Structure of an SspH1-PKN1 Complex Reveals the Basis for Host Substrate Recognition and Mechanism of Activation for a Bacterial E3 Ubiquitin Ligase (original) (raw)
Abstract
IpaH proteins are bacterium-specific E3 enzymes that function as type three secretion system (T3SS) effectors in Salmonella, Shigella, and other Gram-negative bacteria. IpaH enzymes recruit host substrates for ubiquitination via a leucine-rich repeat (LRR) domain, which can inhibit the catalytic domain in the absence of substrate. The basis for substrate recognition and the alleviation of autoinhibition upon substrate binding is unknown. Here, we report the X-ray structure of Salmonella SspH1 in complex with human PKN1. The LRR domain of SspH1 interacts specifically with the HR1b coiled-coil subdomain of PKN1 in a manner that sterically displaces the catalytic domain from the LRR domain, thereby activating catalytic function. SspH1 catalyzes the ubiquitination and proteasome-dependent degradation of PKN1 in cells, which attenuates androgen receptor responsiveness but not NF-κB activity. These regulatory features are conserved in other IpaH-substrate interactions. Our results explain the mechanism whereby substrate recognition and enzyme autoregulation are coupled in this class of bacterial ubiquitin ligases.
INTRODUCTION
The covalent modification of protein substrates by ubiquitin (Ub) is a universal mechanism in eukaryotes for the control of protein stability, activity, and/or localization (1). Ub conjugation to substrates is mediated by a conserved cascade whereby an E1 enzyme activates the C terminus of Ub as a labile thioester, followed by transfer of the Ub thioester to an E2 enzyme and finally E3-mediated transfer of the Ub to one or more free amino groups on the substrate as an isopeptide bond. E3 enzymes govern substrate specificity through dedicated protein interaction domains, such as WD40 repeat domains or leucine-rich repeat (LRR) domains. Over 600 E3 enzymes are encoded by the human genome and are comprised of two main classes, the HECT domain class, which forms a catalytic thioester intermediate, and the RING domain class, which bridges the E2 enzyme to the substrate. Rapid reiteration of the catalytic cycle can generate poly-Ub chains of distinct linkages between lysine residues on Ub itself. Typically, the formation of K48-linked Ub chains leads to substrate recognition and degradation by the 26S proteasome (2). Other chain linkages can dictate the formation of protein complexes, for example, in the signal-dependent activation of the NF-κB response (3). The conjugation of single Ub moieties can also control protein interactions and localization, such as in the secretory system and the DNA damage response.
The ubiquitin-proteasome system (UPS) plays an important role in pathogenic infection (4). Host cells use Ub to activate the innate immune responses via the NF-κB network (3) and as a means to mark cytoplasmic bacteria for destruction by autophagy (5–8). Conversely, pathogenic Gram-negative bacteria turn the UPS against the host by injection of effector proteins into the host cytoplasm by the type III secretion system (T3SS) (9). For example, pathogenic Salmonella organisms secrete the effectors SseL and SopB, which subvert the UPS by distinct mechanisms: SseL attenuates Ub-mediated autophagy through its ability to act as a deubiqutinase (10, 11), while SopB, a phosphoinositide phosphatase, exploits its own ubiquitination to temporally and spatially modulate its substrate repertoire (12).
A group of conserved effectors found in Salmonella, Shigella, and other Gram-negative bacteria, termed IpaH proteins, represent a new bacterium-specific class of E3 enzymes that function in the host cell (13). IpaH enzymes share a conserved tripartite domain architecture consisting of (i) an N-terminal T3SS targeting sequence, (ii) a variable LRR domain for substrate recognition, and (iii) a conserved novel E3 ligase (NEL) catalytic domain (13, 14). The NEL domain forms a Ub thioester intermediate via a catalytic cysteine (13, 15, 21) in a manner analogous to that of the structurally unrelated eukaryotic HECT domain (17, 18). In the absence of substrate, the LRR domain can also inhibit NEL catalytic activity through intramolecular interactions (15, 19, 20). Structural and biochemical analysis of the IpaH family members IpaH3 (21) and SspH2 (20) revealed that IpaH autoinhibition can occur by two distinct mechanisms that disrupt the catalytic domain (19). Removal of the LRR domain results in a constitutively active E3 enzyme that is able to autoubiquitinate and form free polyubiquitin chains (13). Although each different IpaH enzyme targets distinct host substrates, the mechanisms of autoregulation appear to be conserved across Salmonella and Shigella IpaH members (19). Autoregulation likely serves to prevent IpaH autoubiquitination, which would otherwise lead to degradation by the 26S proteasome (19), and/or formation of free polyubiquitin chains, which can elicit the innate immune response (22).
The interaction between the Salmonella SspH1 enzyme and human protein PKN1 is one of the best-characterized IpaH-substrate interactions (13, 23). PKN1 directly interacts with the LRR domain of SspH1 (23) and is ubiquitinated by SspH1 in vitro (13). Nonfunctionally redundant SspH1 and SspH2 isoforms are required for Salmonella virulence in vivo (14). PKN1 is a serine/threonine kinase whose activity is regulated through interactions with Rho family GTPases (24–27) or by proteolytic activation (28), both of which can trigger PKN1 activation during Salmonella infection (29–31). PKN1 is also a target of the bacterial effector YopM from Yersinia (32). PKN1 influences at least three aspects of host immune signaling. First, PKN1 is a potent positive regulator of androgen receptor (AR), mineralocorticoid receptor (MR), and progesterone receptor (PR) signaling (33–35). AR knockout mice exhibit neutropenia, increased susceptibility to bacterial infection, attenuated macrophage activation, and slow accumulation of tumor necrosis factor alpha (TNF-α) at wound sites (36, 37). Macrophages from MR knockout mice exhibit decreased classical activation (antimicrobial functions) and increased alternative activation (tissue repair functions), leading to reduced inflammation (38). Second, PKN1 is a negative regulator of Akt, such that PKN1 knockout mice display increased basal Akt activation, resistance to cell death signals in B cells, and concomitant autoimmune phenotypes (39). With respect to infection, the intracellular growth of pathogenic Salmonella depends on activation of Akt (40). Third, PKN1 activity correlates with suppression of NF-κB signaling (23), a key regulator of innate and adaptive immune function (3).
The structural basis for how the LRR domains of IpaH enzymes target specific substrates and the mechanism whereby substrate recognition is coupled to IpaH enzyme activation are unknown. To provide insight into these issues, we solved the crystal structure of SspH1 in complex with PKN1. We show that the LRR domain of SspH1 interacts specifically with the HR1b antiparallel coiled-coil subdomain of PKN1, and that this interaction releases an inhibitory interaction between the catalytic domain and the LRR domain. We also show that SspH1 catalyzes the ubiquitination and proteasome-dependent degradation of PKN1 in cells, and that this destruction of PKN1 leads to suppression of AR but not NF-κB activity. Finally, we provide evidence that these features can be generalized to other IpaH-substrate interactions.
MATERIALS AND METHODS
Plasmids, antibodies, cell lines, and reporter assay reagents.
SspH1, PKN1, and IpaH9.8 constructs were PCR amplified from previously reported vectors (13, 19) and inserted into pProEx (SspH1) or pGEX-TEV (PKN1 and IpaH9.8). Vectors for Ub, UBE1, and UBE2D3 were previously reported (19). Mammalian expression plasmids for SspH1 and PKN1 were generated from PCR-amplified SspH1 and PKN1 with silent mutations to remove internal EcoRI sites and cloned into either pCMV-Tag 2 (Flag tag) or pCMV-Tag 3 (Myc tag) vector (Stratagene). A mammalian expression vector that expresses constitutively active RhoA was generated by PCR amplifying humanized mouse RhoA (I192V) containing a constitutive activating mutation (G14V) and cloned into pCMV-Tag 2. Mammalian expression vectors for IκBβ and β-galactosidase were a gift from Tony Pawson. The synthetic androgen R1881 and mammalian vectors expressing human AR and mouse mammary tumor virus (MMTV)-driven firefly luciferase were gifts from Ted Brown. _GAL1_-inducible yeast expression vectors containing Flag-tagged SspH1 or IpaH9.8 were as previously reported (19). Site-directed mutagenesis of proteins was carried out by the QuikChange method (Stratagene). Immunoblots were probed with antibody in 3% milk powder (anti-Flag, anti-Myc, anti-Ub, and antitubulin) or in 3% bovine serum albumin (BSA; anti-His5) overnight. Secondary horseradish peroxidase (HRP)-conjugated antibodies were blotted in 3% milk powder for at least 1 h. Anti-His5 was purchased from Qiagen, anti-Flag (F3165) and immobilized anti-Flag (F2426) were purchased from Sigma, anti-Myc (9E10) and immobilized anti-Myc (sc-40 AC) were purchased from Santa Cruz Biotech, anti-Ub (P4D1) was purchased from Covance, and antitubulin (6199) was purchased from Sigma.
Unaltered HEK 293 cells and HEK 293 cells that stably expressed an NF-κB inducible promoter upstream of firefly luciferase (HEK293NF-κB) were gifts from Tony Pawson. Cells were maintained in Dulbecco's modified Eagle medium (DMEM), high glucose, supplemented with antibiotics and 10% fetal bovine serum (Invitrogen).
Yeast experiments were performed with the FUS1pr-HIS3 yeast strain (SY2625 MATa bar1 ade2 his3::FUS1-HIS3 mfa2::FUS1-lacZ leu2 can1 ura3 trp1) (13).
Expression and purification of recombinant proteins.
Protein constructs were transformed and expressed in Escherichia coli BL21(DE3)-RIL CodonPlus cells (Stratagene). Cultures were grown at 37°C to an optical density at 600 nm (OD600) between 0.6 and 0.8 prior to the addition of 0.25 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and shifting the temperature to 17°C. Tagged proteins were affinity purified by nickel or glutathione resin and, unless otherwise indicated, tags were removed by the addition of tobacco etch virus (TEV) protease overnight at 4°C. Purified proteins were buffer exchanged by gel filtration chromatography into sizing buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM dithiothreitol [DTT]) prior to concentration and flash freezing in liquid nitrogen. Fluorescein-conjugated Ub was generated by the addition of a Cys residue at the N terminus to which fluorescein was conjugated prior to purification as described above. For crystallography, SspH1-PKN1 complexes were prepared by mixing GST-HR1b lysate with His-LRR lysate, followed by nickel affinity chromatography, capture of eluates onto glutathione-resin, and TEV cleavage of bound complexes.
Crystallization and data collection.
Purified SspH1LRR(162-405)-PKN1HR1b(122-199) complex was concentrated to 26 mg/ml in 20 mM HEPES (pH 7.5), 150 mM NaCl, and 2 mM DTT. Crystals were obtained at 20°C by hanging-drop diffusion with 20.5 mg/ml protein, 0.18 M triammonium citrate, and 24 to 28% polyethylene glycol 3350 (PEG 3350). Crystals were soaked briefly in a buffer containing the crystallization condition supplemented with 20% 1,6-hexanediol prior to flash freezing in liquid nitrogen. Diffraction data were collected at 93 K at the Canadian Light Source (Saskatchewan, Canada). For the apo-SspH1LRR crystal structure, purified SspH1LRR(162-399) was concentrated to 28 mg/ml in 20 mM HEPES (pH 7.5), 150 mM NaCl, and 2 mM DTT. Crystals were obtained at 20°C by hanging-drop diffusion with 10 mg/ml protein, 0.95 M ammonium sulfate, 0.5% PEG 8000, and 0.1 M HEPES, pH 6.2. Crystals were soaked briefly in a buffer containing the crystallization condition supplemented with 30% glycerol prior to flash freezing in liquid nitrogen. Diffraction data were collected at 93°K at the Argonne Photon Source (Lemont, IL).
Structure determination and refinement.
Diffraction data for the SspH1LRR-PKN1HR1b complex was indexed and scaled using HKL2000 (41), and phases were determined by molecular replacement with PHASER (42) using a homology model of the SspH1LRR domain determined by the PHYRE2 server (45). The final model was generated by iterative rounds of refinement with REFMAC (43) and PHENIX (44) with manual model building using COOT (46). Bound PKN1HR1b was built manually into experimental electron density maps. Diffraction data for SspH1LRR in isolation was indexed and scaled using iMOSFLM (47), and the structure was determined by molecular replacement as described for the complex.
Crystal structure analysis.
Overlapping volume between autoinhibited SspH2 NEL and LRR-bound PKN1 substrate was calculated by superposition of the SspH2 and SspH1 LRR domains via the C-terminal repeats and submitting models generated with COOT processing of the individual NEL domain, HR1b domain, and overlapped NEL-HR1b domains for volume determination using the 3V server (48). Root mean square deviation (RMSD) calculations of reported domains were calculated using the COOT secondary structure matching function.
Protein interaction and ubiquitination assays.
GST fusion proteins were expressed as described above and captured onto fresh glutathione resin in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM DTT, 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 1 h with rotation. Resin was washed once with high-salt lysis buffer (500 mM NaCl) and three times with standard lysis buffer. Equal amounts of bound protein were combined with purified recombinant prey at a concentration of 15 μM for 3 h at 4°C. Unbound proteins were removed by washing the resin three times in lysis buffer. Bound proteins were separated by SDS-PAGE and visualized by Coomassie blue staining.
Ubiquitination reactions were performed as previously described (19), with minor modifications. Reactions were carried out in 25 mM Tris-Cl (pH 7.5), 50 mM NaCl, 10 mM MgCl2, 5 mM ATP (pH 7), 0.25 mM DTT at a final volume of 18 μl at room temperature for 20 to 90 min. Reactions were terminated by the addition of SDS-PAGE loading buffer and boiled for 5 min prior to loading the gel. Gels were scanned with a Typhoon FLA 9500 (GE Healthcare) at 433 nm if Ub-fluorescein was used prior to transfer and immunoblotting with the indicated antibodies. Ub-fluorescein, when used, was mixed with wild-type Ub in equal amounts.
SspH1-PKN1 association and PKN1 ubiquitination in cells.
HEK 293 cells grown in 100-mm petri dishes were transiently transfected at 60% confluence with 1 to 2 μg of each plasmid DNA. Twenty-four hours posttransfection, cells were lysed in lysis buffer (50 mM HEPES, pH 7.2, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 10% glycerol, 5 mM NaF, 20 mM NEM, 1 mM PMSF) and lysates were immunoprecipitated using immobilized anti-Flag or anti-Myc antibodies. Retained proteins were visualized by immunoblotting with the indicated antibodies. Where indicated, 10 μM MG-132 was added to cells 8 to 12 h posttransfection.
Luciferase assays and yeast growth assays.
For luciferase assays, HEK 293 cells were seeded into 6-well plates or 60-mm dishes and grown to 90% confluence prior to transfection with Lipofectamine 2000 (Invitrogen) in Opti-MEM. Medium was replaced after 4 to 6 h with DMEM supplemented with 10% charcoal stripped fetal bovine serum (FBS; Invitrogen). Twenty-four hours posttransfection, cells were stimulated with 0.1 nM R1881 or ethanol carrier solvent. Twenty-four hours poststimulation, cells were washed with phosphate-buffered saline (PBS) and lysates were prepared and assayed according to the luciferase assay kit (Promega). For AR signaling experiments, cells were transfected with vectors bearing MMTV-fluc, human AR, RhoAG14V, β-galactosidase, and the indicated Flag-SspH1, Myc-PKN1, or Flag-IκBβ construct. Luciferase and β-galactosidase activity were measured using an EnVision multilabel reader (PerkinElmer). All experiments were performed at least twice in duplicate.
Yeast viability assays were carried out as previously described (19). Yeast cells were transformed with the wild-type or mutant IpaH9.8-Flag or SspH1-Flag construct under the control of the GAL1 promoter inserted into the pRS425 vector. Transformed strains were grown at 30°C in liquid culture with 2% (wt/vol) raffinose and spotted as a dilution series on 2% (wt/vol) glucose- or galactose-containing synthetic dropout medium lacking histidine and leucine. Plates were imaged after 2 to 3 days of growth at 30°C.
PDB accession codes.
Coordinates and structure factors for SspH1LRR and the SspH1LRR:PKN1HR1b complex have been deposited in the Protein Data Bank (PDB) under codes 4NKH and 4NKG, respectively.
RESULTS
Minimal requirements for PKN1 recognition and ubiquitination by SspH1.
The HR1 domain of PKN1 consists of three consecutive antiparallel coiled-coil subdomains (27), referred to as HR1a, HR1b, and HR1c (Fig. 1a), and it directly interacts with the LRR domain of SspH1 (23). To determine which coiled-coil subdomain(s) meditates the interaction with SspH1, we used fragments of PKN1 fused to GST to capture the purified LRR domain of SspH1 onto glutathione resin. The HR1b subdomain of PKN1 (PKN1HR1b) was both necessary and sufficient for high-affinity interaction with the LRR domain of SspH1, whereas the HR1a and HR1c subdomains did not detectably interact with the LRR domain (Fig. 1b).
FIG 1.

PKN1HR1b is the minimal subdomain both necessary and sufficient to bind and activate SspH1. (a) Domain architecture of SspH1 and PKN1. (b) Capture of soluble SspH1 LRR domain with the indicated GST-PKN1 constructs (a, HR1a; b, HR1b; c, HR1c; abc, HR1abc). Detection was by Coomassie blue stain. (c) In vitro ubiquitination reactions using isolated SspH1 NEL or larger LRR-NEL fragments in the presence and absence of full-length (FL) PKN1 or the indicated fragments thereof. Activity was assessed by anti-Ub antibody. (d) In vitro ubiquitination reactions of His-tagged PKN1 fragments in the presence and absence of SspH1 NEL or LRR-NEL fragments. Fluorescein-labeled Ub (UbFluor) was detected by fluorescence prior to detection of substrate with anti-His5 antibody.
To assess if the interaction of the HR1b subdomain with SspH1 was sufficient to activate SspH1, we performed in vitro ubiquitination assays using an LRR-NEL fragment of SspH1 (SspH1LRR-NEL), which recapitulates the autoinhibitory properties of the full-length protein. As a control, we also tested effects on the activity of the isolated NEL catalytic domain (SspH1NEL). Enzyme activity was measured by anti-Ub immunoblot analysis of the complete reaction mixture, which detects the sum of SspH1 autoubiquitination, substrate ubiquitination, and free polyubiquitin chain formation. All PKN1 fragments that contained the HR1b subdomain activated the ability of SspH1LRR-NEL to form high-molecular-weight Ub conjugates, whereas an HR1a fragment had no effect (Fig. 1c). The size of conjugates stimulated by the HR1b subdomain was increased in the presence of larger fragments harboring either the HR1a subdomain or full-length PKN1, possibly due to differential activation of SspH1, the availability of additional substrate lysine residues, and/or differences in substrate size. None of the PKN1 fragments altered the constitutive ubiquitination activity of the isolated SspH1NEL domain (Fig. 1c), consistent with its inability to bind PKN1. In order to discriminate substrate ubiquitination from free Ub chain assembly, reactions were also performed with His-tagged PKN1 fragments and fluorescein-labeled Ub (Fig. 1d). SspH1NEL predominantly generated free Ub chains, both alone and in the presence of PKN1 fragments, whereas SspH1LRR-NEL predominantly generated PKN1-poly-Ub conjugates. Quantification of total conjugated ubiquitin between PKN1 fragments HR1b and HR1ab (Fig. 1d, lanes 8 and 9) indicated that both fragments yielded comparable amounts of ubiquitination despite differences in chain length distributions. These results establish PKN1HR1b as a minimal substrate of SspH1 and demonstrate that PKN1 ubiquitination is dependent on its recruitment by the LRR domain.
Structure of the SspH1LRR-PKNHR1b complex.
To identify the molecular determinants of the SspH1-PKN1 interaction and to understand the mechanism of substrate-mediated activation, we determined the crystal structure of SspH1LRR alone and in complex with PKN1HR1b by molecular replacement (see Materials and Methods). The structures were determined to 2.75- and 2.9-Å resolution, respectively (Table 1 shows data collection and refinement statistics). The complex cocrystal structure contains two nearly identical copies of the SspH1LRR-PKN1HR1b complex in the asymmetric unit (RMSD of <0.7 Å across all atoms); hence, our subsequent analyses will focus on one copy of the complex corresponding to chain A and chain B (Fig. 2a). The structures of PKN1 and SspH1 proteins were well ordered, with a total of only 16 and 13 residues disordered at the termini of each protein, respectively. The unbound SspH1LRR crystal structure contained six nearly identical copies that are all very similar to the bound SspH1LRR structure, with minor differences in RMSD values (0.5 to 1 Å) attributable to LRR conformational flexibility and mobility of the C-terminal helices.
TABLE 1.
Data collection and refinement statistics for SspH1LRR-PKN1HR1b and SspH1LRR structures
| Parameter | Value(s)a for: | |
|---|---|---|
| Salmonella SspH1(LRR)-human PKN1(HR1b) | Salmonella SspH1(LRR) | |
| Data collection | ||
| Beamline | CLS 08ID-1 | APS 24ID-C |
| Space group | P32 2 1 (#154) | P42 21 2 (#94) |
| Cell dimensions | ||
| a, b, c (Å) | 167.6, 167.6, 89.1 | 114.93, 114.93, 252.17 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 |
| Resolution (Å) | 45–2.9 (3.0–2.9) | 50–2.75 (2.9–2.75) |
| _R_merge | 8.4 (70.7) | 14.2 (70.1) |
| I/σ_I_ | 18.7 (2.1) | 6.9 (2.4) |
| Completeness (%) | 99.4 (99) | 99.7 (100) |
| Redundancy | 4.4 (4.4) | 5.2 (5.3) |
| Refinement | ||
| Resolution (Å) | 40–2.9 (2.98–2.9) | 50–2.75 (2.85–2.75) |
| No. of reflections | 32,003 | 84,316 |
| _R_work/_R_free | 20.9 (32.2)/24.2 (38.4) | 24.6 (34.6)/28.1 (41.2) |
| RMSD | ||
| Bond lengths (Å) | 0.003 | 0.0039 |
| Bond angle (°) | 0.69 | 0.938 |
| Geometry | ||
| Ramachandran outlier (%) | 0 | 0.1 |
| Ramachandran favored (%) | 98 | 96.9 |
| Rotamer outlier | 0.6 | 5.9 |
| Cβ outlier | 0 | 0 |
| Clashscore | 17.02 | 18.69 |
FIG 2.

Structure analysis of the SspH1LRR-PKN1HR1b enzyme-substrate complex. (a) Ribbon representation of PKN1HR1b bound to SspH1LRR. (b) Comparison of SspH1LRR and PKN1HR1b to previously determined structures. Left, SspH1LRR superimposed on IpaH3LRR (PDB code 3CVR); right, SspH1-bound PKN1HR1b (Chain B) superimposed on free PKN1HR1b (PDB code 1URF) and Rac1-bound PKN1HR1b (PDB code 2RMK). (c) Peel-away surface representations showing intermolecular contact residues on SspH1LRR and on PKN1HR1b. (d) Stereo views of interacting residues in the SspH1LRR-PKN1HR1b binding interface. The lower images are magnifications of various contact regions from the upper images. Residues mutated in this study are highlighted with red labels.
The LRR domain in both apo- and complex structures adopted an expected periodic structure consisting of 10 parallel β-sheet repeats and was similar to the existing LRR structure of IpaH3 (21), with a small deviation in the position of the C-terminal capping helices (PDB code 3CVR; RMSD, 1.71 Å) (Fig. 2b). The PKN1HR1b fragment consisted of a single coiled coil and was similar to nuclear magnetic resonance (NMR) structures of free (PDB code 1URF; RMSD, 3.1 Å) (27) and Rac1-bound HR1b (PDB code 2RMK; RMSD, 1.24 Å) (26) (Fig. 2b). SspH1LRR engages PKN1HR1b with 1:1 stoichiometry along a single continuous surface on the concave face of the LRR domain that is composed of repeats 3 to 10 (Fig. 2a). The interaction buried a total of 884 Å2 on the LRR domain (Fig. 2c) as mediated by a mixture of hydrogen bonds, salt bridges, and hydrophobic interactions (Fig. 2c and d provide a comprehensive list of contact residues). Important contacts that were probed by mutational analysis in subsequent experiments included an interaction network between Arg181 and Arg185 on PKN1HR1b and Tyr325, Asp343, Tyr366, and Asp368 on SspH1LRR (Fig. 2c and d, indicated in red). We note that LRR domains are tremendously versatile protein interaction domains, as virtually all solvent-exposed surfaces have been documented to participate in ligand interactions in various known structures (see Fig. S1 in the supplemental material) (49). The SspH1LRR-PKN1HR1b complex falls within a predominant interaction class that exploits the concave face of the LRR domain, similar to that employed by SCFSkp2, one of only two other LRR-containing E3 enzyme structures solved to date, in complex with substrate (50, 51) (Fig. 2c; also see Fig. S1). To our knowledge, the SspH1LRR-PKN1HR1b complex represents the first LRR domain shown to engage a coiled-coil domain.
Residues on SspH1 that contact PKN1 were unique across the IpaH family, with 87% of interaction residues (13 of 15 total) absent from the closest family member, SspH2 (see Fig. S2A in the supplemental material), in agreement with the previous observation that SspH2 does not interact with PKN1 (23). PKN1 engages the LRR domain of SspH1 primarily through its C-terminal helix α2, which provides 10 of the 13 interaction residues (see Fig. S2B). Comparison of contact residues on PKN1HR1b to corresponding positions on HR1a and HR1c shows that 69 and 62% of interaction residues (9 and 8, respectively, out of 13 total) are not conserved in either coiled coil (see Fig. S2B). These results establish the basis for the observed specificity of the SspH1LRR-PKN1HR1b interaction.
Effects of the LRR-HR1b contact surface on SspH1 enzymatic activity.
To probe the binding determinants observed in the crystal structure, we generated mutations on the interaction surface of PKN1 (R181A, R181K, and R185A) and SspH1 (Y325A, D343A, Y366A, and D368A) and examined their effects in GST capture assays. All three PKN1 mutants showed impaired interaction with SspH1LRR relative to the wild type, as the R181A and R185A mutations caused almost complete loss of interaction and the conservative R181K mutation diminished the interaction (Fig. 3a). All four SspH1 mutations impaired the interaction with PKN1HR1b, as the Y325A, D343A, and Y366A mutations virtually eliminated the interaction, whereas the D368A mutation caused a partial reduction (Fig. 3b).
FIG 3.
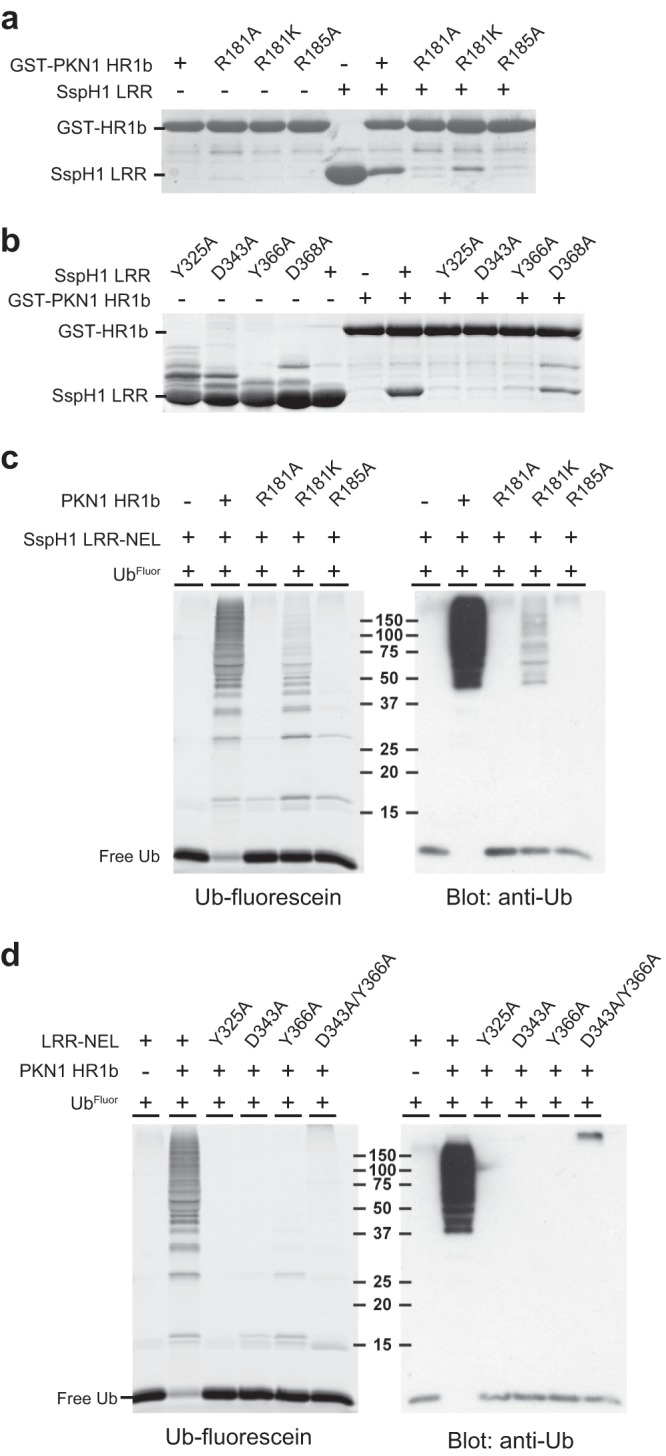
Determinants of substrate recognition by SspH1. (a) Capture of soluble wild-type SspH1LRR by either wild-type GST-PKN1HR1b or the indicated mutants. Detection was by Coomassie blue stain. (b) Capture of soluble wild type or the indicated mutants of SspH1LRR by wild-type GST-PKN1HR1b. Detection was by Coomassie blue stain. (c) In vitro ubiquitination reactions of SspH1LRR-NEL in the presence or absence of wild-type PKN1HR1b or the indicated mutants. UbFluor was detected by fluorescence prior to immunoblotting with anti-Ub antibody. (d) In vitro ubiquitination reactions with wild-type SspH1LRR-NEL or the indicated mutants in the presence or absence of PKN1HR1b. Detection was performed as described for panel c.
We then tested binding interface mutations for effects on SspH1 activity in vitro. Ubiquitination reactions were carried out using a mixture of wild-type and fluorescein-labeled Ub in order to enable orthogonal detection by fluorescence and immunoblotting. Mutations in PKN1 that abrogated the interaction (R181A and R185A) strongly attenuated the formation of Ub conjugates, whereas the intermediate R181K mutation partially attenuated enzymatic activity (Fig. 3c). Quantification of total ubiquitin-conjugated species between the wild-type HR1b fragment and the corresponding R181K mutant (Fig. 3c) revealed that the latter exhibits an approximately 80% decrease in activation activity, consistent with a similar defect observed for its interaction with SspH1 (Fig. 3a). Similarly, the three strongest interface mutations in SspH1 (Y325A, D343A, and Y366A) and a double mutation (D343A/Y366A) severely compromised the activation of SspH1 by wild-type PKN1 (Fig. 3d). Taken together, these results demonstrate that the interaction surface between SspH1 and PKN1 directly controls SspH1 catalytic activity in vitro.
The basis for coupling of substrate recognition with catalytic activation.
The X-ray structures of IpaH3 (21) and SspH2 (20) suggested two distinct modes of autoinhibition, and structure-guided mutational analyses (19) demonstrated that both modes can operate in each enzyme but that one mode dominates in a given enzyme (Fig. 4a). The predominant mode of autoinhibition observed in the SspH2 structure is mediated by LRR-NEL interactions, which in SspH1 are centered on Leu550 of the NEL domain and a hydrophobic patch around Ile391 on the concave face of the LRR domain (Fig. 4b, blue surface). The minor mode of autoinhibition observed in the IpaH3 structure involves the same surface on the SspH1 NEL domain but an opposite convex surface of the LRR domain centered on Leu352 (Fig. 4b, magenta surface). Both modes of autoinhibition appear to operate concurrently to attenuate NEL activity, as mutational disruption of either interaction results in activation, producing unanchored Ub chains and autoubiquitinated species (19). Comparison of the SspH1LRR-PKN1HR1b complex structure to the predominant autoinhibited state observed in the SspH2 structure readily explained how PKN1 binding relieves the major autoinhibited mode of SspH1 (Fig. 4a). The NEL domain interaction residues in this predominant mode lie immediately adjacent to the interaction interface engaged by the PKN1HR1b substrate, with minor overlap at His392 (Fig. 4b, yellow surface). While the direct overlap of contact surfaces is small, when both substrate and NEL domains were simultaneously modeled on the LRR domain of SspH1, steric clashes corresponding to 681 Å3 of atomic overlap between the substrate and NEL domain arise, indicating that the two interactions are mutually exclusive (Fig. 4a, bottom inset). The minor mode of autoinhibition occurs by interaction of the NEL domain with the convex side of the LRR, which is directly opposite and not overlapping the PKN1 engagement site (Fig. 4b, magenta). Comparison of SspH1LRR in free and substrate-bound states revealed no evidence of allosteric coupling between the two physically remote binding sites (Fig. 4c). We conclude that SspH1 activation by PKN1 is mediated solely by disengagement of the predominant autoinhibitory mode visualized in the SspH2 structure (Fig. 4a, inset cartoon).
FIG 4.

Coupling of substrate recognition with catalytic activation by competitive displacement. (a) NEL domain orientations corresponding to major (SspH2 [PDB code 3G06]) and minor (IpaH3 [PDB code 3CVR]) modes of autoinhibition onto the SspH1LRR-PKN1HR1b (orange-green) complex superimposed by their respective LRR domain coordinates (for clarity, only the LRR domain from the SspH1-PKN1 complex is shown). A clash of residues between the major mode NEL domain and PKN1HR1b is shown in the inset (bottom right), which is a cartoon model of substrate-induced E3 ligase activation. (b) Surface view of SspH1LRR highlighting solvent-exposed surface, contact surface with PKN1HR1b, and the projected contact surfaces with NEL domains in major and minor modes of autoinhibition. The overlapping contact residue for PKN1HR1b and the major-mode NEL domain interaction site is colored yellow. (c) Comparison of the six SspH1LRR protomers in the unbound crystal structure (gray) to the two SspH1LRR-PKN1HR1b (orange-green) complexes. Superpositions were performed using the LRR domain coordinates.
Requirements for SspH1-mediated degradation of PKN1 in cells.
To evaluate the role of the SspH1-PKN1 contact surface in cells, we coexpressed epitope-tagged versions of wild-type and mutant alleles of SspH1 and PKN1 in HEK 293 cells. No interaction between wild-type Myc-PKN1 and Flag-SspH1 was detected upon coexpression, which correlated with an absence of detectable PKN1 protein in the cell lysates (Fig. 5a). When PKN1 was instead coexpressed with a catalytically dead allele of SspH1 (C492A), PKN1 was readily detected and also interacted strongly with SspH1C492A (Fig. 5a). This interaction was sensitive to mutations at the binding interface on SspH1 (D343A/Y366A) or PKN1 (R181A, R181K, and R185A) (Fig. 5a). These results suggested that the interaction of wild-type SspH1 and PKN1 in cells led to PKN1 ubiquitination and subsequent destruction by the 26S proteasome. Consistent with this hypothesis, interaction mutants caused stabilization of PKN1 when coexpressed with catalytically competent SspH1 (Fig. 5a). The extent of stabilization for the various PKN1 mutants (R181A, R181K, and R185A) correlated well with the effects of each mutation on PKN1 ubiquitination in vitro (Fig. 3c).
FIG 5.
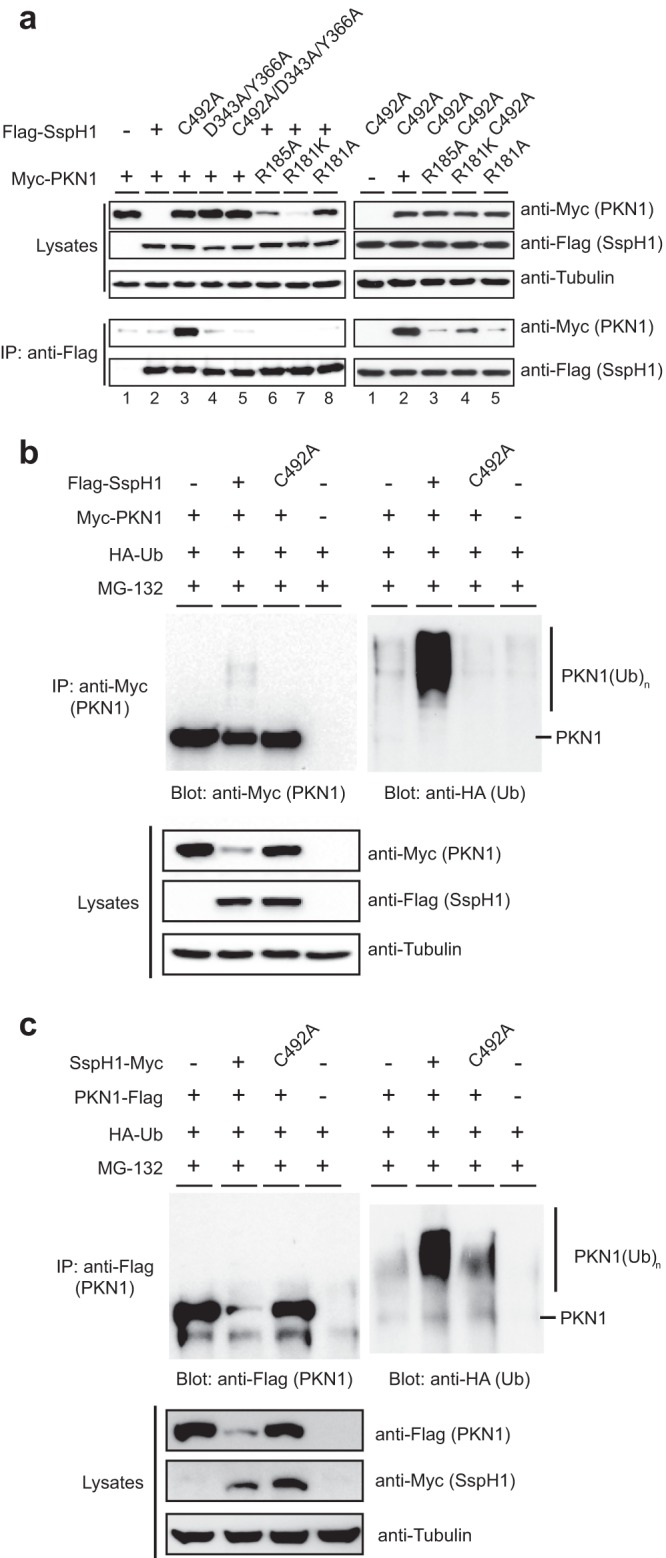
Interaction of SspH1 and PKN1 in human cells. (a) Immunoblot analysis of transiently expressed epitope-tagged (Myc-PKN1 and Flag-SspH1) wild-type protein and the indicated mutant proteins, or endogenous tubulin loading control, in HEK 293 cells. Protein expression was assayed directly from cell lysates (upper), and interaction was assayed by coimmunoprecipitation (IP) with anti-Flag antibody prior to immunoblotting for Flag-SspH1 and Myc-PKN1 (lower). (b) Immunoblot analysis of transiently expressed wild-type and indicated mutant tagged proteins (Myc-PKN1, Flag-SspH1, and HA-Ub) in HEK 293 cells treated with the proteasome inhibitor MG-132. Protein expression was assayed directly from cell lysates (lower) prior to immunoprecipitation with anti-Myc, followed by immunoblotting for Myc-PKN1 (top left) and HA-Ub (top right). (c) The experiment was the same as that for panel b but with the epitope tags swapped and fused at the C terminus, that is, PKN1-Flag and SspH1-Myc.
To demonstrate that PKN1 is a direct substrate for ubiquitination when coexpressed with SspH1, the wild type and the indicated mutants of Flag-SspH1 and Myc-PKN1 were coexpressed with HA-Ub in HEK 293 cells. As PKN1 abundance was not detectable in the presence of wild-type SspH1, cells were treated with the proteasome inhibitor MG-132, which increased PKN1 abundance to detectable levels (Fig. 5b). Under these conditions, PKN1 was heavily ubiquitinated when coexpressed with wild-type SspH1 but not a catalytically dead (C492A) mutant (Fig. 5b). This effect was independent of the position or type of tag on SspH1 or PKN1 (Fig. 5c). Taken together, these results validate the relevance of the SspH1-PKN1 interaction surface observed in the crystal structure and demonstrate that PKN1 can be degraded in an SspH1- and 26S proteasome-dependent manner in a human cell line.
SspH1 attenuates the PKN1-dependent AR response.
In light of the striking effect of SspH1 on PKN1 stability, we sought to establish the biological consequences of SspH1 activity in cells. It has been previously reported that expression of SspH1 in CHO-K1 hamster cells suppresses NF-κB signaling (23). To determine if SspH1 suppresses NF-κB signaling in HEK 293 cells in a manner dependent on its catalytic function and its interaction with PKN1, wild-type and mutant alleles of Flag-tagged SspH1 were transiently expressed prior to determination of endogenous NF-κB activity. Transfection of an SspH1 expression construct suppressed NF-κB signaling in HEK 293 cells that were stimulated either with TNF-α or by overexpression of Nod1 (Fig. 6a and b). However, this suppressive effect was largely insensitive to either catalytic site (SspH1C492A) or PKN1 interaction site (SspH1D343A/Y366A) mutations in SspH1 (Fig. 6a and b). As the expression levels of the SspH1 wild-type and mutant versions were comparable in these experiments (Fig. 5a), NF-κB suppression by SspH1 appeared to be independent of its catalytic activity and its ability to bind PKN1, at least in HEK 293 cells. We infer that SspH1 influences NF-κB activity through sequestration of other cellular targets using one or more surfaces that do not overlap the PKN1 binding surface.
FIG 6.

Catalytic and noncatalytic functions of SspH1 in cells. (a) HEK 293 NF-κB reporter cells were transiently transfected with vectors expressing the wild type and the indicated mutants of Flag-SspH1 and a β-galactosidase reporter prior to stimulation with TNF-α or no stimulation. Luciferase activity in cell lysates was measured and normalized against β-galactosidase activity. (b) HEK 293 NF-κB reporter cells were transiently transfected and assayed as described for panel a, except with the addition of a vector expressing Myc-Nod1. (c) HEK 293 cells were transiently transfected with vectors expressing the wild type and the indicated mutants of Flag-SspH1, Flag-IκBβ, Myc-RhoA(V14), human androgen receptor (AR), MMTV-luciferase reporter, and β-galactosidase prior to treatment with synthetic androgen R1881 or no treatment. Luciferase activity was measured as described for panel a. The expression level of the indicated proteins in lysates was assessed by immunoblotting. (d) HEK 293 cells were transiently transfected and assayed as described for panel c, except with the addition of a vector that expressed either the wild type or the indicated Myc-PKN1 mutants. The expression level of the indicated proteins in lysates was assayed by immunoblotting. (e) Viability of a FUS1pr-HIS3 yeast strain (SY2625) transformed with galactose-inducible vectors (pGAL1) that expressed wild-type and mutant Flag-tagged SspH1. Strains were spotted as a serial dilution series on 2% glucose or galactose and imaged after 2 to 3 days at 30°C. (f) Protein expression in lysates from strains tested in panel e. Cultures were induced in 2% galactose for 2 h before detection of the indicated proteins by immunoblotting. (g) Viability of a FUS1pr-HIS3 yeast strain (SY2625) transformed with vectors that expressed wild-type and mutant Flag-tagged IpaH9.8. (h) Protein expression in lysates from strains tested in panel g. Detection was as described for panel f.
A primary biological function of PKN1 is to superactivate androgen receptor (AR) signaling (33, 34). To investigate whether this function of PKN1 was influenced by SspH1 in a catalytically dependent manner, we first established a functional AR signaling assay in HEK 293 cells, as reported previously (33). Stimulation of HEK 293 cells with the synthetic androgen R1881 gave rise to a robust (>50-fold) increase in signal compared to that of unstimulated cells (Fig. 6c). As explained by the above-described effects of SspH1 on PKN1 abundance, transient expression of wild-type SspH1 but not catalytic (SspH1C492A) or PKN1 interaction mutants (SspH1D343A/Y366A) severely attenuated AR activation (Fig. 6c). To test if PKN1 mutants defective for interaction with SspH1 could rescue AR responsiveness in the presence of wild-type SspH1, wild-type PKN1 and three binding mutants (R181A, R181K, and R185A) were transiently coexpressed prior to assaying for AR superactivation. All three PKN1 mutants restored the AR response (Fig. 6d). However, rescue by the R181A mutant was weaker than that of R185A and R181K mutants, perhaps due to an unintended perturbation of PKN1 function. Nevertheless, these data demonstrate that SspH1-mediated elimination of PKN1 results in the attenuation of the AR response in cells and suggest that the effect of SspH1 on NF-κB signaling is independent of its catalytic function or its interaction with PKN1.
Conservation of IpaH substrate recognition.
Our in vitro and in-cell analyses of the interaction between SspH1 and PKN1 revealed a mechanism whereby substrate interaction relieves autoinhibition by engaging a specific site on the LRR domain that competitively displaces autoinhibitory interactions (Fig. 4a, cartoon). We explored the generality of this enzyme-substrate relationship in other IpaH family members using a previously established yeast surrogate model for IpaH function (13, 19). Expression of SspH1 in yeast causes inviability, presumably because of ubiquitination of one or more yeast substrates, and this effect is dependent on both the catalytic and autoinhibitory functions of SspH1 (19). We tested the effect of SspH1 mutants that are compromised for PKN1 binding in the yeast viability assay and found that the D343A/Y366A double mutant abrogated the toxicity of SspH1, comparable to the catalytically dead version (Fig. 6e). As protein expression levels of wild-type and mutant versions of SspH1 were comparable in yeast (Fig. 6f), this result demonstrates that the same region of the LRR domain is used to target other SspH1 substrates.
Since autoregulation of IpaH enzyme activity relies on structurally conserved features in divergent Salmonella and Shigella enzymes (19), we tested whether a similar mechanism of substrate recognition operates in another autoregulated IpaH family member. We showed previously that expression of wild-type but not catalytically inactive Shigella IpaH9.8 is lethal in a yeast strain that relies on activity of the mating pheromone pathway for viability (13). We assessed the effect of an R163A/F187A/H210A triple mutant of IpaH9.8 predicted to disrupt a spatially analogous surface to the substrate interaction surface of SspH1 (corresponding to W323/D343/Y366 on SspH1). Strikingly, viability of the yeast reporter strain was restored equally well by the triple mutant as by a control catalytic mutant of IpaH9.8 (Fig. 6g). This effect was not due to perturbation of protein abundance, since the mutant proteins were expressed to similar levels as the wild-type protein (Fig. 6h). These results demonstrate that diverse substrates are recognized by spatially equivalent surfaces on distantly related IpaH enzymes that exhibit autoinhibition.
DISCUSSION
Pathogenic Gram-negative bacteria, such as Salmonella enterica serovar Typhimurium and Shigella, inject effector proteins into host cells in order to overcome intracellular barriers to infection (9, 52, 53). Once in the host, the function of these effectors depends both on their ability to interact with specific host factors and to be regulated in an appropriate fashion. Here, we illuminate the structural basis of specific recognition of the substrate PKN1 by the SspH1 enzyme and the mechanism of substrate-dependent activation of SspH1 enzymatic activity. We show that PKN1 engages SspH1 at a site on the LRR domain that lies in close proximity to the major autoinhibitory binding site for the SspH1 NEL domain. This close juxtaposition of binding sites enables substrate interaction to be coupled directly to liberation of the active catalytic domain.
LRR domains can engage structurally diverse ligands using different regions of the LRR domain surface, which can be classified into concave, convex, ascending, and descending contact surfaces (49). Two notable examples of E3-substrate interactions mediated by an LRR domain are recognition of the CDK inhibitor p27 in complex with Cks1 by the F-box protein Skp2 in humans (50), which engages the concave face of the Skp2 LRR, and of the transcription factor ASK1 by the F-box protein TIR1 in Arabidopsis (51), which engages the ascending and terminal end of the TIR1 LRR domain (see Fig. S1 in the supplemental material). In addition, mutagenesis and structural modeling implicates a similar surface in the recognition of the G1 cyclin Cln2 by the LRR domain of the F-box protein Grr1 in budding yeast (54). Another example of a bacterial effector that employs an LRR interaction domain is the Listeria invasion protein InlB, which uses the concave surface of its LRR domain to engage the host Met receptor for pathogen activation and internalization (see Fig. S1) (55). Other LRR surfaces are also exploited in pathogenesis. For example, mutational analysis has recently uncovered a role for the convex surface of the LRR domain of the Yersinia effector YopM in recognition of host caspase-1 (56). In this regard, we note that the PKN1 interaction site on the SspH1 LRR domain lies some distance from a second autoinhibitory site (19), which we speculate represents a secondary substrate-coupled autoinhibitory site. The conservation of activation-coupled substrate binding sites in other autoinhibited IpaH enzymes suggests that these sites will also be substrate interaction surfaces. Small molecules that target such surfaces may have the dual effect of both stabilizing host substrates and causing autocatalytic ubiquitination and degradation of the IpaH enzyme itself. The elucidation of further IpaH enzyme-host substrate structures will be of considerable interest in this regard.
Our mutational analysis based on the SspH1-PKN1 structure has enabled further insights into the mechanism of SspH1 action in cells. SspH1-mediated recognition and ubiquitination of PKN1 results in its destruction by the proteasome, with consequent impairment of the PKN1-dependent AR response. This observation is consistent with a role for steroid hormones in the proper function and activation of neutrophils and macrophages (36–38), which are the physiologically relevant host cell type during Salmonella infection (57). For example, neutrophils and macrophages derived from AR knockout mice produce lower levels of proinflammatory cytokines and TNF-α, respectively (58). Our observation that a Salmonella effector impairs steroid hormone signaling suggests that it is targeted by other pathogenic bacteria. We also note that other PKN1 functions may be modulated by SspH1, such as the suppressive effect of PKN1 on the Akt-dependent prosurvival response, which is known to influence the intracellular growth rate of Salmonella (40). It will be of interest to see if Akt signaling is modulated by the effects of SspH1 on PKN1.
Intriguingly, whereas SspH1 clearly exerts its effect on PKN1 stability through a catalytic mechanism, SspH1 instead appears to influence NF-κB signaling in a catalytic site-independent manner. As the effect of SspH1 on NF-κB signaling was also insensitive to mutations that disable the PKN1 interaction, we infer that other interaction targets of SspH1 in the host must mediate this effect. Noncatalytic functions for other pathogenic LRR-containing E3 enzymes have been observed previously, including for the Salmonella effector SlrP (59) and the plant pathogen effector XopL (60). Moreover, some pathogen-encoded LRR domain proteins, such as YopM from Yersinia (56, 61), lack catalytic domains entirely and presumably act solely by sequestration of host proteins. The autoregulation of enzymatic function may have evolved not only to allow pathogenic E3 enzymes to surgically target key substrates but also to allow the same enzymes to carry out noncatalytic functions through differential engagement of host proteins. The disruption of these distinct functions in IpaH enzymes may be necessary for effective therapeutic interdiction of the complex host-pathogen relationship.
Supplementary Material
Supplemental material
ACKNOWLEDGMENTS
We thank Igor Kurinov at the Argonne National Laboratory for assistance with microdiffraction experiments at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines (supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health and contract DE-AC02-06CH11357 from the U.S. Department of Energy) and the CMCF staff for data collection at beamline 08ID-1 at the Canadian Light Source (supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan). We also thank members of the Trevor Moraes laboratory at the University of Toronto for crystal data collection.
A.F.A.K. is funded by a Natural Sciences and Engineering Research Council postgraduate scholarship. This work was supported by grants to F.S. and M.T. from the Canadian Institutes of Health Research (MOP-57795 and MOP-126129), by a Genome Quebec International Recruitment Award to M.T., by a Canada Research Chair in Structural Biology to F.S., and by a Canada Research Chair in Systems and Synthetic Biology to M.T.
Footnotes
Published ahead of print 18 November 2013
REFERENCES
- 1.Komander D, Rape M. 2012. The ubiquitin code. Annu. Rev. Biochem. 81:203–229. 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- 2.Ravid T, Hochstrasser M. 2008. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9:679–690. 10.1038/nrm2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayden MS, Ghosh S. 2012. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26:203–234. 10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang X, Chen ZJ. 2012. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 12:35–48. 10.1038/nri3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, Xavier RJ. 2012. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella typhimurium. Cell Host Microbe 12:778–790. 10.1016/j.chom.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thurston TLM, Ryzhakov G, Bloor S, Muhlinen von, Randow NF. 2009. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10:1215–1221. 10.1038/ni.1800 [DOI] [PubMed] [Google Scholar]
- 7.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. 2009. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183:5909–5916. 10.4049/jimmunol.0900441 [DOI] [PubMed] [Google Scholar]
- 8.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I. 2011. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333:228–233. 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izoré T, Job V, Dessen A. 2011. Biogenesis, regulation, and targeting of the type III secretion system. Structure 19:603–612. 10.1016/j.str.2011.03.015 [DOI] [PubMed] [Google Scholar]
- 10.Mesquita FS, Thomas M, Sachse M, Santos AJM, Figueira R, Holden DW. 2012. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 8:e1002743. 10.1371/journal.ppat.1002743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rytkönen A, Poh J, Garmendia J, Boyle C, Thompson A, Liu M, Freemont P, Hinton JCD, Holden DW. 2007. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. U. S. A. 104:3502–3507. 10.1073/pnas.0610095104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel JC, Hueffer K, Lam TT, Galán JE. 2009. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell 137:283–294. 10.1016/j.cell.2009.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. 2007. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 1:77–83. 10.1016/j.chom.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 14.Miao EA, Scherer CA, Tsolis RM, Kingsley RA, Adams LG, Bäumler AJ, Miller SI. 1999. Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol. Microbiol. 34:850–864. 10.1046/j.1365-2958.1999.01651.x [DOI] [PubMed] [Google Scholar]
- 15.Singer AU, Rohde JR, Lam R, Skarina T, Kagan O, DiLeo R, Chirgadze NY, Cuff ME, Joachimiak A, Tyers M, Sansonetti PJ, Parsot C, Savchenko A. 2008. Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 15:1293–1301. 10.1038/nsmb.1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reference deleted.
- 17.Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP. 1999. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286:1321–1326. 10.1126/science.286.5443.1321 [DOI] [PubMed] [Google Scholar]
- 18.Scheffner M, Nuber U, Huibregtse JM. 1995. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373:81–83. 10.1038/373081a0 [DOI] [PubMed] [Google Scholar]
- 19.Chou Y-C, Keszei AFA, Rohde JR, Tyers M, Sicheri F. 2012. Conserved structural mechanisms for autoinhibition in IpaH ubiquitin ligases. J. Biol. Chem. 287:268–275. 10.1074/jbc.M111.316265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quezada CM, Hicks SW, Galán JE, Stebbins CE. 2009. A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U. S. A. 106:4864–4869. 10.1073/pnas.0811058106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Li H, Hu L, Wang J, Zhou Y, Pang Z, Liu L, Shao F. 2008. Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 15:1302–1308. 10.1038/nsmb.1517 [DOI] [PubMed] [Google Scholar]
- 22.Xia Z-P, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. 2009. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461:114–119. 10.1038/nature08247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haraga A, Miller SI. 2006. A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cell Microbiol. 8:837–846. 10.1111/j.1462-5822.2005.00670.x [DOI] [PubMed] [Google Scholar]
- 24.Flynn P, Mellor H, Casamassima A, Parker PJ. 2000. Rho GTPase control of protein kinase C-related protein kinase activation by 3-phosphoinositide-dependent protein kinase. J. Biol. Chem. 275:11064–11070. 10.1074/jbc.275.15.11064 [DOI] [PubMed] [Google Scholar]
- 25.Maesaki R, Ihara K, Shimizu T, Kuroda S, Kaibuchi K, Hakoshima T. 1999. The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol. Cell 4:793–803. 10.1016/S1097-2765(00)80389-5 [DOI] [PubMed] [Google Scholar]
- 26.Modha R, Campbell LJ, Nietlispach D, Buhecha HR, Owen D, Mott HR. 2008. The Rac1 polybasic region is required for interaction with its effector PRK1. J. Biol. Chem. 283:1492–1500. 10.1074/jbc.M706760200 [DOI] [PubMed] [Google Scholar]
- 27.Owen D, Lowe P, Nietlispach D, Brosnan C, Chirgadze NY, Parker PJ, Blundell T, Mott HR. 2003. Molecular dissection of the interaction between the small G proteins Rac1 and RhoA and protein kinase C-related kinase 1 (PRK1). J. Biol. Chem. 278:50578–50587. 10.1074/jbc.M304313200 [DOI] [PubMed] [Google Scholar]
- 28.Takahashi M, Mukai H, Toshimori M, Miyamoto M, Ono Y. 1998. Proteolytic activation of PKN by caspase-3 or related protease during apoptosis. Proc. Natl. Acad. Sci. U. S. A. 95:11566–11571. 10.1073/pnas.95.20.11566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srikanth CV, Wall DM, Maldonado-Contreras A, Shi HN, Zhou D, Demma Z, Mumy KL, McCormick BA. 2010. Salmonella pathogenesis and processing of secreted effectors by caspase-3. Science 330:390–393. 10.1126/science.1194598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hänisch J, Kölm R, Wozniczka M, Bumann D, Rottner K, Stradal TEB. 2011. Activation of a RhoA/myosin II-dependent but Arp2/3 complex-independent pathway facilitates Salmonella invasion. Cell Host Microbe 9:273–285. 10.1016/j.chom.2011.03.009 [DOI] [PubMed] [Google Scholar]
- 31.Ohlson MB, Huang Z, Alto NM, Blanc M-P, Dixon JE, Chai J, Miller SI. 2008. Structure and function of Salmonella SifA indicate that its interactions with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host Microbe 4:434–446. 10.1016/j.chom.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hentschke M, Berneking L, Belmar Campos C, Buck F, Ruckdeschel K, Aepfelbacher M. 2010. Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One 5:e13165. 10.1371/journal.pone.0013165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metzger E, Müller JM, Ferrari S, Buettner R, Schüle R. 2003. A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J. 22:270–280. 10.1093/emboj/cdg023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, Patnaik D, Higgins JMG, Potier N, Scheidtmann K-H, Buettner R, Schüle R. 2008. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat. Cell Biol. 10:53–60. 10.1038/ncb1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Müller JM, Greschik H, Kirfel J, Ji S, Kunowska N, Beisenherz-Huss C, Günther T, Buettner R, Schüle R. 2010. Phosphorylation of histone H3T6 by PKCβI controls demethylation at histone H3K4. Nature 464:792–796. 10.1038/nature08839 [DOI] [PubMed] [Google Scholar]
- 36.Chuang K-H, Altuwaijri S, Li G, Lai J-J, Chu C-Y, Lai K-P, Lin H-Y, Hsu J-W, Keng P, Wu M-C, Chang C. 2009. Neutropenia with impaired host defense against microbial infection in mice lacking androgen receptor. J. Exp. Med. 206:1181–1199. 10.1084/jem.20082521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai J-J, Lai K-P, Chuang K-H, Chang P, Yu I-C, Lin W-J, Chang C. 2009. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J. Clin. Investig. 119:3739–3751. 10.1172/JCI39335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. 2010. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Investig. 120:3350–3364. 10.1172/JCI41080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasui T, Sakakibara-Yada K, Nishimura T, Morita K, Tada S, Mosialos G, Kieff E, Kikutani H. 2012. Protein kinase N1, a cell inhibitor of Akt kinase, has a central role in quality control of germinal center formation. Proc. Natl. Acad. Sci. U. S. A. 109:21022–21027. 10.1073/pnas.1218925110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuijl C, Savage NDL, Marsman M, Tuin AW, Janssen L, Egan DA, Ketema M, van den Nieuwendijk R, van den Eeden SJF, Geluk A, Poot A, van der Marel G, Beijersbergen RL, Overkleeft H, Ottenhoff THM, Neefjes J. 2007. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 450:725–730. 10.1038/nature06345 [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67:271–281. 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voss NR, Gerstein M. 2010. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 38:W555–W562. 10.1093/nar/gkq395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bella J, Hindle KL, McEwan PA, Lovell SC. 2008. The leucine-rich repeat structure. Cell. Mol. Life Sci. 65:2307–2333. 10.1007/s00018-008-8019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. 2005. Structural basis of the Cks1-dependent recognition of p27Kip1 by the SCFSkp2 ubiquitin ligase. Mol. Cell 20:9–19. 10.1016/j.molcel.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 51.Tan X, Calderon-Villalobos LIA, Sharon M, Zheng C, Robinson CV, Estelle M, Zheng N. 2007. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446:640–645. 10.1038/nature05731 [DOI] [PubMed] [Google Scholar]
- 52.Ham H, Sreelatha A, Orth K. 2011. Manipulation of host membranes by bacterial effectors. Nature 9:635–646. 10.1038/nrmicro2602 [DOI] [PubMed] [Google Scholar]
- 53.Ashida H, Ogawa M, Kim M, Mimuro H, Sasakawa C. 2012. Bacteria and host interactions in the gut epithelial barrier. Nat. Chem. Biol. 8:36–45. 10.1038/nnano.2012.208 [DOI] [PubMed] [Google Scholar]
- 54.Hsiung YG, Chang HC, Pellequer JL, La Valle R, Lanker S, Wittenberg C. 2001. F-box protein Grr1 interacts with phosphorylated targets via the cationic surface of its leucine-rich repeat. Mol. Cell. Biol. 21:2506–2520. 10.1128/MCB.21.7.2506-2520.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niemann HH, Jäger Butler, VPJG. van den Heuvel J, Schmidt S, Ferraris D, Gherardi E, Heinz DW. 2007. Structure of the human receptor tyrosine kinase met in complex with the Listeria invasion protein InlB. Cell 130:235–246. 10.1016/j.cell.2007.05.037 [DOI] [PubMed] [Google Scholar]
- 56.LaRock CN, Cookson BT. 2012. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12:799–805. 10.1016/j.chom.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geddes K, Cruz F, Heffron F. 2007. Analysis of cells targeted by Salmonella type III secretion in vivo. PLoS Pathog. 3:e196. 10.1371/journal.ppat.0030196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lai J-J, Lai K-P, Zeng W, Chuang K-H, Altuwaijri S, Chang C. 2012. How does the androgen receptor in the innate and adaptive immune system defend the body? Lessons from conditional AR knockout mice. Am. J. Pathol. 181:1504–1512. 10.1016/j.ajpath.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernal-Bayard J, Cardenal-Munoz E, Ramos-Morales F. 2010. The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J. Biol. Chem. 285:16360–16368. 10.1074/jbc.M110.100669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singer AU, Schulze S, Skarina T, Xu X, Cui H, Eschen-Lippold L, Egler M, Srikumar T, Raught B, Lee J, Scheel D, Savchenko A, Bonas U. 2013. A pathogen type III effector with a novel E3 ubiquitin ligase architecture. PLoS Pathog. 9:e1003121. 10.1371/journal.ppat.1003121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Evdokimov AG, Anderson DE, Routzahn KM, Waugh DS. 2001. Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: a leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol. 312:807–821. 10.1006/jmbi.2001.4973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material