From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing (original) (raw)
Abstract
Single-cell RNA-seq mammalian transcriptome studies are at an early stage in uncovering cell-to-cell variation in gene expression, transcript processing and editing, and regulatory module activity. Despite great progress recently, substantial challenges remain, including discriminating biological variation from technical noise. Here we apply the SMART-seq single-cell RNA-seq protocol to study the reference lymphoblastoid cell line GM12878. By using spike-in quantification standards, we estimate the absolute number of RNA molecules per cell for each gene and find significant variation in total mRNA content: between 50,000 and 300,000 transcripts per cell. We directly measure technical stochasticity by a pool/split design and find that there are significant differences in expression between individual cells, over and above technical variation. Specific gene coexpression modules were preferentially expressed in subsets of individual cells, including one enriched for mRNA processing and splicing factors. We assess cell-to-cell variation in alternative splicing and allelic bias and report evidence of significant differences in splice site usage that exceed splice variation in the pool/split comparison. Finally, we show that transcriptomes from small pools of 30–100 cells approach the information content and reproducibility of contemporary RNA-seq from large amounts of input material. Together, our results define an experimental and computational path forward for analyzing gene expression in rare cell types and cell states.
Gene expression levels can differ widely between superficially similar cells. One source of variation is stochastic transcriptional “bursting” (Elowitz et al. 2002; Ozbudak et al. 2002; Blake et al. 2003; Raser and O'Shea 2005; Kaufmann and van Oudenaarden 2007). Those studies mainly used fluorescent protein fusion genes to monitor the expression of one or a few genes. They revealed dynamic fluctuations through time that are seen as “salt-and-pepper” variation across a cell population at any given time. In addition to this bursting behavior, individual cells are expected to display controlled and coordinated differences in the expression of genes engaged in dynamic physiologic processes, such as cell cycle phase progression, paracrine or autocrine signaling response, or stress response. Beyond such already appreciated heterogeneity lie currently unknown cell-to-cell differences with biological implications for defining cell states, metabolic function, and, in complex tissues, cell identity.
Measuring RNA transcripts in single cells is now done in multiple ways, and similar conclusions about variability are emerging from the higher sensitivity methods. For individual genes, single molecule RNA fluorescence in situ hybridization (SM-RNA FISH) is highly informative (Femino et al. 1998; Raj et al. 2008), and multiplexed versions now enable multiple genes to be measured in parallel (Lubeck and Cai 2012). In principle, an advantage of SM-RNA FISH is the ability to accurately count the absolute number of transcripts in a cell. A second and older approach is multiplexed single-cell RT-qPCR (Cornelison and Wold 1997), which has now been advanced to increasingly high-throughput formats (White et al. 2011; Sanchez-Freire et al. 2012, Livak et al. 2013). It produces semiquantitative relative comparisons between individual cells. However, neither SM-RNA FISH nor the current forms of multiplex RT-qPCR cover the entire transcriptome or have the single-nucleotide resolution needed to study fine-structure features of gene expression such as allele specificity, RNA editing, and alternative splicing.
To address these and other limitations, elegant methods have recently been developed for performing RNA-seq with very small amounts of RNA, down to the level of individual cells. These are broadly referred to as “single-cell RNA-seq” (Tang et al. 2009, 2010, 2011; Ozsolak et al. 2010; Islam et al. 2011; Brouilette et al. 2012; Cann et al. 2012; Hashimshony et al. 2012; Pan et al. 2012; Qiu et al. 2012; Ramsköld et al. 2012). Despite these significant advances, there are substantial shortcomings in these methods, and a robust method for comprehensive and accurate measurement of the transcriptome of a single cell is not yet available.
A particular challenge for single-cell methods is the efficiency and uniformity with which each mRNA is copied into cDNA and ultimately represented in the library. This challenge intersects in crucial ways with transcriptome structure. Specifically, thousands of genes are expressed in the range of 1 to 30 mRNA copies per cell, including many essential mRNAs (for example, key transcription factors) (Zenklusen et al. 2008). Even lower transcript levels, averaging <1 mRNA per cell on the population level, are now being reliably detected by RNA-seq. This raises questions whether very rare RNAs represent background biological noise, or alternatively, are functional in only a small fraction of cells. Single-cell RNA-seq has the potential to address these issues, but their resolution depends on how faithfully and efficiently RNAs are captured and represented in sequencing libraries (referred to throughout as the “single-molecule capture efficiency,” psmc). In addition, the uniformity of transcript coverage in early single-cell RNA-seq protocols has typically been heavily biased toward the 3′ end, which affects both gene expression estimates and the ability to analyze alternative splicing, RNA editing, and allelic bias.
A second major use for single-cell RNA-seq is the transcriptomic characterization of rare cells. The human body consists of hundreds of distinct cell types, plus large numbers of neuronal and transient developmental cell types. Many of these are numerically minor components of complex tissues, making them inaccessible to standard methods relying on large RNA inputs. Isolation of single cells based on the cell surface markers or using microdissection coupled with single-cell RNA-seq could fill this gap in complex multicellular organisms. However, the feasibility of this approach also depends on the experimental robustness of single-cell RNA-seq protocols. Alternatively, single-cell resolution may not be absolutely required for this purpose, and small pools of cells may be sufficient to characterize rare cell-type transcriptomes. An open unresolved question is how small such pools can be to adequately meet that goal.
In this study, we address the issues highlighted above. We used the SMART-seq protocol (Ramsköld et al. 2012) to measure the transcriptome of single cells and small cell pools from the GM12878 lymphoblastoid cell line. This line is derived from the NA12878 individual, for which a fully sequenced genome with completely phased heterozygous single nucleotide polymorphisms (SNPs) and indels is available (The 1000 Genomes Project Consortium 2012). GM12878 cells have also been the subject of an extensive functional genomic characterization by the ENCODE Consortium (The ENCODE Project Consortium 2011, 2012) and have been used in prior population-level studies of allele-biased gene expression and transcription factor occupancy (Rozowsky et al. 2011; Reddy et al. 2012).
Using spike-in quantification standards of known abundance (Mortazavi et al. 2008), we derive estimates for the absolute number of transcript copies for each gene in each cell and directly measure the average value of psmc. “Pool/split” experiments (consisting of pooling RNA from multiple single cells, splitting the pool into the same number of separate reactions and building libraries from them) allowed us to measure the extent of and control for technical variation. We find that the psmc value is quite low: ∼0.1. An analysis framework accounting for technical stochasticity is described and used to assess variability in gene expression, allelic bias, and alternative splicing between single cells. Distinct from prior studies, our approach allowed us to parse findings into those that are just as likely to be of technical origins and those that are more likely to be of biological interest.
We report evidence of significant variability in the total number of mRNA molecules per cell, and identify biologically coherent modules of coexpressed genes specifically expressed in individual cells or groups of cells. These include expected variation associated with cell cycle phases, and an unexpected module enriched for mRNA processing and splicing genes. We observe evidence of higher levels of autosomal allelic exclusion on the single-cell level, potentially associated with transcription bursts; however, it is at present difficult to confidently distinguish from technical variability. In contrast, we find much stronger evidence for widespread major splice site usage switches between individual cells. Finally, our analysis of similarly constructed small cell pools (30–100 cells) reveals a high robustness and reproducibility, approaching that of bulk RNA measurements. This presents a reliable path forward toward the future comprehensive transcriptomic characterization of rare cell types.
Results
In silico examination of major variables affecting informativeness of single-cell and small cell-pool RNA-seq
We began this study with two goals: first, to study gene expression heterogeneity in GM12878 cells on the single-cell level, and second, to determine the minimal optimal size of a cell pool that is informative of the characteristics of the larger cell population, with the goal of applying that approach to rare cell types in future studies. How well these goals are achieved depends on several parameters affecting biological and technical stochasticity and detection sensitivity, the values of which were unknown. To understand their influence, we carried out a simulation of single-cell and cell-pool transcriptomes (see Supplemental Methods for details) by varying the following parameters:
- Single-molecule capture efficiency psmc. In contrast to bulk RNA-seq libraries, an individual cell contains a very limited total number of mRNA molecules. Individual genes can be present in single-digit transcript numbers. If only a fraction of mRNAs are successfully represented in a library, a technical stochasticity component is introduced. Depending on its magnitude, data interpretability can be significantly affected due to false negatives and a distortion of relative gene abundance estimates. The psmc parameter is the probability that any given original RNA molecule is captured in the final library. We examined the effect on expression quantification of psmc ranging from 0.01 to 1.
- Total number of mRNA molecules per cell. The impact of low psmc on expression measurements will be more severe if fewer mRNA molecules are present in a cell. The average total number of mRNA molecules in a single cell is not known for most cell types, but it is expected to vary with cell size, metabolic status, and even cell cycle phase. This means that single-cell expression measurements in some cell types are likely to be more robust to technical noise than in others. We varied the total number of mRNAs from 50,000 to 1,000,000 (while keeping the number of genes expressed constant).
- Frequency of expression of individual genes in single cells. From prior studies we expect that some genes will be expressed in all or most cells, while others will be expressed in only a subset of cells. Genes detected at lower levels in bulk RNA-seq are the most obvious candidates to be expressed in a subset of cells in a population, although we do not know what fraction of low-abundance RNAs behave in such a way. This is particularly relevant to cell pools: a gene expressed at 50 copies per cell but only in 10% of cells would still be stochastically represented in a pool of 10 cells even if psmc is high. In the absence of reliable data on this, we modeled the probability of expression in a given single cell with a distribution centered around very high values for genes highly expressed in bulk RNA-seq measurements, and progressively lower values with decreasing expression levels (details in Supplemental Methods).
The simulation results are summarized in Figure 1, A–C and Supplemental Figures 1–25. As expected, low psmc has a profoundly negative impact on gene expression quantification accuracy and reliability, leading to frequent false negatives (Fig. 1A; Supplemental Fig. 1), and to poor estimates of expression levels. For example, in a single cell with 100,000 mRNAs, psmc = 0.1 results in only 40% of genes expressed at 100 FPKM receiving FPKMs within 20% of the true value (Supplemental Fig. 1C), but this fraction rises to nearly 100% if psmc = 0.8 (Supplemental Fig. 1G). The quantification of relative expression levels is similarly affected, with only the most highly expressed genes being consistently well-quantified relative to each other at low psmc (Supplemental Figs. 12–25).
Figure 1.

Simulated and measured transcriptome profiles from individual cells and small cell pools. (A) Number of detected genes in simulated data sets as a function of the number of cells pooled and the single molecule capture efficiency (psmc) (assuming 100,000 mRNA molecules per cell). See Supplemental Figure 1 for full details. (B,C) Accuracy of gene expression estimation as a function of the number of cells pooled and the single molecule capture efficiency; psmc = 0.1 in B and psmc = 0.8 in C, 100,000 mRNA molecules per cell assumed. Shown is the fraction of genes at the indicated expression levels in FPKM, whose estimated expression level in FPKM in simulated libraries was within 20% of their true value, after modeling the stochasticity due to the single-molecule capture efficiency of the library-building protocol. See the Methods section and Supplemental Figures 2–11 for full details. Note that the simulation is intended to illuminate the relative effects of the various parameters studied, and the absolute numbers of genes should not be directly compared to the real-life data shown in G. (D) Experimental design. Single cells are combined with spike-in quantification standards and SMART-seq libraries are generated. In parallel, multiple single cells are pooled together and combined with spikes, then lysed and split into the same number of reactions and converted into SMART-seq libraries. Libraries are then sequenced, data processed computationally, and estimates for the absolute number of copies per cell are derived based on the spikes. Variation in pool/split experiments is due to technical stochasticity, while variation in single-cell libraries is a combination of biological variation and technical noise. (E) Uniformity of transcript coverage. Shown is the average coverage along the length of an mRNA for single cells and pool/split experiments. Only mRNAs longer than 1 kb from genes with a single annotated isoform in the RefSeq annotation set were included. See Supplemental Figure 29 for more details. (F) Number of detected protein-coding genes for libraries built from 10 ng and 100 pg of poly(A) RNA, pools of 100, 30, and 10 cells, representative pool/split experiments (individually and summed across all libraries), and representative single cells (individually and summed across all libraries). (G) Fraction of genes from 100-ng bulk poly(A)+ RNA libraries that were detected in pools of 100, 30, or 10 cells, 100 pg of poly(A)+ RNA, pools/split experiments, and single cells. FPKM is shown on the _x_-axis.
In contrast, our simulation results indicate that cell pools are much more robust to technical noise, with 90% of genes expressed at 10 FPKM receiving FPKM estimates within 20% of their true value (Supplemental Fig. 1C) at psmc = 0.1 in a pool of 100 cells. They also represent the expression profiles of the general population reasonably well (Supplemental Fig. 1), even at low psmc, starting from a size of ∼30 cells (10-cell pools seem not to be sufficient to achieve this). Finally, as expected, the larger the number of total mRNA molecules per cell, the greater is the buffer against technical noise, resulting in more robust quantification (Supplemental Figs. 2–11).
Transcriptome measurements of individual single cells and companion pool/splits
The simulation results informed our experimental design, which aimed to gain a firm grasp on technical stochasticity in two ways (Fig. 1D). First, we generated single-cell RNA-seq libraries and in parallel carried out “pool/split” experiments. In a pool/split, multiple cells are pooled and lysed together, then split into the same number of reactions, from which libraries are built. Variation between these libraries should be purely technical (with stochastic splitting possibly playing a role at the low end). Variation observed at similar levels in both single cells and pool/splits cannot be confidently attributed to biological differences, although the stringency of this criterion may cause some true biological variation to be obscured. However, variation above the pool/split level can be identified and ascribed to biological sources with high confidence.
We generated single-cell RNA-seq libraries from 15 single GM12878 cells and from two pairs of 10-cell pool/split experiments. We also sequenced replicates of pools of multiple cells (10, 30, and 100 cells), as well as 100-pg and 10-ng samples of bulk RNA (corresponding to ∼10 and ∼1000 cells), to assess the stability of measurements as a function of the amount of starting material.
We used the SMART-seq protocol (Supplemental Fig. 12; Ramsköld et al. 2012) to generate our libraries. A detailed description of the protocol, as we implemented it, is presented in Supplemental Methods. We obtained nearly uniform full-length transcript coverage (Fig. 1E; Supplemental Fig. 29). Uniformity of coverage, which depends on the intactness of RNAs and the successful copying of full-length molecules, is highly desirable for several reasons. First, RNA-seq data quantification using the RPKM/FPKM metric (Mortazavi et al. 2008; Trapnell et al. 2010) makes an implicit assumption of full coverage. Second, it enables the analysis of alternative splicing and allelic bias, as read coverage of 5′-proximal splice sites and heterozygous positions is ensured.
We added spike-in quantification standards of known abundance (in absolute number of RNA copies) (Supplemental Table 2) at the very beginning of cDNA synthesis. This allows us to, first, estimate psmc, and second, derive gene expression estimates in absolute numbers of copies per cell. The latter is important because while FPKM is useful for comparing expression levels within a library, it can only be used to compare directly across different libraries when the total amount of RNA in each starting sample is roughly the same (Anders and Huber 2010). This assumption is usually only mildly violated when working with bulk samples, but when single cells are compared, it becomes significantly more problematic as the variation in the total amount of RNA in each cell is expected to be much larger.
Figures 1 and 2 summarize the technical characterization of the SMART-seq protocol applied to GM12878 cells. In addition to the mostly complete coverage along transcript length, sequencing libraries were also highly enriched for exonic sequences (Supplemental Fig. 28), indicating a high efficiency of enrichment for polyadenylated molecules.
Figure 2.

Technical and biological variation in single-cell RNA-seq measurements of gene expression. (A) Correlation between expression levels (in FPKM) between two pools of 100 cells. (B) Correlation between expression levels (in FPKM) between two pools of 10 cells. (C) Correlation between expression levels (in FPKM) between two representative pool/split libraries. A pseudocount of 0.001 was added to each data point in the scatter plots for visualization purposes. (D,E) Hierarchical clustering of estimated copies-per-cell values for protein-coding genes in single-cell (D) and pool/split (E) libraries. Pearson correlation was used as a distance metric, and only genes expressed at a level of at least one estimated copy in at least one library were included. (F,G) Correlation between estimated copies-per-cell values for protein-coding genes in single-cell libraries (F) and pool/split libraries (G). Two sets of pool/split experiments (1 and 2) are shown and “1-2” in the boxplot refers to correlations between the two sets, while “1” and “2” refer to correlation within each experiment. Similar plots, but using the Spearman correlation, are shown in Supplemental Figure 32.
Gene detection in single cells versus pools of varied sizes
We compared single-cell and pool/split libraries, as well as cell pools, with bulk RNA samples from GM12878 cells (Fig. 1F). In bulk RNA libraries, we detect about 12,000 genes expressed at more than 0.1 FPKM. A lower number of genes, between 4000 and 5000, is detected in both single-cell and pool/split libraries. These differences between single cells and bulk libraries are due mostly to genes expressed at low levels. Genes expressed at more than 100 FPKM in 10-ng bulk RNA samples are detected in almost all libraries, while only ∼30% of genes expressed at ∼10 FPKM and 10% of genes expressed at ∼1 FPKM were detected in any given single-cell library (Fig. 1G). Notably, the number of genes detected in both 100-cell and 30-cell pools was similar to that detected in the 10-ng libraries (∼11,000). In contrast, in the 10-cell pools and 100-pg libraries, lower numbers of genes were detected, between 6000 and 7000. This is consistent with simulation results suggesting that 30 cells is the lower limit of cell number at which the transcriptome library complexity begins to approach that of the larger cell population. This is corroborated by the correlation between the expression levels of replicate measurements (Fig. 2A; Supplemental Fig. 50). In contrast, a sizable population of genes present at high levels in one replicate and at very low levels or completely absent in the other appears in 10-cell pools (Fig. 2B) and especially in pool/split libraries (Fig. 2C). Finally, union sets of genes detected in all individual cell libraries and in all pool/split libraries was ∼10,000, which was in the range seen for 30- to 100-cell pools.
Pool/splits measure technical variation and reveal biological variation among single cells
The observed variations in gene expression levels and detection can be explained as a combination of some genes not being expressed in each and every cell and low psmc resulting in large numbers of false negatives. We calculated the average psmc across all libraries based on the detection of spike-ins (details in Methods). This number is in our estimates: ∼0.1. We also estimate that for GM12878 single cells, one transcript copy corresponds to ∼10 FPKM on average. This agrees well with the observation that detection of genes becomes unstable below ∼100 FPKM (Fig. 2B,C), which is also consistent with previous observations (Ramsköld et al. 2012).
We next compared expression measurements in single-cell and pool/split libraries. Hierarchical clustering of each group is shown in Figure 2, D and E (with two independent biological replicate pool/split experiments shown in Fig. 2E). The distances between the expression profiles within the same pool/split experiment were significantly smaller than those for individual single cells (branch lengths in Fig. 2D,E), and average correlations between single cells were, accordingly, lower than those between libraries from the same pool/split (Fig. 2F,G; Supplemental Fig. 32). A notable feature of the data is small clusters of genes present at high levels in only one library. These are more prominent in single cells than in pool/splits, yet they are clearly present in all samples. In single cells, this is due to a mixture of stochastic capture effects and real biological variation. In pool/splits, stochastic capture is the predominant source. It is important to note that given the low psmc, it is difficult to determine the cause of variation for any given gene. Nevertheless, the major conclusion at the transcriptome level is that there are biological differences between single cells because the technical stochasticity in pool/splits is significantly less than variation across single cells.
Estimating absolute transcript levels in single cells
Absolute transcript counts are the biologically relevant values ideally obtained from a single-cell gene expression profiling experiment because, as discussed above, FPKM is a poor metric for comparing gene expression levels between individual cells if the total amount of RNA varies substantially. We derive transcript number estimates for each gene based on the FPKM values of spike-ins. We observed good agreement between the input number of spike-in RNA copies and the corresponding FPKM values in the final libraries (Supplemental Figs. 30, 31).
We use the transcripts-per-cell estimates for all subsequent analyses. Previous studies have reported that genes can be separated into two distinct groups based on their expression levels—one group expressed at high (>1 FPKM) levels and one at very low (<<1 FPKM) levels (Hebenstreit et al. 2011). We examined the distribution of estimated copies per cell in single cells and in pool/splits (Fig. 3A). We find that in individual cells, most protein-coding genes are expressed at levels between 1 and ∼50 copies per cell. The distribution suggests a roughly equal number of genes at each level except for a larger group of transcripts with fractional transcript-per-cell values. Obviously, single-cell determinations are constrained in a way that population level measurements cannot be: One transcript per cell is the minimum nonzero value possible. The lower values likely represent a combination of mapping artifacts (due to high sequence homology of paralogs) and RNAs that were present at low levels to begin with and then poorly represented in the final library (due, for example, to the fragmentation of a single original RNA molecule resulting in artificially low FPKMs as a result of coverage only at the 3′ end). The distribution of estimated copies in pool/split libraries exhibited a more linear decrease in the number of more highly expressed genes, consistent with averaging of variation between cells.
Figure 3.

Absolute expression levels at the single-cell level. FPKM values converted to estimated copies per cell using the spike-in quantification standards are shown. (A) Distribution of expression levels of RefSeq protein-coding genes in estimated copies per cell in single cells and pool/split experiments. (B) Distribution of expression levels of GENCODE v13 lncRNA protein-coding genes in estimated copies per cell in single cells and pool/split experiments. (C) Total number of mRNA copies per cell in single cells. (D) Total number of mRNA copies in pool/split experiments. (E) Expression levels of housekeeping and highly expressed genes (GAPDH, CD74, left panel), and general (CTCF, REST, YY1) and B-cell regulatory (PAX5, EBF1, BCL11A, ETS1, IRF4, IKZF1, PBX3, POU2F2, RUNX3, TCF3, TCF12) transcription factors (right panel). Upper and middle panels show the estimated copies-per-cell numbers for single cells and pool/splits, respectively. The lower panel shows FPKM values for cell pools and bulk RNA libraries. (F_–_H) Distribution of absolute expression levels in copies per cell in single cells for translation initiation, elongation, and termination proteins (F), splicing regulators (G), and transcription factors (H). The list of translation proteins was retrieved from the corresponding GO category annotations downloaded from FuncAssociate 2.0 (Berriz et al. 2009). The list of splicing regulators was obtained from the SpliceAid-F database of human splicing factors (Giulietti et al. 2013). The list of transcription factors used was the one from Vaquerizas et al. (2009). Note that only values ≥0.1 estimated copies per cell were included in these plots, i.e., libraries in which the genes were not detected were excluded.
We also examined the distribution of the expression levels of long noncoding RNAs (lncRNAs) (Guttman et al. 2009). Consistent with previous observations (Ramsköld et al. 2009; Guttman et al. 2010; Djebali et al. 2012), lncRNAs have generally much lower expression levels compared to protein-coding genes (Fig. 3B).
We were also able to directly assess the total number of mRNAs present in each cell (Fig. 3C,D). Based on the average mass of RNA in each cell (derived from bulk RNA samples from a known number of cells) and the average length of mRNAs in the human genome, we estimated that each GM12878 cell contains, on average, ∼80,000 mRNAs. However, we observed striking cell-to-cell differences in the total transcript number of single cells, with some cells expressing <50,000 mRNAs and others almost 300,000. In contrast, pool/split experiments exhibited remarkable uniformity (between 50,000 and 100,000 transcripts) and agree well with prior expectations. It is therefore unlikely that the observed cell-to-cell variability is due to technical noise.
Because transcriptional regulators play a crucial role in defining the gene expression state of cells, we examined the expression of several well-known general transcription factors as well as major regulators of B-cell differentiation (Fig. 3E). Remarkably, except for IRF4, which was usually expressed at several dozen copies, most factors were detected at <10 copies per cell, and were often not detected at all. We stress that this does not mean that they are not expressed. Given the 10% psmc of the protocol, these observations are consistent with simple technical failure to detect them. It is also possible that there are no mRNA copies in some cells at the moment of harvest, especially if they are infrequently transcribed. Extending these observations to other functional groups, we assessed proteins involved in translation (as a major group of genes with housekeeping functions) (Fig. 3F), splicing regulators (Fig. 3G), and all transcription factors (Fig. 3H). The median number of copies per cell was ∼100 for translation proteins, ∼10 for splicing regulators, and strikingly, only ∼3 for transcription factors. Beyond their biological interest, these large expression differences between functional gene categories mean that quantification is inherently less robust and less informative for some biological functions than it is for others.
Identification of modules of coexpressed genes
Cell-to-cell gene expression variability may occur on the level of individual genes, but it can also occur in a coordinated fashion. A well-studied example is cell cycle phase-specific gene expression. In an asynchronous culture, groups of genes expressed highly at specific times during the cell cycle should be present in a fraction of cells that is roughly proportional to the time cells spend in each identified phase. Population data do not, however, predict that most cells will be in a “pure” phase state nor that they will express phase-class genes at peak levels.
To test whether we are able to identify cell cycle-associated variation, and to search for any novel functional modules, we carried out weighted gene coexpression network analysis (WCGNA) (Zhang and Horvath 2005) using the copies per cell estimates for single cells and removing genes that were highly variant in pool/split libraries in order to minimize technical noise (see Methods; Supplemental Figs. 33, 34). We identified 19 coexpression modules containing ≥10 genes each (Supplemental Fig. 35). The expression patterns of these modules were mostly well-differentiated among single cells and were absent from pool/split libraries (Fig. 4B; Supplemental Fig. 34).
Figure 4.

Gene coexpression modules derived from single GM12878 cells. Weighted gene correlation networks were constructed using the WCGNA R package (Langfelder and Horvath 2008). (A) Expression levels and hierarchical clustering of genes within modules (modules are sorted by number, which corresponds to their size) in single cells and pool/split experiments. Only genes are clustered (dendrograms on the left), and the identity of the cells and pool/split experiments is the same in each column (two right panels). The absolute expression values of genes belonging to representative GO categories associated with cell cycle phases (modules 1 and 6) and mRNA processing and splicing (module 2) are also shown. (B) Distribution of cell cycle states in a representative GM12878 cell population, in growth media (GM), and picking media (PM). The fraction of cells in M phase is consistent with one such cell being picked in a sample of 15.
We then determined the Gene Ontology (GO) category enrichment of each module. The largest module (module 1) was highly enriched for GO categories relating to housekeeping and anabolic gene functions (Table 1; Supplemental Table 3). This included some enrichment for the G1- and S-phase GO terms, and also contained most genes that are generally highly expressed (Fig. 4A). Module 6 was enriched for genes involved in the M phase of the cell cycle. A single cell from the sample of 15 showed strong coordinated expression of genes from the M-phase GO categories enriched in this module. Transcripts from these M-phase genes were not similarly coordinated in other individual cells or in pool/split samples. We measured the fraction of unsynchronized GM12878 cells in the G0 + G1, S, and M phases of the cell cycle using flow cytometry (Fig. 4B). About 14% of cells were in M phase, and the probability of capturing exactly one such cell out of 15 is 0.25; that is, these observations are consistent with this cell being in the peak of M phase.
Table 1.
Representative Gene Ontology categories enriched in coexpressed gene modules
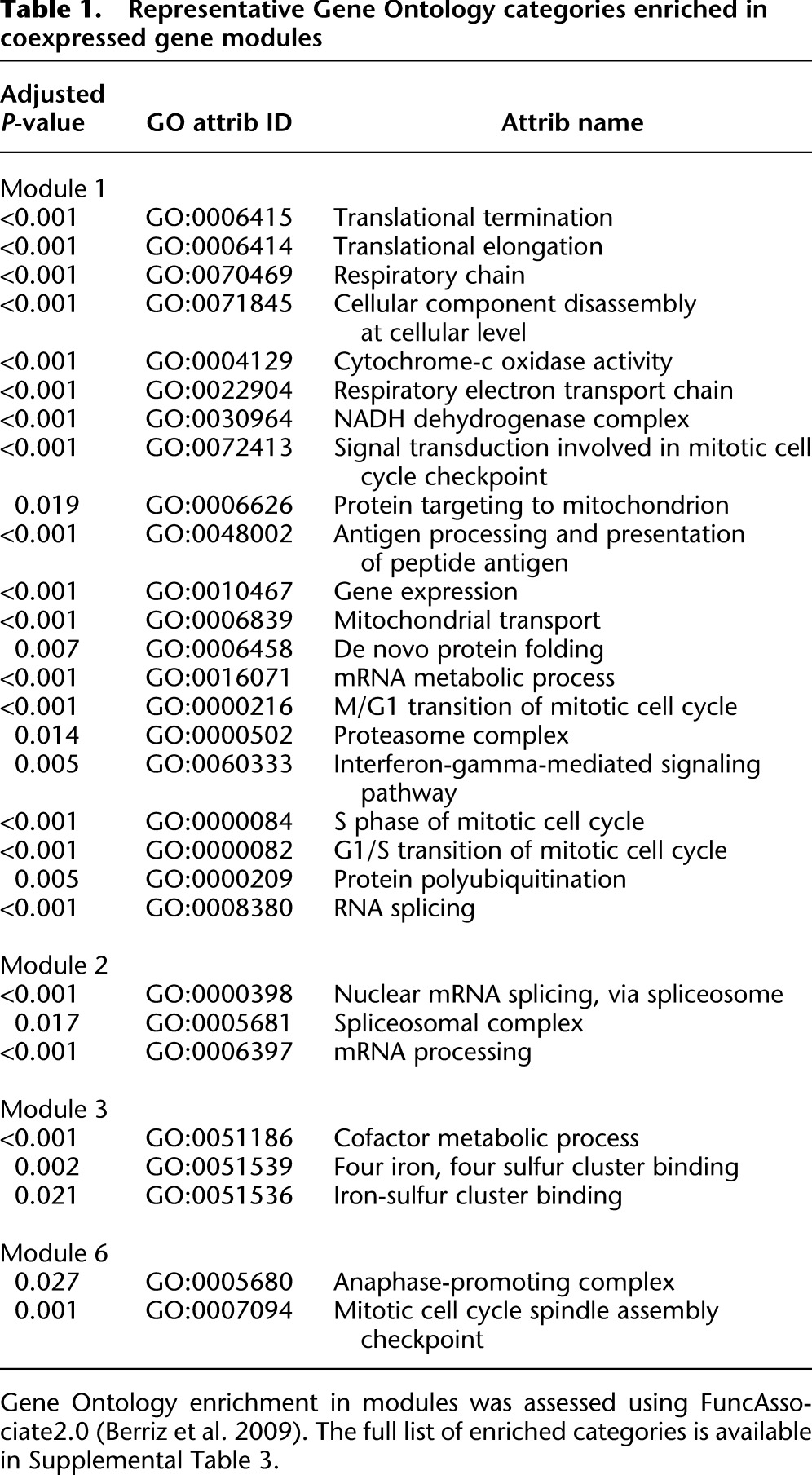
A more surprising observation was that the second largest module (module 2) was enriched for genes involved in splicing and mRNA processing. It is driven by an individual cell and two additional cells with a somewhat similar expression profile. The signature cell, however, was not an outlier when splice site usage patterns were compared between individual cells (data not shown). A simple interpretation of these observations is a general up-regulation of splicing and mRNA processing factors in that cell that does not result in a distinctive alternative splicing program.
Module 3 was enriched for metabolic cofactor and iron-sulfur cluster binding proteins, including proteins involved in mitochondrial respiratory chains. This is an intriguing observation, as module 3 was mostly driven by the two cells exhibiting the highest total number of mRNA molecules per cell (Fig. 3C; fourth and fifth columns in clustergram in Fig. 4A), consistent with a generally elevated metabolic state.
We also carried out a mirrored WCGNA analysis in which the pool/splits were treated as single cells and vice versa. We did not observe significant GO enrichment beyond a few trivial terms in the largest modules (Supplemental Fig. 54; Supplemental Table 4). This is in contrast to the much more specific GO enrichment seen in single cells.
In addition to the coexpression analysis, we also examined the relationship between the expression variability of genes and various genomic data about their promoters, including long-range chromatin interactions, DNA methylation status, histone marks, transcription start site sequence elements, and CpG islands. No robust explanatory correlation was evident (Supplemental Figs. 46–-50), and we expect that data with less technical stochasticity will be needed to illuminate relationships of this kind.
Allele-biased expression at the single-cell level
Allele-specific gene expression (either monoallelic or highly biased toward one autosomal allele) has been previously reported to be widespread (Gimelbrant et al. 2007; Zhang and Borevitz 2009; McManus et al. 2010; Pickrell et al. 2010; Rozowsky et al. 2011; Reddy et al. 2012). An intriguing phenomenon observed for hundreds of genes in clonal lymphoblastoid cell lines (Gimelbrant et al. 2007; Chess 2012) is the random monoallelic expression of autosomal genes. However, those studies were conducted on large pools of cells, producing a snapshot of average allelic bias in the population, and leaving open the possibility that monoallelic expression is even more widespread on the single-cell level.
GM12878 cells are a good system for addressing this issue, as the fully phased heterozygous NA12878 genome sequence is available (The 1000 Genomes Project Consortium 2012). We aligned RNA-seq reads in an allele-specific manner to the heterozygous GM12878 transcriptome and calculated allelic bias for each gene as the fraction of reads mapping to the maternal allele. As detailed in the Methods and Supplemental Methods, we applied very stringent criteria for determining statistically significant allele-biased expression events based on the absolute transcript number estimates and taking into account the challenges presented by the nature of single-cell RNA-seq data. We observed good reproducibility of allelic bias profiles in 10-ng bulk RNA libraries (Supplemental Fig. 37A), with most genes being expressed from both alleles (Supplemental Fig. 37D). Allelic bias was also highly reproducible in 30-cell and 100-cell pools (Supplemental Fig. 51). In contrast, allelic bias profiles of single cells correlated poorly with each other, and a large fraction of genes were apparently monoallelically expressed from different alleles in different cells (Supplemental Fig. 37B). The majority of highly expressed genes (≥100 copies per cell) exhibited biallelic expression, while most genes at low expression levels were measured as monoallelically expressed (Supplemental Fig. 37F). We then compared allelic bias variability for individual genes across individual single cells, focusing only on cells in which statistically significant allelic bias was observed, and observed frequent “switching” between the two alleles (Supplemental Figs. 37G, 38A).
These observations can be explained as a combination of biological and technical factors. First, it has been previously reported that allelic bias at the population level is more common among genes expressed at low levels (Gimelbrant et al. 2007, Reddy et al. 2012). A second explanation is the phenomenon of “transcriptional bursting” (Raj and van Oudenaarden 2008; Dar et al. 2012). A single transcription burst produces several mRNA molecules from a single allele. If all mRNAs from a gene in a given cell at a given moment are the product of one or a linked series of such bursts, all copies would originate from one allele. Finally, stochastic effects due to the low single-molecule capture efficiency of the protocol undoubtedly play a role. The fewer founder molecules are captured, the more likely it is that they come from only one allele. To help parse these sources of variation, we performed the same analyses on pool/split libraries and observed a broadly similar (although always lower) fraction of genes passing all significance tests for allelic bias (Supplemental Figs. 37C,E, 38). The quantitative trend within the pool/split comparison suggests there is a component of allelic RNA bias between cells that is biological in origin but that there is also a large technical variation component. The widespread occurrence of random monoallelic expression at the single-cell level should therefore be viewed as a provisional conclusion.
Alternative splicing at the single-cell level
Previous studies have suggested that most genes in mammalian genomes undergo some alternative splicing (Mortazavi et al. 2008; Wang et al. 2008; Djebali et al. 2012). At present, however, the biological relevance of the majority of these alternative isoforms is still uncertain, and stochastic noise in the splicing machinery is one explanation (Sorek et al. 2004; Melamud and Moult 2009). Characterizing alternative splicing at the single-cell level should bring clarity to the population-based observations, and perhaps offer clues about the mechanistic origin of the multiple isoforms observed within cell types.
We quantified alternative splicing using the intron-centric splice inclusion ψ score approach (Pervouchine et al. 2013). Details of our mapping and analysis pipeline are described in the Supplemental Methods. For reasons given there, we adopted a conservative approach and only analyzed novel splice junctions for which at least one of the donor or acceptor sites has already been annotated in GENCODE v13 (Harrow et al. 2012), thus avoiding library-building artifacts.
We detected between 200 and 2000 novel splice junctions satisfying these criteria in each individual cell (Supplemental Fig. 43). This number is certainly an underestimate, given the low psmc. About 35% of novel junctions connected two annotated exons (Fig. 5A); most of these represent novel exon skipping events. In another 60%, the unannotated donor or acceptor site was internal to the gene. These were concentrated close to already annotated splice sites (Supplemental Fig. 40B,C). In particular, novel acceptor sites peaked at the +3 and −3 position downstream from annotated sites representing mostly instances of NAGNAG tandem acceptor sites (Hiller et al. 2004; Bradley et al. 2012). Novel 5′ donor sites were fewer in number and peaked at +4 and −4 positions relative to annotated donor sites, thus shifting the coding frame of the transcript. This is a phenomenon we have previously also observed in bulk RNA-seq data (observations of the present study's authors), the significance of which is at present not clear. The proportions observed were independent of the read coverage and estimated number of copies per cell thresholds applied (Supplemental Fig. 40A).
Figure 5.

Alternative splicing at the single-cell level. (A) Classification of new junctions connecting known splice sites. (B) Frequency of detection of novel splice junctions. Novel junctions for which neither the donor nor acceptor site has been annotated were excluded for reasons described in the main text in both A and B. A threshold of 10 estimated copies and a coverage of 10 reads was applied, but results are essentially the same, independent of the thresholds used (Supplemental Fig. 40A). (C) Distribution of ψ scores in bulk RNA samples for annotated and novel splice junctions. A threshold of 15 reads combined for all splice junctions in which a donor or acceptor site participates was applied. Note that for each ψ1 score there is at least one matching ψ2 ≤ 1 − ψ1 score corresponding to the other alternative junction; in some cases, more than two alternative donor or acceptor sites exist; thus the relative height of the 0 ≤ ψ ≤ 0.1 bar. (D, upper and lower). Distribution of 5′ ψ scores for annotated splice junctions at two different detection thresholds in single-cell libraries (see Supplemental Fig. 41 for more detail). (E, upper and lower) Distribution of 5′ ψ scores for novel splice junctions at two different detection thresholds in single-cell libraries (see Supplemental Fig. 42 for more detail). (F,G) Frequency of major splice site usage switches between individual cells (F) and individual libraries in a pool/split experiment (G). Note the strong support for major splice site use switching across the collection of single cells.
We also examined the distribution of unannotated splices across individual single cells and found that the majority were detected in only a single cell, with <10% found in two cells, and very few in three or more cells (Fig. 5B). While this result could be greatly affected by psmc issues, it was independent of the read and estimated transcript copies threshold used (Supplemental Fig. 40), suggesting that most novel splices are indeed only present in a small fraction of cells.
We asked how often multiple alternative splice sites are used at individual single cells. In bulk RNA-seq at a threshold of 15 distinct read fragments, a numeric minority of ψ scores was equal to 1 (i.e., exclusive use of only one donor-acceptor pair). The presence of alternative splice sites is thus widespread in the cell population. Nevertheless, in most cases, ψ was close to 1, suggesting quantitative dominance of one isoform. The vast majority of novel splices received very low inclusion scores (Fig. 5C) and would generally be considered to be the result of biological noise in the splicing system. In contrast, in single cells, one dominant splice site was the norm for annotated junctions, except for very highly expressed genes (≥100 copies per cell), for which a wide diversity of splice site usage was seen (Fig. 5D; details in Supplemental Fig. 42). As this observation was true even for genes expressed at ≥50 copies per cell, we believe it is not a psmc artifact. It is an interesting and open question why very highly expressed genes (enriched for genes with housekeeping function) exhibit very high levels of alternative splicing in single cells. These results differ significantly from the same analysis carried out on novel splice junctions (Fig. 5E; Supplemental Fig. 43). Somewhat surprisingly, we found that a significant proportion of novel splices had ψ scores of 1 in single cells. This was true, however, only for genes expressed at lower levels (≤50 copies), where psmc artifacts are a likely cause. In contrast, in highly expressed genes, no novel junctions received a dominant (≥0.5) ψ score. However, the scores were still consistently higher than what is observed for novel splices in bulk RNA samples.
Finally, we evaluated the consistency of splice site usage between individual cells. We applied a statistical framework similar to the one used to analyze allelic bias and derived a list of dominant splice junctions in each cell, taking into account the estimated absolute number of copies and the stochastic capture effects. We asked how often the dominant splice site changes between different cells. We found 282 such genes in single cells, suggesting the phenomenon may be widespread. The genes involved were enriched for ribosomal and translation proteins, and also, intriguingly, for proteins involved in RNA splicing and processing (Supplemental Table 6). We tested this single-cell variation against pool/split experiments, in which we found very few genes with different dominant splice sites across libraries. (Fig. 5F,G; Supplemental Fig. 44). This argues that much alternative splicing variation is in fact due to biological differences between cells, and is in agreement with the bimodality of splicing in individual mouse immune cells reported recently (Shalek et al. 2013).
Discussion
Two major goals for single-cell RNA-seq are to obtain high-resolution transcriptomes for rare cell types or states and to measure the differences in RNA expression and processing between individual cells. Here, we showed that the first goal can be achieved by studying 30- to 100-cell pool samples even in the absence of perfect capture of each and every individual RNA molecule. Our conclusion is consistent with independent 80-cell measurements (Ramsköld et al. 2012). The pools reproduce the expression profiles (Supplemental Fig. 53) and allelic-bias patterns (Supplemental Fig. 51) of the larger population, and similar numbers of genes and splice junctions are detected in them (Supplemental Figs. 52, 53). The approach is applicable to cells collected by laser-capture (to be presented elsewhere), micromanipulation (used here), or cell sorting based on molecular markers or reporter-gene expression. This defines a general and relatively economical path forward for the transcriptomic characterization of many previously inaccessible rare cell types and states, including transient cell types in embryonic development, diverse neuronal types in the brain, and cells in tumors.
In agreement with previous single-cell RNA-seq studies, we observed high cell-to-cell variability in gene expression levels in GM12878 B-cells. We found that some of this variation was attributable to coordinated differences in the expression of biologically coherent sets of genes: for example, genes associated with the M phase of the cell cycle or with mRNA processing and splicing.
Despite good data quality, evidenced by complete and relatively uniform coverage across the mRNA length spectrum, our results were similar to other published data in displaying significant stochasticity. Stochasticity is expected to arise from a combination of biological variation and technical measurement variation. We present experimental and analytical approaches for measuring and accounting for technical stochasticity. We introduced and measured single-molecule capture efficiency, the key parameter influencing technical stochasticity, and found that its value was around 0.1 with the current SMART-seq protocol. This low capture efficiency provides a parsimonious explanation for the level of variation between single-cell measurements that is technical in origin. We also measured technical variation by carrying out pool/split experiments. This empirical test for nonbiological variation in the system is a stringent one, which includes capture efficiency, PCR effects, and any other unspecified sources. We then used the pool/split results to help parse biological variation between cells that is detectable over and above variation in pool/split measurements.
We observed unexpected levels of cell-to-cell variation in autosomal allelic expression bias and alternative splicing. The observation of allele switching between single cells could be explained as a technical artifact, given that a similar, although always lower, level of switching was observed in pool/split libraries. We therefore consider this a provisional result in need of further investigation with improved experimental protocols. The observed frequency of major splice switching in single cells is a stronger effect, and based on comparison with pool/split experiments, it is unlikely to be the sole result of technical stochasticity. It has also been independently reported in a different system (Shalek et al. 2013).
Transcriptional bursting suggests an attractive biological explanation for these observations. If a gene is expressed in a series of infrequent (relative to the half life of its mRNAs) such bursts, at any given time the population of mRNAs in the cell is likely to originate from only one allele. Such bursting could also be the source of cell-to-cell variation in alternative splicing. It is possible that the same set of factors influencing splice-site choice maintain physical association with the gene during a transcriptional burst, leading to a particular splicing pattern being highly favored locally in space and time, even if factors supporting a different splice choice are present within the same nucleus. Alternatively, isoform choice could be driven by temporal switching of factors and would operate regardless of bursting behavior. These are testable alternatives for future studies.
Many specific biological processes, especially regulatory ones, involve genes whose transcript levels are in the range highly affected by technical variation, as shown by our survey of transcription factors. While measurements with current methods can give some important clues about coherent biological variation, especially when large numbers of individual cells are assayed, our results argue that considerable improvement in the single-molecule capture efficiency would profoundly advance the field. Based on our simulations and results from pool/split experiments, we estimate that an increase in psmc from 0.1 to 0.5 would be a major leap forward, while psmc ≥ 0.8 would provide sufficient reliability for virtually any biological use. We anticipate that this empirical and analytical framework will be useful for evaluating future improvements in protocols, such as the recently described SMART-seq2 protocol (Picelli et al. 2013).
Finally, we found that the amount of mRNA per cell is highly variable between individual cells. Beyond biological interest, these differences in mRNA number are important for analysis pipelines. RPKM-type metrics are not reliable when there are large differences in total RNA per cell (Lin et al. 2012; Lovén et al. 2012). At present, the direct relationship between the absolute number of mRNA copies per cell and the number of sequencing reads in a library is lost due to the fragmentation of amplified cDNA molecules that is a common feature of available protocols, resulting in multiple distinct but overlapping sequencing fragments for each founder RNA molecule. mRNA copy number can be estimated back from FPKMs with the help of spike-in sequences. However, this is far from ideal, as it depends on the accuracy of quantification of the spike-ins and assumes the absence of systemic differences between spike-in RNAs and endogenous RNAs. If these assumptions are wrong, we expect a systematic error in the calculated number of mRNAs per cell, although the more interesting and important differences between individual cells versus pool/splits would remain. The above considerations make it very clear that a future ideal single-cell RNA-seq assay would combine a very high single-molecule capture efficiency with an amplification-free, and preferably also reverse transcription-free, direct RNA sequencing method to achieve direct counting of intact transcripts. Emerging sequencing technologies (Branton et al. 2008; Schadt et al. 2010) already hold promise for such radical improvements.
Methods
Cell growth and single-cell RNA-seq library construction
Individual GM12878 cells grown according to standard ENCODE protocols were picked with a glass micropipette, deposited into lysis buffer, and frozen. Cells were later lysed in reaction buffer, and single-cell SMART cDNA was generated following the SMART-seq protocol (Ramsköld et al. 2012) with the following modifications: (1) Carrier yeast tRNA was added in the lysis buffer to reduce handling losses and help maintain the integrity of the mRNA; (2) spikes of known copy number were introduced; and (3) the PCR cycle number was empirically titrated to accommodate the relatively small GM12878 cells. The SMART cDNA was tagmented using Illumina/Nextera reagents as described in Gertz et al. (2012). A detailed description of experimental protocols is provided in the Supplemental Methods.
Sequence alignment and gene expression quantification
Reads were aligned against a combined Bowtie index of the human genome and spike-in sequences using TopHat (Trapnell et al. 2009, 2012). Gene expression was quantified using Cufflinks (Trapnell et al. 2010, 2012). FPKMs were converted to copies-per-cell estimates using the input and measured spike-in abundances.
Single-molecule capture efficiency estimation
We estimated the average psmc based on the number of libraries with 0 FPKM for each spike and the number of input molecules (accounting for the fact that the number of successful captures is not known but only the number of complete failures; a detailed description of the procedure is provided in the Supplemental Methods). The average psmc for all spikes for which libraries with 0 FPKMs were observed was used, which is ∼0.01.
Analysis of allele-biased expression
We used the diploid (May 2011 release) NA12878 genome containing phased SNPs and indels based on the NCBI build 36 (hg18) version of the human genome (downloaded from http://sv.gersteinlab.org/NA12878_diploid/). Heterozygous transcriptomes containing two copies of each transcript were built, and reads were aligned using Bowtie (Langmead et al. 2009) (version 0.12.7) with zero mismatches allowed. Identical reads were collapsed, and reads were assigned to an allele if they mapped only to one of the alleles of a gene. Allele-biased expression was assessed by accounting for all of the following: (1) significance of allelic bias on the level of reads; (2) significance of allelic bias on the level of estimated copies per cell for each allele (derived from the total number of estimated copies for the gene); this is necessary, as a common feature of all current single-cell protocols is the production of multiple overlapping fragments from each original molecule; and (3) the possibility that the observed allelic bias is due to differential stochastic capture of the two alleles. A detailed description of the procedure is provided in the Supplemental Methods.
Alternative splicing analysis
We carried out alternative splicing analysis using the 5′ and 3′ splicing inclusion ψ scores described by Pervouchine et al. (2013), and applying the same statistical procedure we used to assess allelic expression bias to determine statistically significant splice variant exclusion. A detailed description of the splicing analysis procedure is provided in the Supplemental Methods.
Gene expression clustering and weighted correlation network analysis
We used the WGCNA R package (Langfelder and Horvath 2008) to carry out the weighted correlation network analysis. Gene Ontology enrichment in modules was assessed using FuncAssociate2.0 (Berriz et al. 2009). Gene expression clustering was carried out using Cluster 3.0 (de Hoon et al. 2004) and visualized using TreeView (Saldanha 2004).
Data access
BAM files containing aligned and unaligned sequencing reads have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE44618.
Supplementary Material
Supplemental Material
Acknowledgments
We thank Henry Amrhein, Diane Trout, and Sean Upchurch for computational assistance, and members of the Wold laboratory for helpful discussions. This work has been supported by NIH grants U54 HG004576 and U54 HG006998, the Simons Foundation, and the McDonnell Foundation. In addition, G.K.M., B.A.W., and B.J.W. are supported by the Beckman Foundation and the Donald Bren Endowment.
Footnotes
[Supplemental material is available for this article.]
References
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP 2009. Next generation software for functional trend analysis. Bioinformatics 25: 3043–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake WJ, Kaern M, Cantor CR, Collins JJ 2003. Noise in eukaryotic gene expression. Nature 422: 633–637 [DOI] [PubMed] [Google Scholar]
- Bradley RK, Merkin J, Lambert NJ, Burge CB 2012. Alternative splicing of RNA triplets is often regulated and accelerates proteome evolution. PLoS Biol 10: e1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, et al. 2008. The potential and challenges of nanopore sequencing. Nat Biotechnol 26: 1146–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouilette S, Kuersten S, Mein C, Bozek M, Terry A, Dias KR, Bhaw-Rosun L, Shintani Y, Coppen S, Ikebe C, et al. 2012. A simple and novel method for RNA-seq library preparation of single cell cDNA analysis by hyperactive Tn5 transposase. Dev Dyn 241: 1584–1590 [DOI] [PubMed] [Google Scholar]
- Cann GM, Gulzar ZG, Cooper S, Li R, Luo S, Tat M, Stuart S, Schroth G, Srinivas S, Ronaghi M, et al. 2012. mRNA-Seq of single prostate cancer circulating tumor cells reveals recapitulation of gene expression and pathways found in prostate cancer. PLoS ONE 7: e49144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess A 2012. Mechanisms and consequences of widespread random monoallelic expression. Nat Rev Genet 13: 421–428 [DOI] [PubMed] [Google Scholar]
- Cornelison DD, Wold BJ 1997. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol 191: 270–283 [DOI] [PubMed] [Google Scholar]
- Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, Cox CD, Simpson ML, Weinberger LS 2012. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci 109: 17454–17459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S 2004. Open source clustering software. Bioinformatics 20: 1453–1454 [DOI] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS 2002. Stochastic gene expression in a single cell. Science 297: 1183–1186 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2011. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femino AM, Fay FS, Fogarty K, Singer RH 1998. Visualization of single RNA transcripts in situ. Science 280: 585–590 [DOI] [PubMed] [Google Scholar]
- Gertz J, Varley KE, Davis NS, Baas BJ, Goryshin IY, Vaidyanathan R, Kuersten S, Myers RM 2012. Transposase mediated construction of RNA-seq libraries. Genome Res 22: 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A 2007. Widespread monoallelic expression on human autosomes. Science 318: 1136–1140 [DOI] [PubMed] [Google Scholar]
- Giulietti M, Piva F, D'Antonio M, D'Onorio De Meo P, Paoletti D, Castrignanò T, D'Erchia AM, Picardi E, Zambelli F, Principato G, et al. 2013. SpliceAid-F: A database of human splicing factors and their RNA-binding sites. Nucleic Acids Res 41: D125–D131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458: 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, Yanai I 2012. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2: 666–673 [DOI] [PubMed] [Google Scholar]
- Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, Teichmann SA 2011. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol 7: 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Huse K, Szafranski K, Jahn N, Hampe J, Schreiber S, Backofen R, Platzer M 2004. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat Genet 36: 1255–1257 [DOI] [PubMed] [Google Scholar]
- Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, Linnarsson S 2011. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res 21: 1160–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann BB, van Oudenaarden A 2007. Stochastic gene expression: From single molecules to the proteome. Curr Opin Genet Dev 17: 107–112 [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S 2008. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA 2012. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Wills QF, Tipping AJ, Datta K, Mittal R, Goldson AJ, Sexton DW, Holmes CC 2013. Methods for qPCR gene expression profiling applied to 1440 lymphoblastoid single cells. Methods 59: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA 2012. Revisiting global gene expression analysis. Cell 151: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, Cai L 2012. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods 9: 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Moult J 2009. Stochastic noise in splicing machinery. Nucleic Acids Res 37: 4873–4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A 2002. Regulation of noise in the expression of a single gene. Nat Genet 31: 69–73 [DOI] [PubMed] [Google Scholar]
- Ozsolak F, Goren A, Gymrek M, Guttman M, Regev A, Bernstein BE, Milos PM 2010. Digital transcriptome profiling from attomole-level RNA samples. Genome Res 20: 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Durrett RE, Zhu H, Tanaka Y, Li Y, Zi X, Marjani SL, Euskirchen G, Ma C, Lamotte RH, et al. 2012. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc Natl Acad Sci 110: 594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervouchine DD, Knowles DG, Guigó R 2013. Intron-centric estimation of alternative splicing from RNA-seq data. Bioinformatics 29: 273–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R 2013. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods doi: 10.1038/nmeth.2639 [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Luo S, Evgrafov O, Li R, Schroth GP, Levitt P, Knowles JA, Wang K 2012. Single-neuron RNA-Seq: Technical feasibility and reproducibility. Front Genet. 3: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van Oudenaarden A 2008. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 135: 216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Wang ET, Burge CB, Sandberg R 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5: e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. 2012. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30: 777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK 2005. Noise in gene expression: Origins, consequences, and control. Science 309: 2010–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Gertz J, Pauli F, Kucera KS, Varley KE, Newberry KM, Marinov GK, Mortazavi A, Williams BA, Song L, et al. 2012. Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res 22: 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. 2011. AlleleSeq: Analysis of allele-specific expression and binding in a network framework. Mol Syst Biol 7: 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ 2004. Java Treeview–extensible visualization of microarray data. Bioinformatics 20: 3246–3248 [DOI] [PubMed] [Google Scholar]
- Sanchez-Freire V, Ebert AD, Kalisky T, Quake SR, Wu JC 2012. Microfluidic single-cell real-time PCR for comparative analysis of gene expression patterns. Nat Protoc 7: 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Turner S, Kasarskis A 2010. A window into third-generation sequencing. Hum Mol Genet 19: R227–R240 [DOI] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. 2013. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498: 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Shamir R, Ast G 2004. How prevalent is functional alternative splicing in the human genome? Trends Genet 20: 68–71 [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. 2009. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6: 377–382 [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA 2010. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc 5: 516–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Lao K, Surani MA 2011. Development and applications of single-cell transcriptome analysis. Nat Methods (Suppl) 8: S6–S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L 2012. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM 2009. A census of human transcription factors: Function, expression and evolution. Nat Rev Genet 10: 252–263 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AK, VanInsberghe M, Petriv OI, Hamidi M, Sikorski D, Marra MA, Piret J, Aparicio S, Hansen CL 2011. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci 108: 13999–14004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Borevitz JO 2009. Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics 182: 943–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S 2005. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: 17. [DOI] [PubMed] [Google Scholar]
- Zenklusen D, Larson DR, Singer RH 2008. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol 15: 1263–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material