14g39300 | AT1G66160 | CMPG1 | CYS, MET, PRO, and GLY protein 1 | Response to biotic or abiotic stress |
| Glyma15g05810 | AT2G41480 | - | Peroxidase superfamily protein | Response to biotic or abiotic stress |
| Glyma15g06010 | AT5G64260 | EXL2 | Exordium like 2 | - |
| Glyma15g19600 | AT2G05940 | - | Protein kinase superfamily protein | Metabolism |
| Glyma20g38000 | AT1G09090 | RBOHB | Respiratory burst oxidase homolog B | Response to biotic or abiotic stress |
| Down regulated | ||||
| Glyma04g12290 | AT1G71380 | CEL3 | Cellulase 3 | Response to biotic or abiotic stress |
| Glyma09g37290 | AT5G15230 | GASA4 | GAST1 protein homolog 4 | Response to biotic or abiotic stress |
| Glyma12g10670 | AT3G21700 | SGP2 | Ras-related small GTP-binding family protein | Response to biotic or abiotic stress |
| Glyma12g29980 | AT4G39370 | - | Ubiquitin-specific protease 27 | - |
| Glyma13g44210 | AT5G20190 | - | Tetratricopeptide repeat (TPR)-like superfamily protein | - |
| Glyma15g18440 | AT4G29720 | PAO5 | Polyamine oxidase 5 | Metabolism |
| Glyma18g49400 | AT5G15230 | GASA4 | GAST1 protein homolog 4 | Response to biotic or abiotic stress |
Putative functions of DEGs based on gene ontology (GO) analysis
To understand the functional components involved in molecular responses to P. sojae, we annotated the putative products encoded by IIGs, CIGs, and BIGs based on GO analysis (Figure 4; Additional file 6). These DEGs were grouped into seven categories: 1) Response to biotic or abiotic stress; 2) Biological regulation; 3) Growth and development; 4) Transport; 5) Metabolism; 6) Miscellaneous; and 7) Unclassified. For IIGs, the majority of annotated genes fell into the category of “response to biotic stress and abiotc stress” (Figure 4; Additional files 6 and 7). This pattern was also observed for CIGs and BIGs (Figure 4; Additional file 7). Of the 23 IIGs shared by the 10 NILs, 12, 2, and 5, were found to be related to “response to biotic or abiotic stress”, “biological regulation”, and “metabolism”, respectively (Table 3). The remaining four shared genes could not be classified into any functional category.
Figure 4.

Heatmap analysis of three categories of differentially expressed genes based on gene ontology analysis. The values used to draw heatmaps are Log2 (fold change) of expression level of post inoculation to mock inoculation. A: Incompatible interaction genes. The Log2 (fold change) less than 3 in all 10 resistant near isogenic lines were not shown here. B: Compatible interaction genes. C: Core basal interaction genes.
Putative transcription factors (TFs) involved in molecular responses to P. sojae
It is documented that TFs play important roles in plant defense and stress responses. As such, we annotated and analyzed putative TFs in the sets of DEGs detected in this study based on a soybean TF database that is composed of 5,671 predicted TFs [37]. Of the 5,806 non-redundant IIGs, 282 up-regulated and 543 down-regulated genes were annotated as putative TFs. The up-regulated and down-regulated IIG TFs range from 13 to 177 and from 21 to 307, respectively, among the 10 NILs (Additional files 8 and 9). The number of IIG TFs also varied among the three clusters, with 34, 164, and 791 in C-I, C-II, and C-III, respectively. The most abundant ones along these IIG TFs (either up-regulated or down-regulated) were of the WRKY family, accounting for 23.1% of all IIG TFs identified in C-I, 18.5-25.8% in C-II, and 14.0-18.6% in C-III (Additional file 9). Only three up-regulated IIG TFs (1 bHLH and 2 WRKY) and one down-regulated TF (TPR) were found to be shared by all the 10 NILs (Table 3; Additional file 9).
Of the 1,139 CIGs identified in Williams, 43 up-regulated and 149 down-regulated genes were found to be putative TFs (Additional file 9). The largest proportion of up-regulated CIG TFs belonged to the MYB/HD-like family (16.3%), followed by the AP2-EREBP family (14.0%). In contrast, members of the AP2-EREBP family represented the largest number (20.8%) of down-regulated CIG TFs, followed by MYB/HD-like (10.1%) and bHLH (9.4%). None of these 43 up-regulated CIG TFs belonged to WRKY family.
Of the 835 non-redundant BIGs shared by all the 11 soybean lines, 41 up-regulated and 9 down-regulated ones were found to be TFs (Additional file 9). Among the up-regulated BIG TFs, the most abundant ones are of the WRKY family (34%), followed by the MYB/HD-like family (19.5%).
Knowledge-based comparative analysis of Rps gene-mediated defense signaling pathways
The DEGs showing homologies to previously identified protein genes responsible for pathogen recognition and defense, or defense-related phytohormone signaling genes were further analyzed. Overall, these DEGs exhibited diverse patterns of distribution among the three NIL clusters (Figure 5). For example, of the 26 up-regulated IIGs homologous to previously reported defense/stress signaling genes, 24 were found only in NILs within Cluster III. In contrast, only one (Glyma20g38000) of the two RBOH B gene homologs was found to be up-regulated in all 10 NILs. It is notable that the majority of the putative DEGs involved in defense-related phytohormone signaling pathways showed distinct or opposite patterns of variation in gene expression between Williams and NILs, particularly the NILs within the Cluster III (Figure 5).
Figure 5.

Expression of selected IIGs or CIGs involved in defense response and defense related phytohormone signaling pathways. The values and the corresponding depth of different colors in each square indicate Log2 (fold change) of expression level of post inoculation to mock inoculation. aClassification of differentially expressed genes. IIG = Incompatible interaction gene. CIG = Compatible interaction gene. bClustering of Rps genes based on hierarchical cluster analysis of IIGs. C-I = Cluster I. C-II = Cluster II. C-III = Cluster III.
Previous studies indicated that the resistance to P. sojae in soybean was mediated by SA signaling [24-26], with the NPR1 as the key component of SA-mediated signaling [38]. In this study, two _NPR1_-like IIGs (Glyma02g45260 and Glyma14g03510) were identified (Figure 5). However, these two genes differ from the two _NPR1_-like genes reported by Sandhu et al [25]. In the later study, they studied soybean leaves four dpi with P. sojae race 4 while, in the present study, we studied stems 24 hpi with P. sojae race 1. As such, these two studies are less comparable.
As an antagonist of SA, the JA pathway is repressed by JAZ proteins in _Arabidopsis_[39,40]. Several JAZ homologs were identified as IIG and/or CIG, including homologs of JAZ1, JAZ6, JAZ8 and JAZ12 (Figure 5). These homologs showed a consistent expression pattern that was up-regulated NILs and down-regulated in Williams. In contrast to the JAZ proteins, JAR1 (Jasmonate resistant1) appears to be required for the activation of the JA signaling pathway in _Arabidopsis_[41,42]. We found that a soybean JAR1 homolog (Glyma07g06370) was up-regulated in Williams upon inoculation with the P. sojae. These observations suggest that the JA signaling pathway may have played opposite roles in phytohormone signaling between incompatible and compatible interactions.
The ET signaling pathway is generally considered to work cooperatively with JA in response to necrotrophic pathogens [8,22]. In Arabidopsis, ET receptors _ETR2_[43] and _EIN4_[44] were both negative regulators of ET signaling, and an ubiquitin/proteasome pathway for the degradation of EIN3 was mediated by an F-box protein, EBF1, to negatively regulate ET responses [23]. We found that one homolog of ETR2 and two homologs of EIN4 were down-regulated in NILs, while another homolog of ETR2 was up-regulated in Williams. In addition to ETR2 and EIN4, we identified five EBF1 homologs that were down-regulated in all NILs and up-regulated in Williams (Figure 5), suggesting that the ET signaling response to the pathogen was repressed during compatible interactions but activated during incompatible interactions.
In addition to the three major phytohormones described above, BR have also been found to enhance disease resistance against viral and bacterial pathogens in tobacco and rice [45]. BAK1 is a crucial component of BR signaling, and can interact with LRR-RLK (leucine-rich repeat receptor-like protein kinase) genes such as FLS2 to promote their function in plant defense responses [46]. As shown in Figure 5, three BAK1 homologs were identified as IIGs and all were up-regulated upon inoculation with the pathogen, suggesting a positive role of BR signaling in defense response to P. sojae.
Discussion
The nature of distinction in molecular immune response among NILs
One of the most striking observations in this study was the distinction in defense response to inoculation with P. sojae among the ten resistant NILs as revealed by comparative transcriptomic analysis. This was reflected not only by the level of variation in the number of IIGs (ranging from 300 to 3073) detected in individual NILs, but also by the relative small numbers of up-regulated (16) and down-regulated (7) IIGs shared by all NILs (Table 3). In general, different Rps genes recognize distinct Avr genes in a P. sojae population [47], but the Avr determinants of different Rps genes in a same isolate of the pathogen (i.e., race 1 in this study) may not be distinct. One hypothesis that may explain the observed variations of DEGs among different NILs may be differential timing and robustness in defense responses and signaling mediated by different Rps genes/alleles [48]. Although the timing of gene expression mediated by different R genes in response to the same pathogen has not been characterized in plants, dramatic and rapid changes in gene expression after inoculation with, or following infection by a pathogen, have been observed [24,49]. A recent study demonstrated that ~5% of DEGs were shared by soybean lines resistant to bacterial leaf pustule at 6 and 12 hpi with _Xanthomonas axonopodis_[33]. In this study, the majority (>95%) of DEGs showed less than an eight-fold difference in expression levels upon inoculation with the pathogen when given the same cutoff time for each of the 11 lines. Such a low level of expressional changes and uniformed cutoff may have affected the detectability of shared DEGs among different lines. In addition, the NILs remain 1-2% genomic difference (mostly surrounding the Rps loci) from each other, which may have affected expression patterns of a small proportion of genes. Furthermore, inoculated regions of seedling stems, near which layers of cells respond to the pathogen, vary in size among different plants and NILs. Such a variation may affect effective identification of DEGs. Therefore, the numbers of DEGs, and particularly IIGs specific to each NIL could be over-estimated. Nevertheless, the DEGs in each line were detected pre-and post-inoculation with the same pathogen, and thus the influence of genomic difference surrounding Rps genes on our pipeline for detection of DEGs triggered by the pathogen may be minimal.
It is documented that resistance genes recognize pathogens either directly or indirectly by guarding a protein or using a decoy [13]. In Arabidopsis, RIN4 is ‘guarded’ by resistance genes RPM1 and RPS2, which trigger the immune response [14,15]. In soybean, two of four _RIN4_- homologous genes appear to function as a heteromeric complex in mediating RPG1-B and RPM1-derived resistance to Pseudomonas syringae, and silencing GmRIN4a or GmRIN4b in rpg1-b plants enhances basal resistance to virulent strains of P. syringae and the oomycete _P. sojae_[50]. In our study, a single RIN4 homolog was found to be up-regulated in C-III NILs, but not in NILs within C-I and C-II. If this RIN4 homolog did encode the “guard” protein that was recognized by the Rps genes in the C-III NILs, then the relatively low numbers of IIGs identified among NILs in C-I and C-II, as compared with the C-III NILs, may be explained by the distinct “recognition” processes needed for triggering the immune response, which may result in variations in speed, timing, and magnitude of molecular responses to the pathogen [48,51].
Do patterns in transcriptomics distinguish Rps genes from alleles?
An allele is defined as an alternative form of the same gene that is located at a specific position on a specific chromosome. In general, different alleles of the same gene are determined by genetic tests for allelism. However, due to the complexity of phenotypes, such as variation in the proportion of surviving plants of a pure NIL that contains an Rps gene, as observed in this study, such a test may not be effective in determining whether the resistance carried by two individual lines is controlled by different alleles or different genes. Moreover, many NBS-LRR gene models predicted in the Williams82 genome are clustered in a small number of genomic regions [32,52], which makes it more difficult to distinguish genes resistant to the same pathogen by classic genetic analysis, especially if they are physically adjacent or closely linked to each other.
The Rps genes/alleles in the 10 NILs have been previously mapped to three genomic regions: Rps1-a, Rps1-b, Rps1-c, and Rps1-k on chromosome 3 [6,53,54], Rps3-a, Rps3-b and Rps3-c on chromosome 13 [1,54,55] and Rps4, Rps5 and _Rps_6 on chromosome 18 [1,54,55]. However, because limited numbers of molecular markers were available and used when these genes/alleles were identified, the genetic distances between genes or alleles mapped in the same chromosomal regions have not been determined. Actually, the alleles at the Rps1 and Rps3 loci were simply designated based on their disease reaction to a set of P. sojae races. As these three regions are highly enriched with NBS-LRR genes, according to the soybean reference genome, it is possible that some designated alleles of the same gene may be located at different loci.
As different alleles of the same gene are generally more identical to each other than to other genes, people are inclined to speculate that the defense response mediated by different alleles of a same gene may be more similar than those mediated by different genes. At first glance, this hypothesis seems to be supported by the observation that the NILs with Rps3-a and Rps3-b exhibited similar patterns of transcriptional changes upon inoculation with P. sojae, as did the NILs with Rps1-b and Rps1-c (Figure 2). However, the pattern of transcriptional changes mediated by Rps3-c was found to be most similar to those mediated by Rps4, Rps5, and Rps6. Thus, the patterns of transcriptomic changes do not appear to predict genetic similarity and difference among these Rps genes and alleles, although we could not rule out the possibility that Rps3-c and Rps1-a are non-allelic to the Rps3 and Rps1 loci, respectively.
An integrated picture of molecular responses to P. sojae
Based on analysis of putative defense-related genes identified in this study and the patterns of their expressions upon inoculation with the pathogen, we propose a hypothesis that four major phytohormone signaling pathways are involved in defense responses (Figure 6A). During incompatible interaction, the Rps protein recognizes the avirulence effector of P. sojae, initiating signaling transduction that involves the SA, JA, ET and BR pathways. The SA, ET, and BR pathways are activated, whereas the JA pathway is suppressed. Although the specific functions of individual genes encoding TFs belonging to the WRKY family, as well as those genes for reactive oxygen species (ROS) and MAP kinase (MAPK) cascades remain to be elucidated, it is apparent that some of the genes belonging to these families play pivotal roles in defense signaling transduction. During compatible interaction, the avirulence effector of P. sojae induces a set of downstream responses that lead to disease development (Figure 6B). The signatures of the susceptible responses would include up-regulation of the JA pathway, suppression of ET pathway and with no significant changes in SA and BR pathways. It seems possible that the defense responses might be delayed in the compatible interaction due to insufficient activation of the expression of genes related to ROS and phytoalexin biosynthesis, as well as MAPK signaling. Together, these data gained from a unique set of soybean NILs provide a comprehensive picture regarding the molecular mechanisms underlying incompatible and compatible interaction between soybean and P. sojae, which shed light on the nature and timing of molecular responses mediated by individual Rps genes.
Figure 6.
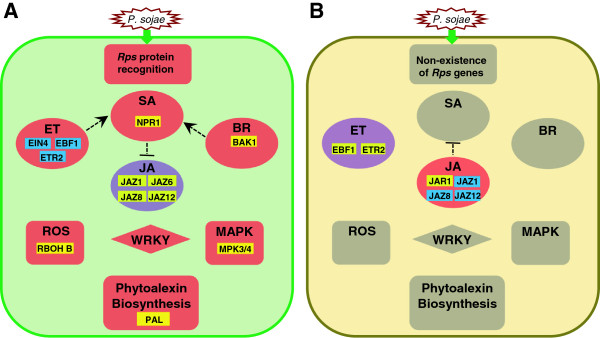
An integrated picture for gene network of interactions between soybean and race1 of Phytophthora sojae. A: Incompatible interaction. B: Compatible interaction. Red = up-regulation of a pathway. Purple = down-regulation of a pathway. Grey = no significant change in pathway. Yellow = up-regulation of genes. Blue = down-regulation of genes. Dotted lines indicate interactions of pathways inferred from Arabidopsis studies.
Conclusions
Comparative transcriptomic analysis of ten resistant NILs and susceptible recurrent line Williams with and without inoculation allowed us to identify DEGs associated with defense responses to P. sojae that were unique in each NIL and common among all these NILs, and thus depicted functional “fingerprints” of individual _Rps_-mediated resistance. Further analysis revealed multiple branches of putative ET, JA, ROS, and MAPK regulatory networks underlying the defense responses. Such responses exhibited dramatic variations among the soybean NILs containing distinct Rps genes/alleles. We propose different timing of individual _Rps_-mediated defense signaling to the same pathogen may be largely responsible for such variations.
Methods
Plant materials and isolates of P. sojae
The susceptible cultivar Williams and its ten NILs, each containing a unique Rps gene/allele, were used this study (Additional file 1). Each NIL was generated by backcrossing the donor for at least five generations with Williams. For inoculation tests, the isolate pmg(1)-3 (pathotype race1) of P. sojae was used.
Treatment of soybean materials
Seeds of each soybean line were planted in sterilized sand in 10.1 cm clay pots and placed in a greenhouse. Approximately seven days after planting, at the VC growth stage (unifoliate leave fully expanded), the seedlings for each line were separated into four groups, each containing ~20 seedlings. Two groups were challenged with P. sojae using a standard hypototyl inoculation method [56], while the other two groups were mock inoculated (wounded) without the pathogen. At 24 hpi, stems were harvested from one set of inoculated and one set of wounded seedlings by excising 2 to 3 cm across the wounded site and storing immediately in liquid nitrogen. The seedlings in the remaining two groups were kept for evaluating symptom development, which was recorded 7 dpi. Experiments were performed a total of three times.
RNA sequencing and analysis
Total RNAs were isolated using the RNeasy Plant Mini Kit (Qiagen Inc., Valencia, CA) according to the manufacturer’s instructions in conjunction with DNase. The quality of total RNA was determined using a Nanodrop spectrometer (Thermo Fisher Scientific Inc., Wilmington, DE) and 1% formaldehyde gel electrophoresis. Total RNA samples were then sent to the Genomics Center at Purdue University for sequencing and 101 bp paired-end reads were generated with the Illumina HiScanSQ system. Since samples were a mixture of RNA from both soybean and P. sojae, to exclude the effect of P. sojae sequences, all reads that aligned to the P. sojae genome sequence were eliminated [57]. All remaining reads were then aligned to G. max reference genome (v8.0, http://www.phytozome.net) using TopHat software [58] with parameters set to allow only one mismatch, and 30 and 100,000 bp of the minimum and maximum intron length based on the current gene annotation. Moreover, only the uniquely mapped reads or fragments were kept for further analyses. The raw count of each gene was calculated by HTSeq (http://www-huber.embl.de/users/anders/HTSeq) with the “intersection_nonempty” mode, and preceded to edgeR package [59] in the R-Bioconductor tools.
To detect variability, we estimated the over dispersion from 40 highly confident soybean housekeeping genes collected from previously reported papers (Additional file 10) [24,33,60-63], considering different soybean lines as replicates of control and inoculation groups. The data were then modeled using a Negative Binomial model in edgeR to identify differentially expressed genes after inoculation using the dispersion estimated from the housekeeping genes as a common dispersion. Any genes with coverage read count less than one count per million of two paired samples were removed in later analyses. Differential expression between the inoculation group and the mock-inoculation group for each line was tested for false discovery rate (FDR) that was controlled at 0.05 [64] using edgeR package [59].
Hierarchical cluster and heatmap analysis
Pvclust and hclust were performed using the log2FC of the 5,806 IIGs with default parameters using distance method “correlation” for complete linkage clustering analysis. Pvclust provides two types of _p_-values to assess the uncertainty for each cluster: approximately unbiased (AU) p-value and bootstrap probability (BP) value, via multi-scale bootstrap re-sampling [35]. The heatmap representing the expression intensity and direction was drawn using pheatmap R package with the distance method “euclidean” for both rows and columns [65].
Gene Ontology, heatmap and homologous gene functional analysis
Annotations of GO terms were obtained from the AgriGO website based on the G. max model [66], and GO biological process categories were used. These terms were manually classified into six broad functional groups, ‘response to biotic or abiotic stress’, ‘biological regulation’, ‘growth and development’, ‘transport’, ‘metabolism’, and ‘miscellaneous’, while genes without GO annotations were grouped as ‘unclassified’. For genes with multiple GO categories, only one category was selected based on priority. Homologous genes were searched against annotated Arabidopsis gene protein database (The Arabidopsis Information Resouces, http://www.arabidopsis.org) using BLASTP to verify gene functions manually.
Quantitative real-time PCR analysis
The qRT-PCR was carried out using StepOnePlus™ Real Time PCR system (Applied Biosystems). The RNA samples used as templates for qRT-PCR were the same as those used for RNA-Seq. The cons4 gene [60] was used as internal control for normalization of qRT-PCR data. For each gene, three experimental replicates were performed. Pearson correlations were calculated between RNA-Seq and qRT-PCR methods using six out of seven randomly selected genes for Log2 fold change of inoculated and mock-inoculated treatments in each soybean line (Additional file 3).
Availability of supporting data
The transcriptome sequences presented in this study have been deposited in NCBI Gene Expression Omnibus under accession numbers GSE48524.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TJH and JM designed the research; FL, MZ, RWD, TJH, and JM interpreted the data; FL, MZ, DDB, JP, LS, YL, BZ, ZT performed the research; FL, MZ, RWD, TJH, and JM drafted and revised the manuscript; FL, MZ, DDB, JP, LS, YL, BZ, ZT, RWD, TJH, and JM approved the submission of the version. All authors read and approved the final manuscript.
Supplementary Material
Additional file 1
Breeding pedigree of soybean NILs used in this study.
Additional file 2
Reactions of soybean NILs to Phytophthora sojae .
Additional file 3
List of primers used in qPCR.
Additional file 4
Comparison in expression of six soybean genes in Williams and 10 NILs, each containing a single Rps gene, as determined by RNA-Seq analysis or qRT-PCR. Y axis indicates differential expression of selected genes for each soybean line. X axis indicates selected genes used for qRT-PCR. These genes are Glyma02g47940 (1), Glyma04g20330 (2), Glyma05g24770 (3), Glyma07g07270 (4), Glyma09g37290 (5), Glyma10g44170 (6), and Glyma11g04130 (7). Pearson’s corrleation coefficient (r).
Additional file 5
Heatmap (hcluster) analysis of 5,806 IIGs for 10 soybean NILs, each containing a single Rps gene. The values used to draw heatmap is Log2 (fold change) of expression level of post inoculation to mock inoculation.
Additional file 6
GO analysis of DEGs in different categories.
Additional file 7
Gene ontology categories for IIGs identified in soybean NILs, each containing a single Rps gene. Left bar = up-regulated genes. Right bar = down-regulated genes.
Additional file 8
Comparison of differentially expressed transcription factors (DETFs) between Williams and 10 NILs each containing a single Rps gene. A: up-regulated DETFs. B: down-regulated DETFs. Red and purple represent the number of DETFs specific to individual Rps genes and collectively referred to as incompatible interaction transcription factors. Brown and dark purple represent the number of DETFs shared by an individual NIL and Williams. Green and light blue represent the number of DETFs specifically expressed in Williams when compared to a specific NIL. The central green and light blue circles represent the common proportion of Williams-specific DETFs that are collectively referred to as compatible interaction transcription factors.
Additional file 9
Counts of transcription factors (TFs) in different groups.
Additional file 10
Housekeeping genes selected in this study.
Contributor Information
Feng Lin, Email: feng.lin@pioneer.com.
Meixia Zhao, Email: zhao185@purdue.edu.
Douglas D Baumann, Email: dbaumann@uwlax.edu.
Jieqing Ping, Email: ping@purdue.edu.
Lianjun Sun, Email: sun259@purdue.edu.
Yunfeng Liu, Email: liu969@purdue.edu.
Biao Zhang, Email: zb380345709@126.com.
Zongxiang Tang, Email: zxtang@sicau.edu.cn.
Elisa Hughes, Email: elisadh@umich.edu.
Rebecca W Doerge, Email: doerge@purdue.edu.
Teresa J Hughes, Email: hughestj@purdue.edu.
Jianxin Ma, Email: maj@purdue.edu.
Acknowledgements
This work was mainly supported by soybean checkoff funds from the Indiana Soybean Alliance (Grant No. 205267), and partially supported by “Partnership for Research & Education in Plant Breeding and Genetics” program funded by the U.S. Department of Agriculture’s National Institute of Food and Agriculture, and corporate partners Ag Alumni Seed, AgReliant Genetics, Beck’s Hybrids, ConAgraFoods, Dow AgroSciences, Indiana Crop Improvement Association and Pioneer Hi-Bred International. We also thank Brittany Radke, Tomara J. Fleury and Brian Foss for their help in inoculation and preparation of inoculum.
References
- Sandhu D, Schallock KG, Rivera-Velez N, Lundeen P, Cianzio S, Bhattacharyya MK. Soybean Phytophthora resistance gene Rps8 maps closely to the Rps3 region. J Hered. 2005;96:536–541. doi: 10.1093/jhered/esi081. [DOI] [PubMed] [Google Scholar]
- Dorrance AE, Mills D, Robertson AE, Draper MA, Giesler L, Tenuta A. Phytophthora root and stem rot of soybean. The Plant Health Instructor. 2007. 10.1094/PHI-I-2007-0830-07.
- Tyler BM. Phytophthora sojae: root rot pathogen of soybean and model oomycete. Mol Plant Pathol. 2007;8:1–8. doi: 10.1111/j.1364-3703.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Kato M, Yoshida S, Matsumoto I, Kobayashi T, Kaga A, Hajika M, Yamamoto R, Watanabe K, Aino M. et al. Pathogenic diversity of Phytophthora sojae and breeding strategies to develop Phytophthora-resistant soybeans. Breeding Sci. 2012;61:511–522. doi: 10.1270/jsbbs.61.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Zhao M, Ping J, Johnson A, Zhang B, Abney TS, Hughes TJ, Ma J. Molecular mapping of two genes conferring resistance to Phytophthora sojae in a soybean landrace PI 567139B. Theor Appl Genet. 2013;126:2177–2185. doi: 10.1007/s00122-013-2127-4. [DOI] [PubMed] [Google Scholar]
- Gao H, Narayanan NN, Ellison L, Hattacharyya MK. Two classes of highly similar coiled coil-nucleotide binding-leucine rich repeat genes isolated from the Rps1-k locus encode Phytophthora resistance in soybean. Mol Plant-Microbe Interact. 2005;18:1035–1045. doi: 10.1094/MPMI-18-1035. [DOI] [PubMed] [Google Scholar]
- Dangl JL, Jones JDG. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet. 2010;11:539–548. doi: 10.1038/nrg2812. [DOI] [PubMed] [Google Scholar]
- Fliegmann J, Mithofer A, Wanner G, Ebel J. An ancient enzyme domain hidden in the putative beta-glucan elicitor receptor of soybean may play an active part in the perception of pathogen-associated molecular patterns during broad host resistance. J Biol Chem. 2004;279:1132–1140. doi: 10.1074/jbc.M308552200. [DOI] [PubMed] [Google Scholar]
- Dodds PN, Lawrence GJ, Catanzariti AM, Ayliffe MA, Ellis JG. The Melampsora lini AvrL567 avirulence genes are expressed in haustoria and their products are recognized inside plant cells. Plant Cell. 2004;16:755–768. doi: 10.1105/tpc.020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Biezen EA, Jones JDG. Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem Sci. 1998;23:454–456. doi: 10.1016/S0968-0004(98)01311-5. [DOI] [PubMed] [Google Scholar]
- Van der Hoorn RAL, Kamoun S. From guard to decoy: a new model for perception of plant pathogen effectors. Plant Cell. 2008;20:2009–2017. doi: 10.1105/tpc.108.060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkhadir Y, Nimchuk Z, Hubert DA, Mackey D, Dangl JL. Arabidopsis RIN4 negatively regulates disease resistance mediated by RPS2 and RPM1 downstream or independent of the NDR1 signal modulator and is not required for the virulence functions of bacterial type III effectors AvrRpt2 or AvrRpm1. Plant Cell. 2004;16:2822–2835. doi: 10.1105/tpc.104.024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey D, Belkhadir Y, Alonso JM, Ecker JR, Dangl JL. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell. 2003;112:379–389. doi: 10.1016/S0092-8674(03)00040-0. [DOI] [PubMed] [Google Scholar]
- Glazebrook J, Rogers EE, Ausubel FM. Isolation of Arabidopsis mutants with enhanced disease susceptibility by direct screening. Genetics. 1996;143:973–982. doi: 10.1093/genetics/143.2.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarts N, Metz M, Holub E, Staskawicz BJ, Daniels MJ, Parker JE. Different requirements for EDS1 and NDR1 by disease resistance genes define at least two R gene-mediated signaling pathways in Arabidopsis. Proc Nat Acad Sci USA. 1998;95:10306–10311. doi: 10.1073/pnas.95.17.10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N, Tootle TL, Tsui F, Klessig DF, Glazebrook J. PAD4 functions upstream from salicylic acid to control defense responses in Arabidopsis. Plant Cell. 1998;10:1021–1030. doi: 10.1105/tpc.10.6.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A, Feys BJ, Frost LN, Jones JDG, Daniels MJ, Parker JE. EDS1, an essential component of R gene-mediated disease resistance in Arabidopsis has homology to eukaryotic lipases. Proc Nat Acad Sci USA. 1999;96:3292–3297. doi: 10.1073/pnas.96.6.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiermer M, Feys BJ, Parker JE. Plant immunity: the EDS1 regulatory node. Curr Opin Plant Biol. 2005;8:383–389. doi: 10.1016/j.pbi.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee S, Halane MK, Kim SH, Gassmann W. Pathogen effectors target Arabidopsis EDS1 and alter its interactions with immune regulators. Science. 2011;334:1405–1408. doi: 10.1126/science.1211592. [DOI] [PubMed] [Google Scholar]
- Glazebrook J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 2005;43:205–227. doi: 10.1146/annurev.phyto.43.040204.135923. [DOI] [PubMed] [Google Scholar]
- Robert-Seilaniantz A, Grant M, Jones JDG. Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu Rev Phytopathol. 2011;49:317–343. doi: 10.1146/annurev-phyto-073009-114447. [DOI] [PubMed] [Google Scholar]
- Moy P, Qutob D, Chapman BP, Atkinson I, Gijzen M. Patterns of gene expression upon infection of soybean plants by Phytophthora sojae. Mol Plant-Microbe Interact. 2004;17:1051–1062. doi: 10.1094/MPMI.2004.17.10.1051. [DOI] [PubMed] [Google Scholar]
- Sandhu D, Tasma IM, Frasch R, Bhattacharyya MK. Systemic acquired resistance in soybean is regulated by two proteins, Orthologous to Arabidopsis NPR1. BMC Plant Biol. 2009;9:105. doi: 10.1186/1471-2229-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugano S, Sugimoto T, Takatsuji H, Jiang CJ. Induction of resistance to Phytophthora sojae in soyabean (Glycine max) by salicylic acid and ethylene. Plant Pathol. 2013;62:1048–1056. doi: 10.1111/ppa.12011. [DOI] [Google Scholar]
- Narayanan NN, Grosic S, Tasma IM, Grant D, Shoemaker R, Bhattacharyya M. Identification of candidate signaling genes including regulators of chromosome condensation 1 protein family differentially expressed in the soybean–Phytophthora sojae interaction. Theor Appl Genet. 2009;118:399–412. doi: 10.1007/s00122-008-0895-z. [DOI] [PubMed] [Google Scholar]
- Ward EWB, Cahill DM, Bhattacharyya M. Abscisic acid suppression of phenylalanine ammonialyase activity and mRNA, and resistance of soybeans to Phytophthora megasperma f. sp. glycinea. Plant Physiol. 1989;91:23–27. doi: 10.1104/pp.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KL, Cahill DM. Influence of abscisic acid and the abscisic acid biosynthesis inhibitor, norflurazon, on interactions between Phytophthora sojae and soybean (Glycine max) Eur J Plant Pathol. 1999;105:651–658. doi: 10.1023/A:1008705321113. [DOI] [Google Scholar]
- Zhou L, Mideros SX, Bao L, Hanlon R, Arredondo FD, Tripathy S, Krampis K, Jerauld A, Evans C, St Martin SK. et al. Infection and genotype remodel the entire soybean transcriptome. BMC Genomics. 2009;10:49. doi: 10.1186/1471-2164-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhao J, Xiang Y, Bian X, Zuo Q, Shen Q, Gai J, Xing H. Proteomics study of changes in soybean lines resistant and sensitive to Phytophthora sojae. Proteome Sci. 2011;9:52. doi: 10.1186/1477-5956-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, Ma JX, Mitros T, Nelson W, Hyten DL, Song QJ, Thelen JJ, Cheng JL. et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- Kim KH, Kang YJ, Kim DH, Yoon MY, Moon JK, Kim MY, Van K, Lee SH. RNA-Seq analysis of a soybean near-isogenic line carrying bacterial leaf pustule-resistant and -susceptible alleles. DNA Res. 2011;18:483–497. doi: 10.1093/dnares/dsr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrance AE, Berry SA, Anderson TR, Meharg C. Isolation, storage, pathotype characterization, and evaluation of resistance for Phytophthora sojae in soybean. Plant Health Progress. 2008. 10.1094/PHP-2008-0118-01-DG.
- Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Nat Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Libault M, Joshi T, Valliyodan B, Nguyen HT, Xu D, Stacey G, Cheng JL. SoyDB: a knowledge database of soybean transcription factors. BMC Plant Biol. 2010;10:14. doi: 10.1186/1471-2229-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X. NPR1, all things considered. Curr Opin Plant Biol. 2004;7:547–552. doi: 10.1016/j.pbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Gfeller A, Liechti R, Farmer EE. Arabidopsis jasmonate signaling pathway. Science. 2010;3:1–2. doi: 10.1126/scisignal.3109cm4. [DOI] [PubMed] [Google Scholar]
- Pauwels L, Goossens A. The JAZ proteins: a crucial interface in the jasmonate signaling cascade. Plant Cell. 2011;23:3089–3100. doi: 10.1105/tpc.111.089300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staswick PE, Tiryaki I, Rowe ML. Jasmonate response locus JAR1 and several related Arabidopsis genes encode enzymes of the firefly luciferase superfamily that show activity on jasmonic, salicylic, and indole-3-acetic acids in an assay for adenylation. Plant Cell. 2002;14:1405–1415. doi: 10.1105/tpc.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staswick PE, Tiryaki I. The oxylipin signal jasmonic acid is activated by an enzyme that conjugates it to isoleucine in Arabidopsis. Plant Cell. 2004;16:2117–2127. doi: 10.1105/tpc.104.023549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Hua J, Chen QG, Chang C, Medrano LJ, Bleecker AB, Meyerowitz EM. ETR2 is an ETR1-like gene involved in ethylene signaling in Arabidopsis. Proc Nat Acad Sci USA. 1998;95:5812–5817. doi: 10.1073/pnas.95.10.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J, Sakai H, Nourizadeh S, Chen QG, Bleecker AB, Ecker JR, Meyerowitz EM. EIN4 and ERS2 are members of the putative ethylene receptor gene family in Arabidopsis. Plant Cell. 1998;10:1321–1332. doi: 10.1105/tpc.10.8.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashita H, Yasuda M, Nitta T, Asami T, Fujioka S, Arai Y, Sekimata K, Takatsuto S, Yamaguchi I, Yoshida S. Brassinosteroid functions in a broad range of disease resistance in tobacco and rice. Plant J. 2003;33:887–898. doi: 10.1046/j.1365-313X.2003.01675.x. [DOI] [PubMed] [Google Scholar]
- Clouse SD. Brassinosteroid signal transduction: from receptor kinase activation to transcriptional networks regulating plant development. Plant Cell. 2011;23:1219–1230. doi: 10.1105/tpc.111.084475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May KJ, Whisson SC, Zwart RS, Searle IR, Irwin JAG, Maclean DJ, Carroll BJ, Drenth A. Inheritance and mapping of 11 avirulence genes in Phytophthora sojae. Fungal Genet Biol. 2002;37:1–12. doi: 10.1016/S1087-1845(02)00027-0. [DOI] [PubMed] [Google Scholar]
- Tao Y, Xie Z, Chen W, Glazebrook J, Chang HS, Han B, Zhu T, Zou G, Katagiri F. Quantitative nature of Arabidopsis responses during compatible and incompatible interactions with the bacterial pathogen Pseudomonas syringae. Plant Cell. 2003;15:317–330. doi: 10.1105/tpc.007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KB, Foley RC, Onate-Sanchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5:430–436. doi: 10.1016/S1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- Selote D, Kachroo A. RPG1-B-derived resistance to AvrB-expressing Pseudomonas syringae requires RIN4-like proteins in soybean. Plant Physiol. 2010;53:1199–1211. doi: 10.1104/pp.110.158147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman MB, de Figueiredo P. Comparative pathobiology of fungal pathogens of plants and animals. Plos Pathog. 2011;7:e1002324. doi: 10.1371/journal.ppat.1002324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale LK, Haun WJ, Xu WW, Bhaskar PB, Anderson JE, Hyten DL, Gerhardt DJ, Jeddeloh JA, Stupar RM. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 2012;159:1295–1308. doi: 10.1104/pp.112.194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng C, Yu K, Anderson TR, Poysa V. Mapping genes conferring resistance to Phytophthora root rot of soybean, Rps1a and Rps7. J Hered. 2001;92:442–446. doi: 10.1093/jhered/92.5.442. [DOI] [PubMed] [Google Scholar]
- Demirbas A, Rector BG, Lohnes DG, Fioritto RJ, Graef GL, Cregan PB, Shoemaker RC, Specht JE. Simple sequence repeat markers linked to the soybean Rps genes for Phytophthora resistance. Crop Sci. 2001;41:1220–1227. doi: 10.2135/cropsci2001.4141220x. [DOI] [Google Scholar]
- Gordon SG, Martin SKS, Dorrance AE. Rps8 maps to a resistance gene rich region on soybean molecular linkage group F. Crop Sci. 2006;46:168–173. doi: 10.2135/cropsci2004.04-0024. [DOI] [Google Scholar]
- Abney TS, Melgar JC, Richards TL, Scott DH, Grogan J, Young J. New races of Phytophthora sojae with Rps1-d virulence. Plant Dis. 1997;81:653–655. doi: 10.1094/PDIS.1997.81.6.653. [DOI] [PubMed] [Google Scholar]
- Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RHY, Aerts A, Arredondo FD, Baxter L, Bensasson D, Beynon JL. et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006;313:1261–1266. doi: 10.1126/science.1128796. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libault M, Thibivilliers S, Bilgin DD, Radwan O, Benitez M, Clough SJ, Stacey G. Identification of four soybean reference genes for gene expression normalization. Plant Genome. 2008;1:44–54. doi: 10.3835/plantgenome2008.02.0091. [DOI] [Google Scholar]
- Jian B, Liu B, Bi Y, Hou W, Wu C, Han T. Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol Biol. 2008;9:59. doi: 10.1186/1471-2199-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Fan C, Li H, Zhang Q, Fu YF. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol Biol. 2009;10:93. doi: 10.1186/1471-2199-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolf-Moreira R, Lemos EGDM, Abdelnoor RV, Beneventi MA, Rolla AAP, Pereira SD, de Oliveira MCN, Nepomuceno AL, Marcelino-Guimaraes FC. Identification of reference genes for expression analysis by real-time quantitative PCR in drought-stressed soybean. Pesqui Agropecu Bras. 2011;46:58–65. doi: 10.1590/S0100-204X2011000100008. [DOI] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Kolde R. pheatmap: pretty heatmap. R package version 061. 2012.
- Du Z, Zhou X, Ling Y, Zhang ZH, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64–W70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1
Breeding pedigree of soybean NILs used in this study.
Additional file 2
Reactions of soybean NILs to Phytophthora sojae .
Additional file 3
List of primers used in qPCR.
Additional file 4
Comparison in expression of six soybean genes in Williams and 10 NILs, each containing a single Rps gene, as determined by RNA-Seq analysis or qRT-PCR. Y axis indicates differential expression of selected genes for each soybean line. X axis indicates selected genes used for qRT-PCR. These genes are Glyma02g47940 (1), Glyma04g20330 (2), Glyma05g24770 (3), Glyma07g07270 (4), Glyma09g37290 (5), Glyma10g44170 (6), and Glyma11g04130 (7). Pearson’s corrleation coefficient (r).
Additional file 5
Heatmap (hcluster) analysis of 5,806 IIGs for 10 soybean NILs, each containing a single Rps gene. The values used to draw heatmap is Log2 (fold change) of expression level of post inoculation to mock inoculation.
Additional file 6
GO analysis of DEGs in different categories.
Additional file 7
Gene ontology categories for IIGs identified in soybean NILs, each containing a single Rps gene. Left bar = up-regulated genes. Right bar = down-regulated genes.
Additional file 8
Comparison of differentially expressed transcription factors (DETFs) between Williams and 10 NILs each containing a single Rps gene. A: up-regulated DETFs. B: down-regulated DETFs. Red and purple represent the number of DETFs specific to individual Rps genes and collectively referred to as incompatible interaction transcription factors. Brown and dark purple represent the number of DETFs shared by an individual NIL and Williams. Green and light blue represent the number of DETFs specifically expressed in Williams when compared to a specific NIL. The central green and light blue circles represent the common proportion of Williams-specific DETFs that are collectively referred to as compatible interaction transcription factors.
Additional file 9
Counts of transcription factors (TFs) in different groups.
Additional file 10
Housekeeping genes selected in this study.