Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission (original) (raw)
Significance
A growing body of evidence has determined that somatic mutations in acute myeloid leukemia (AML) accumulate in self-renewing hematopoietic stem cells (HSCs). Thus, at the time of diagnosis, AML patients harbor preleukemic HSCs containing some, but not all, of the mutations in the downstream leukemia. Despite these findings, common patterns of preleukemic clonal evolution have not been determined, nor has the response of preleukemic HSCs to standard therapy been identified. This report addresses both of these questions determining that there are common patterns of preleukemic clonal evolution in AML, and that these preleukemic HSCs often survive standard induction chemotherapy. This study is of interest to the AML field, and broadly in cancer genomics as the principle that stem cells acquire initial cancer-initiating mutations is likely to extend beyond AML.
Keywords: preleukemia, clonal evolution
Abstract
Cancer is widely characterized by the sequential acquisition of genetic lesions in a single lineage of cells. Our previous studies have shown that, in acute myeloid leukemia (AML), mutation acquisition occurs in functionally normal hematopoietic stem cells (HSCs). These preleukemic HSCs harbor some, but not all, of the mutations found in the leukemic cells. We report here the identification of patterns of mutation acquisition in human AML. Our findings support a model in which mutations in “landscaping” genes, involved in global chromatin changes such as DNA methylation, histone modification, and chromatin looping, occur early in the evolution of AML, whereas mutations in “proliferative” genes occur late. Additionally, we analyze the persistence of preleukemic mutations in patients in remission and find CD34+ progenitor cells and various mature cells that harbor preleukemic mutations. These findings indicate that preleukemic HSCs can survive induction chemotherapy, identifying these cells as a reservoir for the reevolution of relapsed disease. Finally, through the study of several cases of relapsed AML, we demonstrate various evolutionary patterns for the generation of relapsed disease and show that some of these patterns are consistent with involvement of preleukemic HSCs. These findings provide key insights into the monitoring of minimal residual disease and the identification of therapeutic targets in human AML.
Acute myeloid leukemia (AML) is an aggressive malignancy of the bone marrow characterized by uncontrolled proliferation of immature myeloid lineage cells (1, 2). Recent advances in high-throughput sequencing have led to the identification of many recurrently mutated genes implicated in the pathogenesis of AML (3–10). The coding genomes of over 200 AML patients have been sequenced by The Cancer Genome Atlas (TCGA) consortium, identifying ∼30 genes that are mutated in greater than 2% of patients (11). In addition, several studies have investigated the genetic changes occurring between diagnosis and relapse in the same patient (12, 13). Despite this extensive characterization of the genetic variation in AML, the evolutionary processes that precede the onset of frank leukemia and shape the architecture of the disease have not been well elucidated.
These sequencing efforts have demonstrated that an individual AML case is associated with an average of five different recurrent mutations (11), raising the question of how these multiple mutations accumulate in a single lineage of cells given the generally low spontaneous mutation rate in hematopoietic cells (14) and the lack of hypermutator phenotypes in this disease (15). To explain these observations, we proposed a model in which the majority of AML mutations are sequentially acquired in successive clones of self-renewing hematopoietic stem cells (HSCs), unless the mutation confers self-renewal potential on a more differentiated cell (16, 17). Early evidence consistent with this model comes from studies of AML patients harboring the AML1-ETO translocation (18). In these patients, HSCs isolated during durable remission produced normal myeloid colonies in vitro with detectable AML1-ETO transcripts. Additionally, in chronic myeloid leukemia, the breakpoint cluster region–Abelson tyrosine-protein kinase 1 (BCR-ABL) translocation occurs in HSCs, causing chronic disease, but subsequent progression to blast crisis involves mutation evolution at the level of granulocyte–macrophage progenitors (19, 20). More recently, a study of elderly women with clonal hematopoiesis indicated by X-inactivation skewing showed that these individuals harbored somatic ten eleven translocation methylcytosine dioxygenase 2 (TET2) mutations, occurring at the level of the HSC, that may predispose them to hematologic malignancies (21).
Recent work from our laboratory has provided direct evidence supporting this model by demonstrating that, in a small subset of AML patients harboring mutations in the FMS-related tyrosine kinase 3 (FLT3) receptor tyrosine kinase, multiple genetic lesions serially accumulate in functionally normal HSCs (22). We defined these HSCs as “preleukemic” because they are genetically distinct from germ-line HSCs in that they harbor only a subset of the leukemia-specific mutations which identifies them as the evolutionary ancestors of the frankly leukemic cells. These preleukemic mutations included mutations in recurrently mutated genes that are known drivers of leukemogenesis, such as TET2, SMC1A, nucleophosmin 1 (NPM1), and CTCF, implicating preleukemic mutations as functional components of AML evolution. Interestingly, mutation of FLT3 never occurred in preleukemic cells, leading to the hypothesis that genetic lesions in AML may occur in a nonrandom pattern. These data are supported by studies of mutation stability between diagnosis and relapse in AML, where mutations in FLT3 were found to often be unstable, indicating that these mutations are late events (13). Additionally, in certain patients, only one recurrent coding mutation distinguished the functionally normal HSCs from the frankly leukemic cells, suggesting that these preleukemic HSCs may be a putative reservoir for relapsed disease by surviving chemotherapy and acquiring additional mutations that transform them into AML.
From these observations, we proposed a model of AML evolution where preleukemic HSCs can persist during clinical remission and have the potential to give rise to relapsed disease (16). More specifically, we proposed that relapsed disease can originate from multiple sources including (i) treatment refractory primary disease, (ii) further evolution of a dominant clone present at diagnosis, (iii) outgrowth of a subclone present at diagnosis, or (iv) further evolution of disease from a preleukemic HSC. Here, through the genomic and functional analysis of de novo AML and patient-matched HSCs, we provide evidence supporting these mechanisms by determining patterns of preleukemic mutation acquisition at diagnosis, and by tracking preleukemic cells and mutations in clinical remission and relapse.
Results
Identification of Preleukemic Mutations and HSCs in a Diverse Cohort of Human AML Patients.
Our previous studies identified preleukemic mutations and HSCs in a small cohort of AML patients. We aimed to expand upon these studies to investigate preleukemic clonal evolution in a larger cohort and broader spectrum of AML patients (Tables S1 and S2). Exome sequencing (23, 24) of FACS-purified leukemia cells (CD99+ TIM3+) (22, 25) and patient-matched CD3+ T cells was used to identify leukemia-specific mutations in 10 patients. Sequencing was performed to a median coverage depth of 56-fold (Fig. S1_A_) and all mutations were validated using Sanger sequencing of genomic DNA (gDNA) (Table S3).
All leukemia-specific mutations were then sequenced at high depth (Fig. S1_E_) using targeted amplicon sequencing of FACS-purified leukemia cells, CD3+ T cells, and Lin−CD34+CD38−TIM3−CD99− HSCs (Fig. S2). Importantly, apart from surface marker expression, these HSCs were also functionally defined by their ability to generate long-term myeloid and lymphoid engraftment in immunodeficient NOD/SCID/IL2R-γ (NSG) mice, as previously described (Fig. S3_A_) (22, 25). Preleukemic mutations were identified as those mutations occurring at >1% variant allele frequency in the purified HSC subpopulation, a threshold identified by control sequencing reactions of known admixtures of leukemic and normal DNA (Fig. S4). As expected, deep sequencing of the HSC subpopulation for leukemia-specific mutations never detected all leukemia-specific mutations, which would be an indicator of leukemia cell contamination. Leukemia-specific mutations found to be absent from purified HSCs were classified as late events. Six of 10 patients had detectable preleukemic mutations in purified HSCs (Fig. 1 A–F). These preleukemic mutations included recurrent mutations in isocitrate dehydrogenase 2 (IDH2), DNA (cytosine-5-) methyltransferase 3 alpha (DNMT3A), additional sex combs like 1 (ASXL1), and IKAROS family zinc finger 1 (IKZF1), and inversion of chromosome 16 resulting in the core-binding factor beta subunit–myosin heavy chain 11 (CBFB-MYH11) fusion gene. In a select number of cases, leukemic mutations were detectable not only in purified HSCs, but also in a fraction of sorted B and T lymphocytes at diagnosis, indicating a clear contribution of preleukemic HSCs to hematopoiesis in humans (Fig. S5). Interestingly, multiple patients that lacked detectable preleukemic mutations in purified HSCs showed preleukemic mutation burden in CD19+ lymphoid cells that developed in NSG mice transplanted with those same purified HSCs (SU067, SU072; Fig. S4). This implies that, in these cases, rare preleukemic HSCs exist that have a competitive advantage in the xenotransplantation setting (Table S1). Of the 10 samples analyzed, 2 (SU290, SU336) showed no detectable preleukemic mutations.
Fig. 1.

Sequencing and single-cell genotyping assays identify preleukemic mutations and HSCs in a diverse cohort of human AML patients. (A–F) Targeted amplicon sequencing was performed on all leukemic mutations identified by exome sequencing in six AML patients. Targeted sequencing was performed on gDNA isolated from FACS-purified leukemia cells (red, CD99+ TIM3+), T cells (yellow, CD3+), and HSCs (blue, CD34+/CD38−/CD99−/TIM3−) for each patient. The dotted line represents the threshold of detection. Gene names highlighted in bold indicate recurrently mutated genes in AML. (G and I) Single HSC-derived colony genotyping assays were performed for patient samples SU320 (G) and SU353 (I) using custom TaqMan SNP genotyping assays specific for all recurrent mutations. Each colony was classified as wild type or mutant for each mutation based on comparison with leukemia (red), T-cell (yellow), or no template (black) control reactions. (H and J) The single-cell data from G and I were used to diagram the evolutionary history of cases SU320 (H) and SU353 (J), where preleukemic mutations are sequentially acquired followed by late mutations never found in colonies derived from HSCs.
To validate these findings and determine the order of mutation acquisition in a subset of samples, colonies derived from single HSCs were genotyped using custom TaqMan SNP genotyping assays for all recurrent mutations, and a select subset of the other leukemia-specific mutations (Fig. 1 G and I). In case SU320, 104 colonies were tested for the presence of mutations in IDH2, deleted in primary ciliary dyskinesia homolog (DPCD), microtubule associated monooxygenase, calponin and lim domain containing 3 (MICAL3), mitochondrially encoded NADH dehydrogenase 5 (MTND5), NPM1, and FLT3 (Fig. 1_G_). Of these colonies, 61 were found to be wild type for all mutations profiled, 1 was found to harbor mutations in IDH2 and DPCD only, 30 were found with mutations in IDH2, DPCD, and MICAL3, and 12 were found to harbor mutations in IDH2, DPCD, MICAL3, and MTND5. Importantly, none of the colonies contained mutations in NPM1 or FLT3, suspected late mutations based on amplicon sequencing (Fig. 1_E_). These single-cell–based assays determined the stepwise pattern of mutation acquisition in case SU320 (Fig. 1_H_). Intriguingly, mutation of MTND5, a mitochondrially encoded gene, showed a progressive increase in mutation burden in preleukemic cells, indicated by a somewhat stepwise increase in mutant allele frequency at the single-cell level (Fig. 1_G_).
Similarly, in case SU353, 192 single-cell–derived myeloid colonies were genotyped for mutations in DNMT3A, MAU2, ASXL1, poly (ADP-Ribose) polymerase family member 6 (PARP6), dynein axonemal heavy chain 10 (DNAH10), NPM1, and FLT3 (Fig. 1_I_). Of these 192 colonies, only 1 was found to be wild type for all mutations profiled, indicating that the preleukemic cells in this patient have strongly outcompeted their genetically normal counterparts. Of the remaining 191 colonies, 1 was found to harbor only the DNMT3A mutation, 5 were found with both the DNMT3A and maternally affected uncoordination (MAU2) mutations, 182 were found to harbor mutations in DNTM3A, MAU2, ASXL1, and PARP6, and 3 were found to contain mutations in DNMT3A, MAU2, ASXL1, PARP6, and DNAH10. Importantly, none of the colonies harbored mutations in NPM1 or FLT3. These single-cell–based assays determined the order of acquisition of mutations in case SU353 (Fig. 1_J_).
Mutation Acquisition in AML Occurs in Patterns.
The identification of preleukemic mutations and late mutations in a diverse cohort of AML patients led us to investigate the possibility that there are patterns of mutation acquisition in human AML. Preleukemic mutation analysis from 16 patients [10 described here and 6 previously reported (22)] was used to identify early and late mutations in recurrently mutated genes (Fig. 2_A_). These recurrently mutated genes were subdivided into categories based on the classification system described by the TCGA AML consortium (Table S4). Of 74 total mutations in recurrently mutated genes, 36 (49%) were found to be preleukemic and 38 (51%) were found to be late. Mutations in genes involved in DNA methylation, chromatin modification, and chromatin topology (cohesin complex) were significantly overrepresented in the subset of preleukemic mutations and underrepresented in the subset of late mutations. In contrast, mutations in genes involved in activated signaling were found to be significantly overrepresented in the subset of late mutations and underrepresented in the subset of preleukemic mutations.
Fig. 2.

Mutation acquisition in AML occurs in patterns with preleukemic landscaping mutations followed by late proliferative mutations. (A) Exome sequencing and targeted amplicon sequencing data identifying preleukemic and late mutations from 16 patients (Table S1) were pooled. Recurrent mutations (74 total) were stratified according to the classification system established by the TCGA, and assigned to be preleukemic or late based on detection in HSCs as described in Table S4. The number of mutations in each category is indicated on the x axis. (B) Additional AML cases were selected for the presence of recurrent mutations in IDH1/IDH2 (n = 15), DNMT3A (n = 4), NPM1 (n = 17), FLT3 (n = 13), or KRAS/NRAS (n = 4). Each dot represents one case (see Table S2 for patient information). The dotted line represents the threshold of detection. The ratio of cases with preleukemic mutation of a given gene to the total number of cases with that mutation analyzed is shown below the plot. (C) A model for the acquisition of mutations in AML. (**P < 0.001, *P < 0.05; NS, not significant, χ2 test).
Additional patient samples were selected based on prior knowledge of somatic mutations in IDH1/IDH2, DNMT3A, NPM1, FLT3, or Kirsten rat sarcoma viral oncogene homolog (KRAS)/neuroblastoma rat sarcoma viral oncogene homolog (NRAS). Targeted amplicon sequencing of gDNA from purified HSCs was used to determine whether these mutations were preleukemic (Fig. 2_B_ and Fig. S6). Mutations in IDH1 and IDH2 were found to be preleukemic in 80% of patients. Similarly, mutations in DNMT3A were found to be preleukemic in 75% of patients. In contrast, only 12% of patients with mutation of NPM1 and 0% of patients with mutation in FLT3 or KRAS/NRAS exhibited detectable preleukemic burden of these mutations in purified HSCs. From this analysis, we developed a model of mutation acquisition (Fig. 2_C_) where the earliest founding mutations occur in “landscaping” genes, involved in global regulation of gene expression through epigenetic mechanisms, whereas late progressor mutations occur in genes that generally lead to an increase in activated signaling and cellular proliferation such as RAS and FLT3.
Preleukemic HSCs Survive Induction Chemotherapy and Persist in Remission.
We hypothesized that preleukemic HSCs may persist in remission and contribute to relapse through the acquisition of new mutations. In most AML patients, some normal HSCs survive induction chemotherapy and eventually repopulate the bone marrow, leading to hematologic remissions. To investigate the chemosensitivity of preleukemic HSCs, we analyzed bone marrow samples from the patients investigated above for the presence of leukemia-specific mutations at the time of first hematologic complete remission. CD34+ hematopoietic progenitors, CD19+ B lymphoid cells, and CD14+/CD11+/CD56+ mature monocytes, macrophages, and natural killer (NK) cells were FACS purified (Fig. S7), and gDNA from these populations was analyzed for leukemia-specific mutations by targeted amplicon sequencing. In four of six patients where preleukemic mutations were detected in purified HSCs at diagnosis, nearly all of these preleukemic mutations were also detected in both the CD34+ progenitors and other mature cells during remission (Fig. 3 A–D). Moreover, the presence of these preleukemic mutations in mature cells indicates that these HSCs retain normal function and actively contribute to bone marrow reconstitution. In the remaining two patients, preleukemic burden was not detected in remission, an observation that is consistent with the inability of preleukemic HSCs from these two samples to engraft in NSG mice (Fig. S4). In the four patients in whom preleukemic mutations were not detected in FACS-purified HSCs at diagnosis, preleukemic mutations were, unsurprisingly, not detected in any cells at remission (Fig. S8). Together, these data indicate that preleukemic HSCs are not eradicated by induction chemotherapy and constitute a cellular reservoir that has the potential to seed relapsed disease.
Fig. 3.

Preleukemic mutations and HSCs persist in remission. (A–D) Bone marrow mononuclear cells from patient remission samples were sorted for CD19+ B cells, CD14+/CD11b+/CD56+ mature monocytes, macrophages, and NK cells, and CD34+ hematopoietic progenitors. Targeted amplicon sequencing was performed for all leukemic mutations in all sorted populations. The dotted line represents the threshold of detection. All mutations identified as being preleukemic at diagnosis are highlighted in blue.
Relapsed AML Follows Diverse Evolutionary Patterns.
The persistence of these preleukemic HSCs in remission led us to investigate whether these cells can contribute to relapsed disease. Some of the patients profiled above at diagnosis went on to relapse including cases SU067, SU320, and SU353. Whole-exome sequencing was performed on gDNA isolated from FACS-purified leukemia cells from the relapsed disease. Variant allele frequencies of putative relapse-specific mutations were determined by targeted amplicon sequencing of leukemic gDNA at diagnosis and at relapse (Fig. 4 A, C, and E). Some mutations discovered at relapse were found to be present at low levels at diagnosis, indicating the existence of a minor subclone at diagnosis. Other mutations were undetectable at diagnosis indicating a newly acquired relapse-specific mutation, or a mutation present below the threshold of detection of this assay.
Fig. 4.
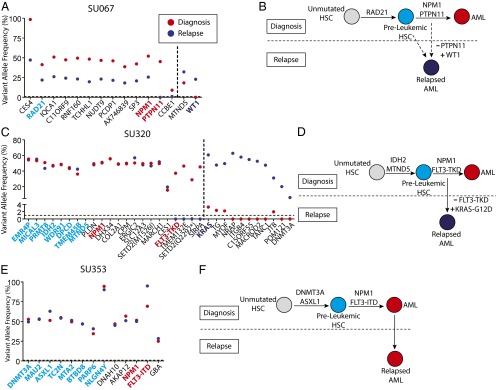
Preleukemic mutations are retained at relapse. (A, C, and E) Exome sequencing was performed to identify mutations detectable at relapse that were not detected at diagnosis. Targeted amplicon sequencing of all diagnosis-specific and relapse-specific mutations was performed in FACS-purified leukemic cells (CD99+ TIM3+) from the disease at diagnosis (red) and at relapse (dark blue). Diagnosis-specific mutations are depicted to the left of the dotted line, whereas relapse-specific mutations are depicted to the right. All mutations identified as being preleukemic at diagnosis are highlighted in light blue, whereas recurrent mutations identified as being late mutations at diagnosis are highlighted in red. Recurrent mutations identified at relapse are highlighted in dark blue. (B, D, and F) Evolutionary models for the progression of disease in each case are presented, with depiction of recurrently mutated genes in each model. The solid arrows represent determined evolutionary steps, and the dotted arrows represent inferred evolutionary steps.
In case SU067, mutations in two genes present at diagnosis, CCBE1 and protein tyrosine phosphatase, non-receptor type 11 (PTPN11), were absent at relapse, and a mutation in Wilms tumor 1 (WT1) was absent at diagnosis and present in the major clone at relapse (Fig. 4_A_). Loss of a mutation in PTPN11, a recurrent mutation that leads to increased proliferation, indicated a step back to a clone ancestral to the dominant diagnostic clone. Subsequent acquisition of a mutation in WT1, another recurrently mutated gene, indicated a step forward in clonal evolution (Fig. 4_B_). Given that the WT1 mutation was undetectable at diagnosis, it is possible that the relapsed disease originated from a rare antecedent preleukemic HSC.
In case SU320, four mutations present at diagnosis, including a recurrent mutation in FLT3, were absent at relapse. Of the 11 new mutations found at relapse, 4 were detected at low levels at diagnosis, including a recurrent mutation in KRAS, indicating the presence of a minor subclone at diagnosis (Fig. 4_C_). These sequencing results indicate that the relapsed disease originated from a minor subclone present at diagnosis and not from a preleukemic HSC (Fig. 4_D_).
In case SU353, no mutation differences were found between the disease at diagnosis and the disease at relapse (Fig. 4_E_). This observation is consistent with the clinical course of this patient who, at the time of complete hematologic remission, harbored evidence of residual disease based on the presence of an abnormal immunophenotypic pattern in the CD34+ compartment (Fig. S7). This was shown by targeted amplicon sequencing to be the result of residual leukemic cells from the diagnostic clone (Fig. 3_D_). Moreover, this patient remained in remission for just 2 mo before presenting with relapsed disease. These results indicate that the SU353 relapsed disease was, in fact, refractory disease that was not fully eliminated by induction chemotherapy (Fig. 4_F_). Collectively, these three patients illustrate multiple distinct mechanisms for relapse, some of which do not preclude the involvement of preleukemic HSCs.
Discussion
We report here the identification of patterns of mutation acquisition in AML and the persistence of preleukemic HSCs in patients in complete remission. Sequencing studies of 10 AML cases identified preleukemic mutations in multiple diverse subgroups. Preleukemic mutations were identified in multiple recurrently mutated genes with clear prognostic impact in AML, including DNMT3A, IDH1, IDH2, ASXL1, IKZF1, and the fusion gene CBFB-MYH11. Incorporating these 10 cases with our 6 previously published cases (22), we identified patterns in mutation acquisition in AML where the earliest mutations occur in landscaping genes, involved primarily in regulation of the epigenome, whereas late mutations occur primarily in “proliferative” genes, involved in activated signaling. These data were supported by directed profiling of 16 additional patients harboring mutations in NPM1, IDH1/IDH2, DNMT3A, FLT3, or KRAS/NRAS. Analysis of the 10 cases reported here during remission demonstrated that preleukemic HSCs can survive induction chemotherapy. Moreover, these cells contributed to myeloid and lymphoid hematopoiesis during remission, generating mature B cells and myeloid cells that harbored preleukemic mutations. Finally, we investigated paired diagnostic and relapse samples from several of these patients and found that relapsed AML follows diverse evolutionary patterns, some of which are consistent with involvement of preleukemic HSCs. These results provide key insights into the evolutionary processes that govern leukemia onset and progression and identify preleukemic HSCs as a cellular reservoir in remission that is poised to generate relapsed disease.
The results presented here demonstrate the characterization of broad patterns of mutation acquisition in AML. Our data identified mutations in landscaping genes as significantly enriched in preleukemic cells. These landscaping genes are involved in processes such as DNA methylation, histone modification, and chromatin looping. In contrast, mutations in NPM1 or in genes involved in activated signaling were significantly absent in preleukemic cells. These observations support a model for leukemogenesis where mutations in landscaping genes occur in HSCs early in evolutionary time, which prepares these preleukemic HSCs, or, more likely, their downstream progenitors, to transform upon the acquisition of additional proliferative mutations. This hypothesis generates multiple testable questions regarding the evolutionary processes governing leukemogenesis. First, can different late proliferative mutations substitute for one another? This question has been indirectly addressed by sequencing studies that show relapsed disease that differs from the disease at diagnosis (12, 13). These previous studies have shown that mutations in FLT3 and NRAS are often lost or gained at relapse, suggesting substitution of one proliferative mutation for another. Here, we present one example in which a mutation in KRAS at relapse substitutes for a mutation in FLT3 at diagnosis (Fig. 4 C and D). Nevertheless, functional studies will be necessary to definitively address this question. Second, are preleukemic mutations absolutely necessary for continued survival of leukemic cells? In chronic myelogenous leukemia harboring the BCR-ABL translocation, it is known that targeting this early mutation has a profound effect on the leukemic cells, indicating that the BCR-ABL translocation does not function via a “hit-and-run” mechanism.
The persistence of preleukemic cells during remission identifies a relatively unexplored aspect of AML biology. Although the presence and contribution of these cells to remission hematopoiesis were variable, patients with high preleukemic burden seemed to maintain similar or elevated levels of preleukemic burden during remission, indicating that these cells are not targeted by induction chemotherapy. Although formal proof of the involvement of preleukemic HSCs in generation of relapsed disease has not yet been obtained, we believe that these cells are poised to acquire additional mutations and transform, once again, into leukemic cells. As more studies are performed on preleukemia in AML, it will be important to rigorously determine whether patients with preleukemic cells at remission develop relapsed disease at an increased frequency. Moreover, the identification of preleukemic cells in remission has important implications for the monitoring of minimal residual disease (MRD) by molecular analyses that might instead detect residual preleukemic cells. As we have identified patterns of mutation acquisition, it will be important to probe for the presence of the earliest founder mutations so as to capture both preleukemic and leukemic contributions during remission. MRD studies using recurrent mutations in NPM1 and FLT3, shown here to be frequently late events in AML evolution, undoubtedly underestimate the contribution of preleukemic cells and leukemic subclones with different progressor mutations.
We have previously hypothesized that relapsed disease in AML can originate from multiple sources including reevolution of disease from a poised preleukemic HSC (16). If a role for these preleukemic cells in relapse disease evolution can be established, it will be necessary to target these cells. Such a result would shift the focus of drug development from late leukemogenic events, such as mutation of FLT3, to the earliest events, such as mutation of IDH1/IDH2, DNMT3A, or members of the cohesin complex. Even so, drugs that are designed to target these mutations in leukemic cells may not be similarly capable of targeting preleukemic HSCs. Therefore, it will be important to identify functional differences between preleukemic HSCs and wild-type HSCs that may provide actionable drug targets to selectively eradicate preleukemic HSCs and allow for more durable remissions and the eventual cure of AML.
Materials and Methods
Human Samples.
Human AML samples were obtained from patients at the Stanford Medical Center with informed consent, according to Institutional Review Board (IRB)-approved protocols (Stanford IRB no. 18329 and 6453). Mononuclear cells from each sample were isolated by Ficoll separation and cryopreserved in liquid nitrogen. All analyses conducted here used freshly thawed cells. Individual case information is presented in Table S2.
Animal Care.
Experiments were conducted under a Stanford University Institutional Animal Care and Use Committee-approved protocol and in adherence to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (26).
Flow Cytometry Analysis and Cell Sorting.
A panel of antibodies was used for analysis and sorting of HSCs from diagnosis and relapse AML samples as previously described (25). In addition to the antibodies used previously, the following antibodies were used: CD11b antibody clone ICRF44, CD14 antibody clone MφP9, CD56 antibody clone B159, CD235a antibody clone GA-R2 (all BD Pharmingen), and CD45 antibody clone J.33 (Beckman Coulter).
Exome Sequencing.
Exome sequencing was performed using the SeqCap EZ Exome SR kit, version 3.0, per the manufacturer’s instructions (Roche/Nimblegen) as described previously on an Illumina HiSEq. 2000 (22).
Targeted Amplicon Sequencing of Leukemia-Associated Mutations.
Targeted Amplicon Sequencing was performed as described previously (22). The variant allele frequency was defined as follows: (mutant read no.)/(germ-line read no. + mutant read no.). Read counts and primer pairs from all assays are available upon request. Each locus was sequenced to high depth (>500-fold coverage for over 99% of assays; 19,551 median fold coverage).
To determine the approximate threshold of detection for each PCR assay, control experiments were conducted on admixtures of gDNA from the given leukemia patient and from normal control DNA from unrelated individuals. Admixtures of 2%, 1%, and 0.5% were used. Assays that did not closely follow a linear increase in variant allele frequency in these assays were either redesigned or ignored (in the case of a small number of likely passenger mutations).
NSG Xenotransplantation Assay.
FACS-purified cells from each sample were transplanted into three newborn NSG mice conditioned with 100 rad of irradiation as described previously (27). After 12 wk, mice were euthanized and the bone marrow was analyzed for bilineage human engraftment (hCD45+, CD33+/CD19+).
Single HSC-Derived Colony Genotyping Assay.
Colony genotyping assays were performed as described previously (22).
Supplementary Material
Supporting Information
Acknowledgments
We acknowledge Feifei Zhao, Serena Tseng, Vivian Zhang, and Julie Koenig for laboratory management, and Max Jan and Dan Webster for review of the manuscript. We acknowledge the Hematology Division Tissue Bank and the patients for donating their samples. M.R.C.-Z. is supported by the Smith Fellowship, the National Science Foundation Graduate Research Fellowship Program, and the National Institutes of Health (NIH) F31 Predoctoral Fellowship. W.-J.H. is supported by the California Institute of Regenerative Medicine Stanford Training Program (TG2-01159) and the Stanford Training Program in Investigative Oncology (T32CA009287-35). R.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a New York Stem Cell Foundation Robertson Investigator. This research was supported by grants from the Stinehardt–Reed Foundation and Ludwig Foundation, and NIH Grant U01HL099999.
Footnotes
The authors declare no conflict of interest.
References
- 1.Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368(9550):1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341(14):1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 3.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan X-J, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43(4):309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 8.Nikoloski G, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 9.Ernst T, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 10.Abdel-Wahab O, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krönke J, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–108. doi: 10.1182/blood-2013-01-479188. [DOI] [PubMed] [Google Scholar]
- 14.Araten DJ, et al. A quantitative measurement of the human somatic mutation rate. Cancer Res. 2005;65(18):8111–8117. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- 15.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135–140. doi: 10.1038/onc.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissman IL. Stem cell research: Paths to cancer therapies and regenerative medicine. JAMA. 2005;294(11):1359–1366. doi: 10.1001/jama.294.11.1359. [DOI] [PubMed] [Google Scholar]
- 18.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97(13):7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamieson CHM, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 20.Abrahamsson AE, et al. Glycogen synthase kinase 3β missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA. 2009;106(10):3925–3929. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busque L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jan M, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi M, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jan M, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci USA. 2011;108(12):5009–5014. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee on Care and Use of Laboratory Animals (1996) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85–23.
- 27.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1(6):635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information