Macroglia-Microglia Interactions via TSPO Signaling Regulates Microglial Activation in the Mouse Retina (original) (raw)
Abstract
Chronic retinal inflammation in the form of activated microglia and macrophages are implicated in the etiology of neurodegenerative diseases of the retina, including age-related macular degeneration, diabetic retinopathy, and glaucoma. However, molecular biomarkers and targeted therapies for immune cell activation in these disorders are currently lacking. To address this, we investigated the involvement and role of translocator protein (TSPO), a biomarker of microglial and astrocyte gliosis in brain degeneration, in the context of retinal inflammation. Here, we find that TSPO is acutely and specifically upregulated in retinal microglia in separate mouse models of retinal inflammation and injury. Concomitantly, its endogenous ligand, diazepam-binding inhibitor (DBI), is upregulated in the macroglia of the mouse retina such as astrocytes and Müller cells. In addition, we discover that TSPO-mediated signaling in microglia via DBI-derived ligands negatively regulates features of microglial activation, including reactive oxygen species production, TNF-α expression and secretion, and microglial proliferation. The inducibility and effects of DBI-TSPO signaling in the retina reveal a mechanism of coordinated macroglia-microglia interactions, the function of which is to limit the magnitude of inflammatory responses after their initiation, facilitating a return to baseline quiescence. Our results indicate that TSPO is a promising molecular marker for imaging inflammatory cell activation in the retina and highlight DBI-TSPO signaling as a potential target for immodulatory therapies.
Keywords: DBI, gliosis, microglia, Müller cells, retina, TSPO
Introduction
Neurodegenerative diseases of the retina, including age-related macular degeneration, diabetic retinopathy, and glaucoma, involve chronic and dysregulated neuroinflammation (Buschini et al., 2011; Vohra et al., 2013). In these disorders, retinal microglia and macroglia populations demonstrate activation and gliosis (Karlstetter et al., 2010; Bringmann and Wiedemann, 2012), which contribute to neuronal degeneration and vision loss (Langmann, 2007). Although these conditions are the major cause of legal blindness in the developed world (Congdon et al., 2004), underlying disease mechanisms are incompletely understood and current preventative and treatment measures remain only partially effective. Understanding the mechanisms regulating chronic neuroinflammation in the retina and how they can be therapeutically modulated are of key importance. In addition, molecular biomarkers that reflect the level of inflammatory change in the retina may serve as clinical outcome measures and aid the discovery of therapies (Capozzi et al., 2013).
Translocator protein (TSPO) is a mitochondrial molecule that regulates cholesterol transport across the outer mitochondrial membrane, the rate-limiting step in steroidogenesis (Papadopoulos et al., 2006; Rone et al., 2009). In the brain and spinal cord, TSPO is expressed in activated gliotic microglial and macroglial cells (Chen and Guilarte, 2008) and serves as a biomarker for disease activity in Alzheimer's and Parkinson's disease (Politis et al., 2012; Venneti et al., 2013). Synthetic ligands to TSPO are also investigated as therapeutic agents for various CNS and PNS disorders (Nothdurfter et al., 2012). However, in the retina, the significance and function of TSPO under normal and pathological conditions have not been studied previously.
Despite its practical utility, TSPO's endogenous functions in the CNS have not been elucidated. Primary endogenous ligands to TSPO include a polypeptide called diazepam-binding inhibitor (DBI) and its shorter peptide cleavage products called endozepines (Ferrero et al., 1986). These molecules, expressed and secreted by macroglial populations in the CNS (Yanase et al., 2002), bind TSPO with high affinity (Guidotti et al., 1983; Slobodyansky et al., 1989), stimulating steroidogenesis (Papadopoulos et al., 1991; Do-Rego et al., 1998). Although DBI has been linked to Alzheimer's disease (Tokay et al., 2005), its functional significance with regard to TSPO signaling is unknown.
In this study, we examined the expression of TSPO in the developing and adult mouse retina under normal conditions and in a variety of injury models to establish its potential utility as a biomarker for retinal disease. We also investigated the functional role of TSPO in the retina by examining the expression pattern of its endogenous ligand, DBI, and the effect of triakontatetraneuropeptide (TTN), its biologically active cleavage product, on microglial activation and function. Our results demonstrate that DBI-TSPO signaling occurs as a form of macroglia-microglia interaction that is activated during retinal inflammation and injury and serves to limit the magnitude of microglial activation responses by negative regulation. These observations indicate the presence of dynamic interactions between microglia and macroglia populations that can shape the resolution of inflammatory responses and highlight TSPO as a molecular target for therapies aimed at modulating chronic neuroinflammation in CNS degenerations.
Materials and Methods
Experimental animals.
Heterozygous CX3CR1+/GFP transgenic animals were created by breeding CX3CR1GFP/GFP mice (Jung et al., 2000) to C57BL/6J mice (The Jackson Laboratory). Developing (0–28 d of age) and young adult (2–4 months of age) mice of either sex were used. Experimental protocols, approved by a local Institutional Animal Care and Use Committee, adhered to the Association for Research in Vision and Ophthalmology statement for animal use.
Cell culture.
Microglia were cultured from retinas of C57BL/6 mice at postnatal day 10–30 (P10–P30) as described previously (Ma et al., 2009). Enucleated globes were incubated in HBSS buffer containing 2% dispase (Roche) at 37°C for 45–60 min and then rinsed three times with DMEM containing 10% fetal bovine serum (FBS; Invitrogen). Retinas were dissected and triturated in culture medium (DMEM, 10% FBS, 1× MEM nonessential amino acids solution; Sigma) and transferred into 75 cm2 flasks at 37°C. The culture medium was changed after 24 h and then at 4–5 d intervals. When confluent, culture flasks were shaken gently to detach microglial cells, which were subcultured at low density in 150 mm dishes. Isolated cell clusters formed from the proliferation of individual microglia cluster (2–3 weeks) were picked off and subcultured in a new 75 cm2 flask. BV-2 cells, a murine microglial cell line, were maintained in DMEM containing 5% FBS and plated in multiwell plates at the density of 1–4 × 104/well for 24 h before further use. For DBI expression and secretion experiments, primary Müller cells were isolated from the retina and plated out in 24-well plates as described previously (Wang et al., 2011). Müller cells were subsequently exposed to conditioned media from either unactivated microglia or microglia that had been pretreated with lipopolysaccharide (LPS; 1 μg/ml). After 6 h of exposure, the existing media was replaced with fresh DMEM medium and allowed to condition for 24 h. The resulting cell lysates and conditioned media were analyzed for DBI protein levels using ELISA.
Reagents.
TTN (sequence TQPTDEEMLFIYSHFKQATVGDVNTDRPGLLDLK) was custom synthesized (CHI Scientific). In vitro effects of TTN were evaluated by preincubation of retinal microglia and BV2 microglia in TTN (0.1 or 10 μm) for 2 h before activation with LPS (0.5 μg/ml for 6 h). Control cultures were preincubated with control culture media containing equivalent levels of DMSO (0.001–0.1%) alone.
Quantitative reverse transcription PCR.
Cultured cells or retinas were lysed by trituration and homogenized using QIAshredder spin columns (Qiagen). Total RNA was isolated using the RNeasy Mini kit (Qiagen) according to the manufacturer's specifications. First-strand cDNA synthesis from mRNA was performed using qScript cDNA SuperMix (Quanta Biosciences) using oligo-dT as a primer. Quantitative reverse transcription PCR (qRT-PCR) was performed using a SYBR green RT-PCR kit (Affymetrix) and the 7900HT Fast Real-Time PCR System (Applied Biosystems) under the following conditions: denaturation at 95°C for 5 min, followed by 40 cycles of 95°C for 10 s and then 60°C for 45 s. Threshold cycle (CT) values were calculated and are expressed as the fold induction determined using the comparative CT (2−ΔΔCT) method. GAPDH, β-actin, and hypoxanthine guanine phosphoribosyl transferase (HPRT) were used as internal controls.
Measurements of protein expression.
Cultured cells or retinas were lysed by trituration in RIPA buffer (Sigma) containing 1:100 proteinase inhibitor (Calbiochem). Lysates were denatured in boiling water at 100°C for 3 min and then loaded onto NuPAGE 4–12% Bis-Tris Gel (Novex). After electrophoresis, proteins were transferred onto nitrocellulose membranes (iBlot Gel Transfer Stacks; Invitrogen). Membranes were first incubated in blocking solution (Western Blocking Reagent; Roche) for 2 h and then in blocking solutions containing primary antibodies overnight at 4°C. The following primary antibodies were used: TSPO (1:4000; Abcam) and DBI (1:2000; Frontier Institute, Hokkaido, Japan). Membranes were then washed three times with Tris-buffered saline with 0.05% Tween 20 (Quality Biological) and incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG as the secondary antibody (1:4000; Cell Signaling Technology). HRP-conjugated β-actin (1:50,000; Sigma) was used as an endogenous control. Resulting blots were developed using a chemiluminescence system (SuperSignal West Femto Chemiluminescent Substrate; Thermo Fisher Scientific). Images were taken using Fujifilm LAS-3000 Imager and protein levels were quantitated using imaging analysis software (ImageJ). TNF-α and DBI protein levels in conditioned media and cell lysate from cultured cells and lysates of retinal tissue were assessed using ELISA kits (R&D Scientific and Cloud-Clone, respectively).
Immunohistochemistry.
Immunohistochemistry was performed on cultured microglial cells, mouse retinal sections (30 μm-thick cryosections and 100 μm-thick vibratome sections) and retinal flat mounts. Cultured cells, retinal sections, or retinal flat mounts were preincubated in blocking buffer (consisting of 10% normal goat serum, 5% bovine serum, and 0.5% Triton X-100 (all from Sigma) in 1× PBS for 2 h at room temperature for cultured cells and cryosections and for overnight at 4°C for flat mounts and vibratome sections. These were then incubated in primary antibody (diluted in the blocking buffer) at 4°C either overnight (for cultured cells and retinal cryosections) or for 48 h (for flat mounts and vibratome sections). Primary antibodies targeting the following molecules were used: glutamine synthetase (GS; 1:200) and NeuN (1:200) (both from Millipore); glial fibrillary acidic protein (GFAP; 1:800) and isolectin GS-IB4 (1:200) (both from Invitrogen); CD11b (1:200), F4/80 (1:200), CD68 (1:200) and CD31 (1:200) (all from AbD Serotec); ionized calcium binding adaptor molecule-1 (Iba1; 1:800; Wako); TSPO (ab109497; 1:200) and vimentin (1:400) (both from Abcam); DBI (1:200; Frontier Institute, Hokkaido, Japan); and Brn-3 (1:50; Santa Cruz Biotechnology). Samples were incubated in secondary antibodies at a 1:400 dilution (1 h at room temperature for cells and cryosections, 4 h at 4°C for flat mounts and vibratome sections). The specificities of primary antibodies to TSPO and DBI were confirmed by their generation of a single strong band of expected size on Western blotting, which in the case of TSPO was decreased in intensity in “knock-down” experiments. In immunohistochemical analyses, negative control experiments were performed with only the secondary antibody and positive control experiments were performed with tissue with known expression patterns for the antigen.
Animal procedures.
Before procedures, experimental mice were anesthetized with intraperitoneal ketamine (90 mg/kg) and xylazine (8 mg/kg). The optic nerve crush injury was performed as described previously (Tang et al., 2011) and retinas were analyzed after 7 d. Intravitreal injections were performed directly behind the limbus using a 30-gauge needle on a model 701 Hamilton syringe. In the endotoxin-induced model of retinal inflammation, LPS (1.0–1.5 μl of 1 mg/ml LPS in 1×PBS) was injected and retinas were harvested 1, 3, 7, 14, and 28 d afterward. In the excitotoxic model of retinal injury, an intravitreal injection of NMDA (1.0 μl, 40 mm in 1× PBS) was performed and retinal tissue was harvested 1 d later. To assess the effect of intravitreal TTN, 4 experimental groups were given 1.0 μl intravitreal injections containing the following: (1) 1× PBS containing 10% DMSO, (2) 1× PBS containing 1 mm TTN and 10% DMSO, (3) 1× PBS containing 1 mg/ml LPS and 10% DMSO, and (4) 1× PBS containing 1 mm TTN, 1 mg/ml LPS, and 10% DMSO. The subretinal hemorrhage model of injury was performed as described previously (Zhao et al., 2011) and retinas were harvested 2 d later.
Measurement of reactive oxygen species and oxidative stress.
Cellular reactive oxygen species (ROS) content was measured using the 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) assay (Invitrogen). Cultured retinal microglial or BV2 microglia in 96-well plates (30,000 cells/well) were pretreated with TTN for 1 h before activation with LPS (0.5 μg/ml for 6 h). Controls included the following: (1) cells that were exposed to culture medium lacking TTN or LPS, (2) cells pretreated with TTN but not activated with LPS, and (3) cells that were pretreated with culture medium lacking TTN and activated with LPS. Cells were then incubated in 20 μm H2DCFDA in PBS for 20–30 min at 37°C, washed, and analyzed spectrally (SpectraMax M5; Molecular Devices). Retina oxidative stress was evaluated by assaying lipid peroxidation levels using a thiobarbituric acid-reactive substances (TBARS) assay kit (Cayman Chemical).
Confocal microscopy and live-cell imaging.
After immunohistochemical staining, cultured cells, retinal sections, and flat mounts were imaged with a confocal microscope (FluoView 1000; Olympus America). Multiplane _z_-series were collected using a 40× oil-immersion objective. Confocal image _z_-series were imported into ImageJ and Adobe Photoshop for image analysis. Mitochondrial superoxide levels were measured using the MitoSox Red indicator dye (Invitrogen). Cultured microglia were plated in 6 cell plates and activated by LPS (0.5 μg/ml LPS for 6 h) with and without pretreatment with TTN (10 μm for 2 h). These cells were then incubated in PBS containing 1.25 μm MitoSox Red for 20 min at 37°C. MitoSox Red labeling in living microglial cells was immediately imaged using a 40× immersion objective in a Leica SP2 confocal microscope.
‘Knock-down’ of TSPO expression.
Small hairpin RNAs (shRNAs) in the pLKO.1 lentiviral vector (5′-TTCTGCAAGCTAGCATACCAC −3′) against mouse TSPO mRNA (NM_009775) (Thermo Fisher Scientific) was electroporated into microglial cells (Amaxa Mouse Macrophage Nucleofector Kit; Lonza) following the manufacturer's protocol. The efficiency of each shRNA knock-down was assessed by quantitative Western blotting for TSPO.
Assays of cellular proliferation.
Microglia proliferation was measured by detecting the incorporation of the modified nucleoside 5-ethynyl-2′-deoxyuridine (EdU) in dividing cells (Click-iT Edu Detection assay; Invitrogen). EdU-incorporating cells were imaged by epifluorescence microscopy (Axiovert 200M; Zeiss). The numbers of EdU+ and DAPI+ cells in eight 20× fields were counted and analyzed.
Measurement of neurosteroids by HPLC.
BV2 microglia were cultured in the absence and the presence of TTN (10 and 50 μm) and harvested after 7 h. Cell lysates were lyophilized and resuspended in 50 μl of water. An aliquot (10 μl) was removed for protein quantification (BCA protein assay kit; Thermo Fisher Scientific) and the remainder added to 10 μl of 10% trichloroacetic acid and 20 μl of 20 mm 2,4-dinitrophenylhydrazine hydrochloric acid solution (Sigma). After 24 h at room temperature, 200 μl of water and 200 μl of diethyl ether were added. Ether extraction was repeated three times and the resulting extract was dried, resuspended in 50 μl of _N_-dimethylformamide (DMF), and injected into a HPLC device (Model 2695, with a 2996 photodiode array detector; Waters). A triphasic gradient was used to separate the steroid-hydrozones using a Luna 5μ C-8(2) 100A column (2 × 250 mm) (Phenomenex) at 0.25 ml/min. The initial condition used was 50:50 water:acetonitrile, followed by a linear gradient to 100% acetonitrile over 15 min and held at 100% acetonitrile for an additional 5 min. A second linear gradient was started from 100% acetonitrile to 100% methanol over 10 min and then held for another 5 min at 100% methanol. Data collection was terminated at 35 min and the column was reequilibrated to initial conditions for 15 min before the next injection. Absoption spectra were collected between 190 and 600 nm and data were analyzed using Empower 2 software (Waters). Standards (10 mm) for dehydroepiandrosterone (DHEA) and pregnenolone were prepared in 45% hydroxypropyl-β-cyclodextrin. The final ether extracts were suspended in 1 ml of DMF. The extinction coefficients for DHEA-DNPH (3825 area U/pmol) and pregnenolone-DNPH (3910 area U/pmol) were determined by peak area integration at 370 nm. Quantification of the androstenolone-DNPH and pregnenolone-DNPH in the cell lysates (n = 4–6 replicates per condition) was performed using peak area integration and normalized to protein content (pmol/μg).
Statistical analysis.
Statistical analyses were performed using statistical software (GraphPad). For comparisons involving two data columns, t tests (paired or unpaired) or nonparametric tests (Mann–Whitney) were used, depending on whether the data followed a Gaussian distribution as determined by normality tests. For comparison involving three or more data columns, a one-way ANOVA (with Dunnett's multiple-comparison test) was used if the data followed a Gaussian distribution and a nonparametric Kruskal–Wallis test (with Dunn's multiple-comparison test) was used if it did not. p < 0.05 was set as the basis for rejecting the null hypothesis. Error bars in graphs indicate SE.
Results
TSPO expression in the developing and adult mouse retina
Although TSPO expression has been primarily localized to microglia, astrocytes, and blood vessels in the normal and diseased brain (Stephenson et al., 1995; Ji et al., 2008; Cosenza-Nashat et al., 2009) and spinal cord (Daugherty et al., 2013), to our knowledge, its expression patterns in the developing and adult retina have not been described previously. Immunohistochemical analyses in CX3CR1GFP/+ mice, in which retinal microglia are specifically marked by GFP, demonstrated that TSPO immunopositivity in the P0 retina was exclusively localized to inner retinal migratory GFP+ microglia, which have an amoeboid morphology (Fig. 1A). At P3 and P7, developing microglia migrated into the neuroblastic and outer plexiform layers of the outer retina, maintaining their TSPO immunopositivity. At P14, when retinal microglia attained their final laminar positions in the inner and outer plexiform layers and developed mature ramified morphologies, TSPO immunopositivity diminished in microglia, whereas it increased in developing retinal blood vessels. In the young adult retina (P28), TSPO immunopositivity was found solely in retinal blood vessels and was not detected in other retinal cells, including microglia and astrocytes. qRT-PCR analyses of the neural retina demonstrated that TSPO mRNA expression was highest in the P0 retina, decreased during the first postnatal week, and then increased again in the adult retina. Retinal levels of TSPO protein, monitored by Western blotting, showed a slowing accumulating level from P0 to adulthood.
Figure 1.

Expression of TSPO in the developing and adult mouse retina. A, Immunohistochemical analyses in retinal sections showed TSPO expression in microglia and inner retinal blood vessels. In the developing P0 retina of CX3CR1GFP/+ mice, TSPO immunopositivity was found exclusively in rounded, deramified microglia marked by GFP expression (arrowheads). In the P3 and P7 retina, GFP+ microglia, which migrate into the outer retina, maintained immunopositivity for TSPO. In the P14 retina, retinal microglia acquired ramified morphologies and decreased in TSPO immunopositivity (arrowheads), whereas inner retinal vessels became immunopositive (arrow). In the P28 adult retina, TSPO immunopositivity was absent in microglia and was found only in retinal vessels. B, qRT-PCR analyses showed varying levels of TSPO mRNA expression in the retina of C57BL6 mice during postnatal development. Expression levels, normalized to that in P0 retina, demonstrated a relative downregulation in the first postnatal week and then increased slightly in the young (P28) adult. C, Protein expression levels of TSPO in the retina, as demonstrated by quantitative Western blot analyses, were low at P0 and then gradually increased. *Comparisons with P0 of p < 0.05, 1-way ANOVA with Dunnett's multiple-comparisons test, n ≥ 4 animals per group. Scale bar indicates 50 μm.
DBI expression in the developing and adult mouse retina
DBI, a protein that is proteolytically cleaved to generate peptide ligands to TSPO, is an evolutionarily conserved protein widely expressed in the CNS and peripheral tissues (Yanase et al., 2002). To investigate the locus of endogenous TSPO signaling in the retina, we characterized the expression of DBI in the developing and adult mouse retina. Immunohistochemical analyses in retinal sections indicated that DBI expression was confined to the astrocytic layer of the inner retina at P0 (Fig. 2A). From P3 onward, DBI immunopositivity emerged in somata in the inner nuclear layer and also in vertically oriented processes spanning the thickness of the retina. In retinal sections from CX3CR1GFP/+ mice, these DBI+ cells colocalized with GS, a marker of Müller glia, but not with GFP expression, indicating that DBI was expressed in Müller cells but absent in microglia (Fig. 2B). Analyses in flat-mounted adult retina revealed DBI immunopositivity in the inner retinal surface, colocalizing with GS+ Müller glia end-foot processes and GFAP+ astrocytic processes (Fig. 2C), indicating DBI to be constitutively and specifically expressed in both retinal macroglial populations. qRT-PCR analyses in the neural retina showed constitutive DBI mRNA expression during development (Fig. 2D). Quantitative Western blot analysis demonstrated increasing levels of DBI protein expression in the retina from P0 to adulthood (Fig. 2E).
Figure 2.

Expression of DBI in the developing and adult mouse retina. A, Immunohistochemical analyses in the retinal sections from developing wild-type C57BL6 mice showed immunopositivity for DBI in the astrocytic layer of the inner retina at P0. At P3, P7, and P14, DBI immunopositivity was found additionally in vertically oriented processes extending through the thickness of the retina and in somata in the inner nuclear layer. B, Immunopositivity for DBI (red) in the adult (P28) retina of CX3CR1GFP/+ mice colocalized with the Müller cell marker, glutamine synthetase (GS, green, middle), but not with GFP-labeled microglia (green, right). C, Immunohistochemical analyses in retinal flat mounts showed that GFAP-expressing retinal astrocytes (blue) in the inner retina were weakly positive for DBI (red), whereas GS+ Müller cell end-foot processes (green) were strongly positive. D, qRT-PCR analyses showing relative levels of DBI mRNA expression in the retina of postnatal and young adult wild-type C57BL6 mice. E, Protein expression levels of DBI as demonstrated by quantitative Western blot analyses showed an increasing trend from P0 to adulthood. *Comparisons with P0 of p < 0.05, 1-way ANOVA with Dunnett's multiple-comparisons test, n ≥ 4 animals per group. Scale bar indicates 50 μm.
Regulation of TSPO and DBI expression by LPS-induced retinal inflammation
To determine how TSPO expression in retinal microglia is regulated as a function of microglial activation, we monitored TSPO expression levels in microglia in vitro and in vivo after activation by LPS. We found that cultured retinal microglia demonstrated increased mRNA expression of TSPO after LPS activation relative to untreated controls (Fig. 3A). This upregulation of TSPO expression after LPS treatment was also evident on the protein level in both cultured retinal microglia and BV2 microglia as assessed by Western blot analysis (Fig. 3B). Immunohistochemical analyses showed that increased TSPO expression was concentrated in the perinuclear region in cultured retinal microglia (Fig. 3C). In parallel with in vitro observations, mRNA (Fig. 3D) and protein (Fig. 3E) expression levels of TSPO in vivo in the retina after intravitreal LPS injection were significantly elevated 1 and 3 d after injection relative to controls, but decreased back to control levels beyond 7 d. Immunopositivity for TSPO, while absent from microglia in PBS-injected control eyes (Fig. 3F), was induced in retinal microglia 1–3 d after activation by LPS injection (Fig. 3G,H) as monitored in flat-mounted retina specimens. Separate analyses in retinal sections showed that the induction of TSPO immunopositivity occurred in activated, F4/80+ microglia located in separate retinal layers, ranging from the ganglion cell layer to the outer plexiform layer (Fig. 3K). This upregulation of TSPO expression was transient; at 28 d after LPS injection, when microglia reverted back to a “resting” state as evidenced by the reacquisition of ramified morphologies and a decrease in F4/80+ immunopositivity, TSPO immunopositivity concurrently diminished in microglia, with a weak residual punctate labeling persisting in the perinuclear region (Fig. 3I). Immunohistochemical analyses confirmed that LPS-induced TSPO upregulation was restricted to microglia; retinal macroglia such as GFAP-labeled astrocytes (Fig. 3J) and GS-labeled Müller cells (Fig. 3K) were immunonegative for TSPO at all time points.
Figure 3.

TSPO expression is upregulated in microglia after LPS-mediated activation in vitro and in vivo. A, qRT-PCR analysis in retinal microglia demonstrated increased TSPO mRNA expression after LPS treatment. B, Top, Western blot analysis demonstrated increased TSPO protein expression in both retinal microglia and BV2 microglia 24 h after LPS treatment. Bottom, Quantitative Western blot analyses in BV2 microglia confirmed increased TSPO protein expression (graphs in A and B are normalized relative to control). *p < 0.05, unpaired t test with Welch's correction, n = 7–13. C, CD11b+ retinal microglia (green) demonstrated TSPO immunopositivity (red) in a perinuclear distribution 6 h after LPS exposure. Scale bar indicates 50 μm. D, qRT-PCR analyses of retinal TSPO mRNA levels after intravitreal LPS injection (black bars) relative to PBS-injected control eyes (white bars), showed a transient increase in TSPO mRNA 1 d after LPS injection. *p < 0.05, unpaired t test with Welch's correction, n = 3–15 eyes. E, Quantitative Western blot analysis of TSPO protein demonstrated similar dynamics. *p < 0.05, paired t test, n = 4–8 eyes. F–I) Retinal flat mounts from adult CX3CR1GFP/+ mice that had been injected intravitreally with LPS or with a PBS control at various time-points. After control (PBS) injections (F), GFP+ retinal microglia, which maintained their “resting” activation state, as evidenced by their ramified morphology (left, green) and the absence of F4/80 immunopositivity (right, blue), remained negative for TSPO (middle, red). One to 3 d after LPS injections (G, H), retinal microglia demonstrated increased activation (appearance of amoeboid morphologies and F4/80 immunopositivity). TSPO immunopositivity (G, H, middle, red) in the retina emerged in GFP+ microglia (green) and amoeboid F4/80+ (blue) activated microglia and macrophages 1 d after LPS injection and increased in intensity at 3 d after injection (inset shows TSPO colocalization with GFP and F4/80 labeling at high magnification). TSPO expression developed in microglia found at various layers of the retina (K). GCL, Ganglion cell layer; IPL, inner plexiform layer; OPL, outer plexiform layer. At day 28 after LPS injection (I), GFP+ microglia reverted to a ramified “resting” morphology, with TSPO immunopositivity decreasing to a weak residual punctate pattern (inset). Scale bar indicates 100 μm. J, K, In eyes injected with intravitreal LPS 3 d prior, immunohistochemical analyses of retinal flat mounts demonstrated that TSPO immunoreactivity colocalized with microglia but not in GFAP+ astrocytes (J), whereas those performed in retinal sections demonstrated that TSPO immunoreactivity was absent in GS+ Müller cells (K). Scale bar indicates 50 μm.
We monitored the expression of DBI in the retina in response to intravitreal LPS injection. DBI mRNA levels were upregulated 1 d after LPS injection and decreased to control levels by 3 d after injection (Fig. 4A). DBI immunopositivity in the retina, which was detectable in Müller cells and astrocytes in control eyes (Fig. 4B,C), increased in intensity in both GS+ Müller cells (Fig. 4D,F) and GFAP+ astrocytes (Fig. 4E,G) after intravitreal LPS, indicating that these cell types, and not activated microglia (Fig. 4H), are the sources of elevated DBI expression during retinal inflammation. Experiments involving primary cultured retinal Müller cells also confirmed macroglial expression and secretion of DBI; after exposure to conditioned media from activated retinal microglia, DBI protein levels in Müller cell lysates and Müller cell conditional media were significantly increased relative to those exposed to media from unactivated microglia (p < 0.05, _n_ = >4 replicates; data not shown).
Figure 4.
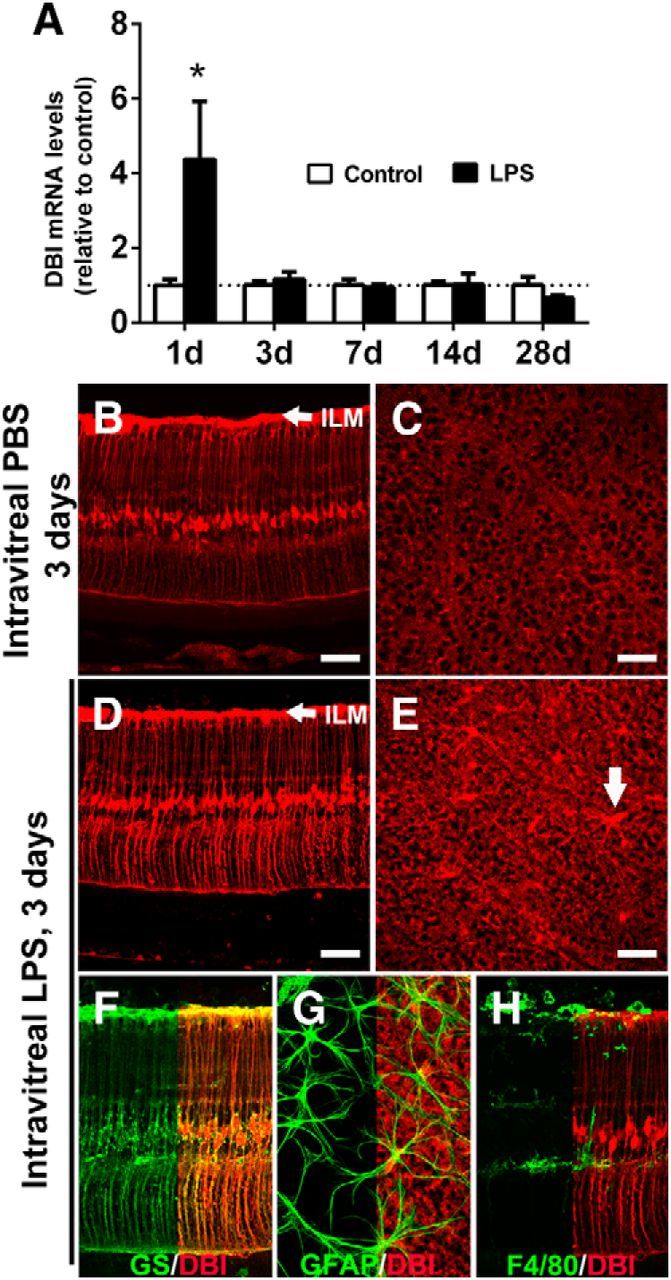
DBI expression in the retina is increased in Müller cells and astrocytes after LPS-mediated activation. A, qRT-PCR analyses of DBI mRNA levels in the retina at various time points after intravitreal LPS injection (black bars) relative to PBS injected controls (white bars). DBI mRNA levels were increased 1 d after LPS injection and returned to control levels by 3 d. *Comparison with control for which p < 0.05, Mann–Whitney test, n = 6–10 eyes. Under control conditions, Müller cell somata and processes, as well as retinal astrocytes, were immunopositive for DBI as observed in retinal sections (B) and retinal flat mounts imaged at the plane of the inner limiting membrane (ILM, indicated by arrow in B, D) at the inner surface of the retina, which revealed Müller cell end-foot processes and astrocytes (C). Three days after intravitreal LPS injection, increased DBI immunopositivity was observed in both radial Müller cell processes (D) and astrocytes (E, arrow). The cellular localization of increased DBI expression was confirmed by immunolocalization with GS for Müller cells (F) and GFAP for astrocytes (G). DBI expression was not observed in F4/80+ activated microglia (H). Scale bar indicates 100 μm.
TSPO expression is upregulated in retinal microglia in models of retinal injury
We examined changes in the retinal expression of TSPO in the contexts of retinal injury in the following animal models: (1) an optic nerve crush model, (2) an NMDA-induced excitotoxicity model, and (3) a subretinal hemorrhage model. In the optic nerve crush model, in which retinal ganglion cells are injured by axonal damage, TSPO immunopositivity was absent in neurons and glia in sham-manipulated controls, but was induced in retinal microglia after nerve crush injury (Fig. 5A–D). In a model of excitotoxic injury induced by the intravitreal injection of NMDA, microglia activated in response to the injury rapidly acquired TSPO immunopositivity (Fig. 5E,F). In a model of subretinal hemorrhage, a model for hemorrhagic exudative age-related macular degeneration (Zhao et al., 2011) in which outer retinal damage is accompanied by local microglia/macrophage infiltration into the outer nuclear layer and subretinal space, activated, CD68+ microglia/macrophages in the outer retina demonstrated prominent TSPO immunopositivity (Fig. 5G,H). The common development of TSPO immunopositivity in retinal microglia across different injury models indicates that TSPO upregulation is a general feature of microglial activation responses in retinal injury and inflammation.
Figure 5.
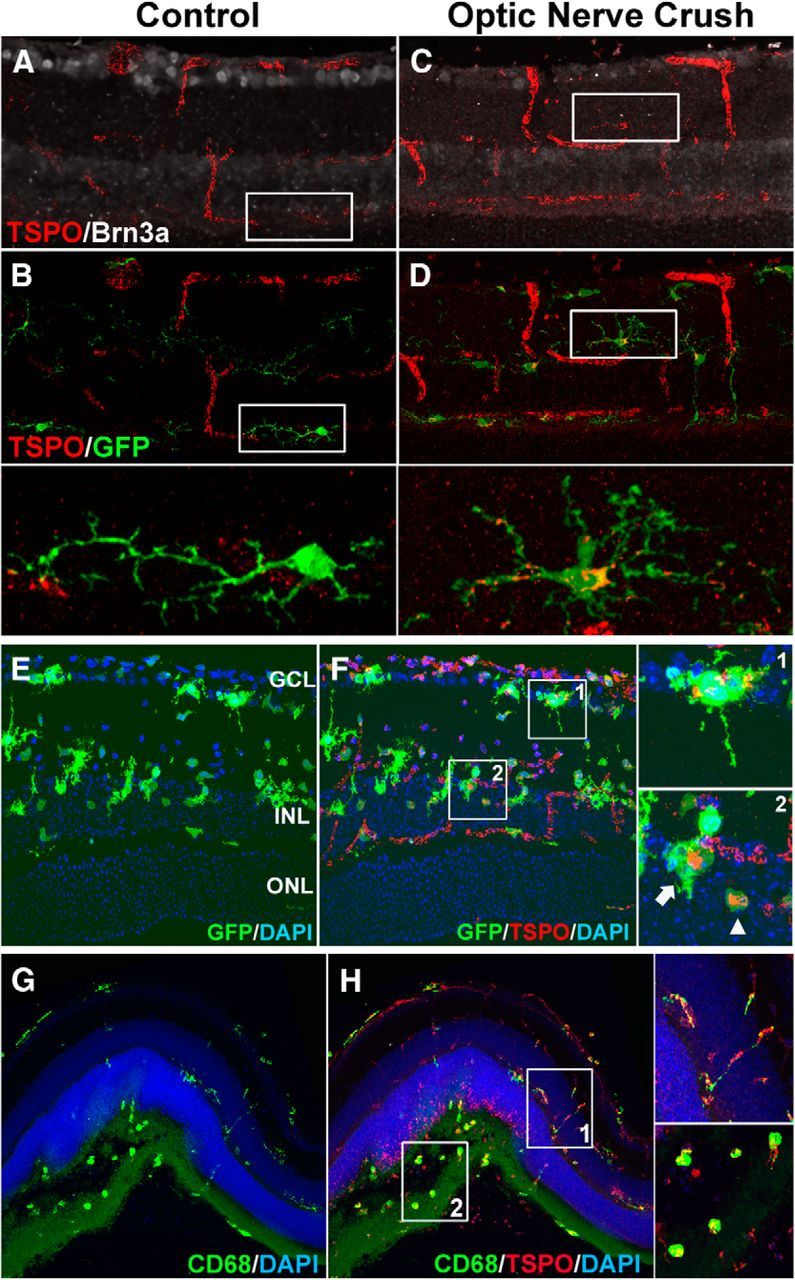
TSPO expression is induced in retinal microglia in models of retinal injury. A–D, TSPO expression was evaluated in a model of optic nerve crush injury in adult CX3CR1GFP/+ mice. Animals were subjected to optic nerve crush and their retinas harvested and examined in cryosections 7 d after injury. In sham-manipulated control mice (A), the ganglion cell marker Brn3 (white) showed prominent labeling in the ganglion cell layer (GCL) and TSPO immunolabeling (red) was present only in retinal vessels and absent in GFP+ retinal microglia (B, inset, green). In treated animals (C), Brn3+ ganglion cells were depleted, punctate, extravascular TSPO-labeling developed in the inner retina (C, D, red) that localized to the processes and perinuclear areas of retinal microglia (D, inset). E, F, Excitotoxic retinal injury was induced in CX3CR1GFP/+ mice with an intravitreal injection of NMDA (1 μl of 40 mm) and their retinas analyzed in cryosections 1 d later. GFP+ microglia in the inner retina (E, green) demonstrated activated morphologies with shortened processes and amoeboid morphologies. Punctate, extravascular TSPO immunostaining also developed in the inner retina (F, red) that colocalized with GFP+ cells (F, arrows, insets). G, H, Outer retinal injury was induced in a model of subretinal hemorrhage in adult C57BL6 mice with a subretinal injection of autologous blood (1.5 μl) and retinas analyzed in vibratome sections 2 d later. Activated, CD68+ microglia with amoeboid morphologies were found aggregated in the outer retina in the region of subretinal hemorrhage (G, green). Punctate, extravascular TSPO immunostaining (H, red) was observed throughout the retina, colocalizing with CD68+ microglial processes (H, inset 1), and microglia somata (H, inset 2). Scale bar indicates 50 μm.
TTN, a DBI-derived peptide and TSPO ligand, inhibits microglial activation in vitro
In previous work, we characterized the ability of interactions between macroglia and microglia to modulate inflammatory responses in the retina (Wang et al., 2011). Our observations here, showing that retinal inflammation induced concurrent increases in microglia-derived TSPO and in macroglia-derived DBI, suggested that DBI-to-TSPO signaling may be a candidate mechanism for this functional interaction. To explore the functional consequences of increased endogenous TSPO signaling in microglia, we evaluated the effects of TTN, a DBI-derived peptide previously shown to function as a TSPO ligand (Slobodyansky et al., 1989; Papadopoulos et al., 1991), on microglial physiology in vitro. In cultured retinal microglia, preincubation in TTN for 2 h significantly suppressed microglial production of ROS, both in the absence of and in the presence of LPS-induced activation (Fig. 6A). In BV2 microglia, TTN preincubation similarly reduced LPS-induced ROS production (Fig. 6B). The effect of TTN on microglial ROS production is likely to be mediated by the suppression of ROS production in mitochondria; live-cell imaging with MitoSOX, a fluorescent marker of superoxide production targeted specifically to mitochondria, revealed that although LPS treatment induced MitoSOX labeling in cultured retinal microglia, preincubation of TTN (10 μm for 2 h) attenuated this response (Fig. 6C). In addition, TTN preincubation significantly reduced mRNA expression of the proinflammatory cytokine TNF-α in cultured microglia (Fig. 6D) and BV2 cells (Fig. 6E) and the secretion of TNF-α protein (Fig. 6F). Proliferation of BV2 cells in culture, as determined by EdU incorporation, was also significantly reduced by TTN relative to untreated cultures both in the absence and the presence of LPS. Because TTN-mediated TSPO signaling exerted a negative regulatory effect on these aspects of activated microglial physiology, the increase in TSPO signaling in microglia after inflammation and injury may serve to limit the magnitude of inflammatory responses and to return activated microglia back to a quiescent “resting” state. These effects of TTN were somewhat similar to those found for the synthetic TSPO ligands PK 11195 and Ro5-4864 in cultured retinal microglia; both PK 11195 and Ro5-4864 significantly decreased ROS production, whereas only Ro5-4864 significantly decreased TNF-α expression (Fig. 7). HPLC analyses of untreated versus TTN-treated BV2 microglial lysates demonstrated that, although pregnenolone levels were not found to be significantly different between the two groups, DHEA, a neurosteroid found to repress microglial activation (Saijo et al., 2011), was significantly increased after TTN treatment (50.0 ± 38.5 vs 260 ± 4.8 pmol/mmol of protein, p < 0.05).
Figure 6.
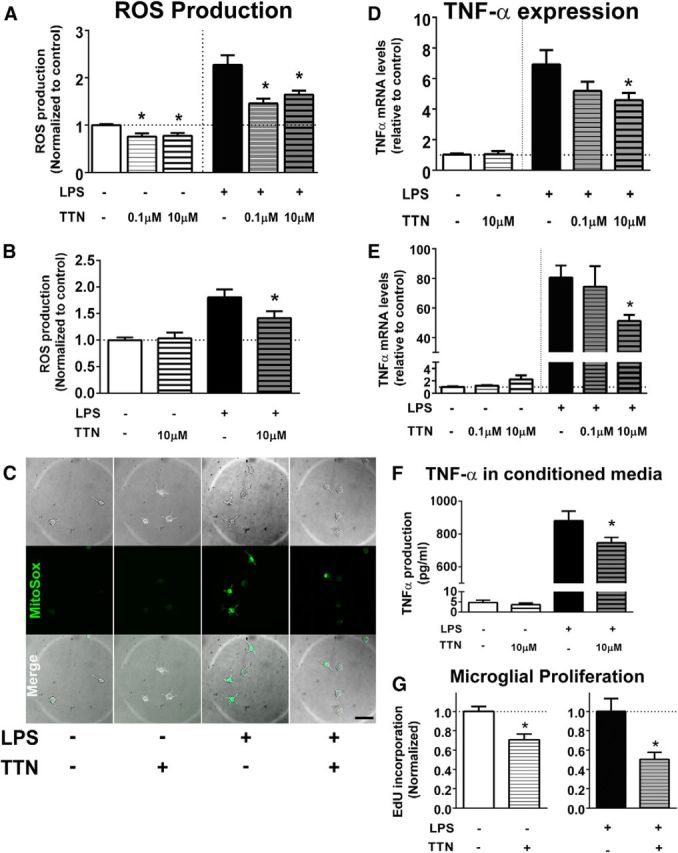
Effects of TTN, an endogenous DBI-derived TSPO ligand, on microglia function in vitro. A, B, The effect of TTN on ROS production in cultured retinal microglia and BV2 microglia was evaluated using the H2DCFDA assay. In cultured microglia (A), ROS production was significantly increased from control conditions (white bar) by LPS treatment (0.5 μg/ml for 6 h, black bar). Pretreatment with TTN at 0.1 and 10 μm for 2 h significantly reduced ROS production in microglia in the absence and presence of LPS. In BV2 microglia (B), TTN pretreatment (10 μm) similarly significantly suppressed ROS production in LPS-treated cells. n = 14–21 replicates per condition. C, Production of mitochondrial superoxide in cultured retinal microglia, as monitored with a MitoSOX indicator. Minimal fluorescence was detected under control conditions but was increased after LPS application. Pretreatment with 10 μm TTN reduced the number of Mito SOX+ cells produced by LPS treatment. D, E, Effect of TTN on TNF-α mRNA expression in cultured retinal microglia (D) and BV2 microglia (E). Although TTN pretreatment did not influence TNF-α mRNA expression in microglia in the absence of LPS treatment (white and unshaded bars), it significantly suppressed it in LPS-treated cells (black and shaded bars). n = 6–12 replicates. F, ELISA analyses demonstrate that although TTN pretreatment did not affect TNF-α secretion in BV2 cells in the absence of LPS treatment (white and unshaded bars), it significantly suppressed the TNF-α secretion in LPS-treated cells (black and shaded bars). n = 8–16 replicates. G, Effect of TTN on BV2 microglia proliferation was measured by the incorporation of EdU. TTN pretreatment (10 μm) significantly suppressed microglial proliferation in both control and LPS-stimulated culture conditions. *Comparison with non-TTN-treated controls for which p < 0.05, Mann–Whitney test, n = 38–50 replicates. Data in A, B, D, E, and F were analyzed with the Kruskal-Wallis one-way ANOVA test with Dunn's multiple-comparisons test. *Comparison with non-TTN-treated controls for which p < 0.05.
Figure 7.

Effects of the synthetic TSPO ligands PK 11195 and Ro5–4864 on microglia function in vitro. A, B, The effect of synthetic ligands in cultured retinal microglia on ROS production was evaluated using the H2DCFDA assay. Pretreatment with either PK 11195 (A) or Ro5–4864 (B) significantly reduced ROS production in microglia in the absence and presence of LPS. C, D, The effect of synthetic ligand pretreatment on TNF-α mRNA expression was evaluated using qRT-PCR. Decrease in TNF-α expression in the presence of LPS was nonsignificant with PK 11195 (C) but significant with Ro5-4864 (D). Data were analyzed with the Kruskal–Wallis one-way ANOVA test with Dunn's multiple-comparisons test. *Comparison with non-ligand-treated controls for which p < 0.05, n = 4–18 replicates per condition.
Downregulation of TSPO expression in microglia increases microglial activation
We assessed the function of TSPO signaling in microglia by evaluating the consequences of knocking down TSPO expression using a lentiviral shRNA vector targeting the TSPO gene. Quantitative Western blot analyses showed that the protein expression of TSPO was effectively decreased in BV2 cells by 30–40% relative to control cultures both in the absence and the presence of LPS-induced activation (Fig. 8A). Knock-down of TSPO expression in BV2 microglia resulted in significantly increased ROS production (Fig. 8B), TNF-α mRNA expression (Fig. 8C), TNF-α protein secretion (Fig. 8D), and proliferation rate (Fig. 8E) in response to LPS activation. For BV2 cells not activated with LPS, the knock-down of TSPO resulted in smaller, but nonsignificant increases in ROS production and TNF-α expression. Together, these data provide corroborating evidence for the role of TSPO signaling in the negative regulation of microglial activation.
Figure 8.

Effects of knock-down of TSPO on microglia function in vitro. A, Knock-down of TSPO protein expression in BV2 microglia. BV2 microglia were transfected with either an shRNA vector targeting TSPO or a control/scramble vector. In BV2 microglia cultured under normal conditions (white bar) or stimulated with LPS (0.5 μg/ml) for 6 h (black bar), shRNA transfection was able to significantly reduce the protein expression of TSPO by 30–40% relative to control. *Comparison with controls for which p < 0.05, Mann–Whitney test, n = 6–13 replicates for each condition. B, Effect of TSPO knock-down on ROS production was evaluated using the H2DCFDA assay. TSPO knock-down in BV2 cells significantly increased ROS production induced by LPS treatment. *Comparison with control for which p < 0.05, Mann–Whitney test, n = 28–33 replicates per condition. C, TSPO knock-down significantly increased TNF-α mRNA expression in BV2 cells treated with LPS (black bars). *Comparison with control for which p < 0.05, Mann–Whitney test, n = 8 replicates for each condition. D, Effect of TSPO knock-down on TNF-α protein production was evaluated in conditioned media using ELISA and normalized to protein concentration of cell lysates. TSPO knock-down significantly increased TNF-α protein production in BV2 cells treated with LPS (black bars). *p < 0.05, unpaired t test, n = 8–16 replicates for each condition. E, Effect of TSPO knock-down on BV2 microglia proliferation was measured by the incorporation of EdU. Although TSPO knock-down did not increase BV2 microglia proliferation under normal conditions, it significantly increased BV2 microglia proliferation in LPS-treated cells. *p < 0.05, Mann–Whitney test, n = 24–27 replicates per condition. Scale bar indicates 50 μm.
TTN-mediated TSPO signaling reduces retinal inflammatory responses in vivo
To assess the effect of TTN-mediated TSPO signaling in vivo, we induced retinal inflammation in experimental animals using an intravitreal injection of LPS (estimated final vitreous concentration of 0.05 mg/ml) and compared the effects in eyes that were either coinjected with TTN (estimated final vitreous concentration: 50 μm) or injected with LPS alone. The controls included eyes that were injected with PBS (containing the equivalent concentration of DMSO solvent) and eyes injected with TTN alone (without added LPS). Experimental animals were euthanized and retinal tissue analyzed 2 d after injection. The production of ROS in the retina was evaluated by measuring TBARS formation, which reflects lipid peroxidation induced by oxidative stress. Although TTN injected alone in the absence of LPS-induced inflammation did not alter lipid peroxidation relative to PBS-injected controls, TTN injection in the presence of LPS significantly reduced retinal lipid peroxidation to close to control levels (Fig. 9A). Although LPS injection alone markedly increased TNF-α protein levels, coinjection of TTN significantly reduced them (Fig. 9B). These data indicate that TTN-mediated TSPO signaling effectively reduced retinal inflammatory responses in vivo, likely by the downregulation of microglial activation responses, as was observed in vitro.
Figure 9.

Effects of TTN in vivo. A, ROS produced in retina was evaluated by measurement of lipid peroxidation using a TBARS assay. Two days after intravitreal LPS injection (1 mg/ml, 1 μl), significantly increased lipid peroxidation in the retina was observed. Although the intravitreal injection of TTN did not affect the lipid peroxidation alone, it effectively suppressed the increase of lipid peroxidation induced by LPS. *p < 0.05, paired t test, n = 8–12 replicates for each condition. B, Two days after intravitreal LPS injection (1 mg/ml, 1 μl), the protein level of TNF-α significantly increased (black bar). The intravitreal injection of TTN significantly suppressed the increase of LPS-induced TNF-α in the retina. *p < 0.05, paired t test, n = 3–7 replicates for each condition.
Discussion
Retinal expression of TSPO during development and injury
Inflammatory and neurodegenerative diseases of the retina share common features with those afflicting the brain (Ohno-Matsui, 2011; Sivak, 2013), including chronic neuroinflammation and microglial activation (Gupta et al., 2003; Karlstetter et al., 2010). However, inflammatory mechanisms may also demonstrate specializations that are unique to different CNS regions (Glass et al., 2010). Although TSPO is a key protein marker of neurodegeneration-associated gliosis in the brain (Chen and Guilarte, 2008), its expression has not been previously examined in the retina. Therefore, we were interested in examining the expression of TSPO within the retina in healthy development, in adulthood, and in the context of injury and elucidating the endogenous functions of TSPO. In our study, we discovered that TSPO expression in the retina demonstrates developmental regulation; at P0, TSPO was expressed in amoeboid, deramified microglia extravasating into the neural retina (Santos et al., 2008), but was subsequently turned off as microglia acquired a mature ramified morphology in adulthood. This transient TSPO expression may be related to developmental functions of retinal microglia, such as complement-mediated synapse elimination (Stevens et al., 2007) and apoptosis regulation (Thanos, 1991). Because TSPO has been linked to phagocytic function (Choi et al., 2011), early TSPO expression may regulate the microglial engulfment of synapses and apoptotic neurons. TSPO may also confer transient migratory capabilities to microglia colonizing the retina during development (Cuadros and Navascués, 2001) and become downregulated when microglia attain their final positions.
In the uninjured adult retina, TSPO mRNA and protein were undetectable in glia and neurons on immunohistochemistry, resembling findings in the uninjured brain parenchyma (Cosenza-Nashat et al., 2009). We found here that TSPO expression was markedly but transiently increased by retinal inflammation in separate animal injury models and declined with time as markers of inflammation subsided after injury. In all models examined, TSPO upregulation was present in F4/80+, CD68+ activated microglia/macrophages, but absent in macroglia (astrocytes and Müller glia). In comparison, TSPO upregulation in the injured brain was observed in both microglia and astrocytes to varying extents, depending on the nature of the injury and the final severity of associated neurodegeneration (Maeda et al., 2007; Ji et al., 2008; Cosenza-Nashat et al., 2009). The absence of TSPO upregulation in retinal macroglia may be related to unique specializations of Müller radial glia that regulate gliosis using mechanisms distinct from those in brain astrocytes (Bringmann and Wiedemann, 2012). The microglial predominance of TSPO expression in retinal injury and its temporal correlation with active inflammation make TSPO a suitable molecular biomarker for microglial activation in retinal diseases. Compared with molecular imaging techniques used in the brain (Cagnin et al., 2007; Venneti et al., 2013), the unique accessibility of the retina to direct visualization creates opportunities for monitoring TSPO expression as a biomarker of microglial activation at high spatial and temporal resolutions. Current in vivo retinal imaging techniques capable of visualizing apoptotic biomarkers on the level of individual cells (Barnett et al., 2009; Galvao et al., 2013) may be used to closely monitor activation in individual microglia in specific retinal locations. These observations may be instrumental in discerning inflammatory mechanisms in retinal disease and may provide useful outcome measures for the clinical evaluation of immunomodulatory treatments.
TTN, a DBI-derived peptide, negatively regulates microglial activation via TSPO
In addition to its use as a biomarker, TSPO has been explored as a therapeutic target for neuroinflammatory and neurodegenerative diseases (Rupprecht et al., 2010). Synthetic ligands to TSPO (PK-11195, Ro5–4864, and etifoxine) have been shown to decrease microglia/macrophage activation in vitro (Choi et al., 2002; Choi et al., 2011) and in animal models of disease, including excitotoxic injury (Ryu et al., 2005), axonal injury (Girard et al., 2008), multiple sclerosis (Daugherty et al., 2013), and Alzheimer's disease (Barron et al., 2013). However, the functional significance of increased microglial TSPO expression and the signaling mechanisms involved in this response are unclear.
We examined the expression and distribution of DBI, an endogenous ligand to TSPO, in the retina under normal conditions and after the induction of retinal inflammation. Previous studies have found DBI expression in the brain, primarily in glial populations (Alho et al., 1991; Yanase et al., 2002). Previous studies have also demonstrated that primary macroglia in culture can secrete DBI into the extracellular DBI secretion medium via a brefeldin A-insensitive unconventional secretion pathway (Lafon-Cazal et al., 2003; Loomis et al., 2010) in response to a number of stimuli, including neural activity (Barmack et al., 2004), neuromediators such as GABA (Patte et al., 1999), somatostatin (Masmoudi et al., 2005), and steroids (Loomis et al., 2010). Specifically, primary Müller cells can express and secrete DBI in response to retinal inflammation, as shown here, and in response to depolarizing stimuli (Qian et al., 2008). In the retina, we found DBI to be expressed during development and adulthood in both retinal macroglial populations, astrocytes and Müller glia. In our endotoxin-induced model of retinal inflammation, we discovered that DBI expression after LPS administration was transiently upregulated in retinal macroglia, akin to TSPO expression in retinal microglia. These concordant “waves” of increased expression of this ligand-protein pair suggested a coordinated form of retinal macroglia-microglia signaling after the induction of retinal inflammation.
Although DBI has been demonstrated to bind to TSPO and activate its signaling directly (Guidotti et al., 1983; Garnier et al., 1993), it is also proteolytically cleaved into several biologically active peptides, including TTN (Slobodyansky et al., 1989), in the extracellular space by extracellular trypsin-like protease activity (Loomis et al., 2010). TTN selectively binds to TSPO (Berkovich et al., 1990), activating TSPO-mediated effects, including neurosteroid biosynthesis (Papadopoulos et al., 1991; Do-Rego et al., 1998; Lesouhaitier et al., 1998). We found that TTN decreased multiple markers of microglial activation induced by LPS in vitro and reduced markers of retinal inflammation in vivo. Converse phenotypes were found when microglial TSPO expression was knocked-down, supporting the notion that TTN-mediated effects on microglial suppression were mediated via TSPO, likely at the level of mitochondria. We have further found that similar effects were obtained when TTN was substituted with the synthetic ligands,PK11195 or Ro5–44864, which have been implicated as sharing a common binding mode within the ligand-binding site (Cinone et al., 2000; Lacapère and Papadopoulos, 2003). We found that PK11195 differed from Ro5-44864 (and TTN) in being not as effective in decreasing TNFα expression in cultured microglia, a difference analogous to those previously documented in vivo (Veiga et al., 2005; Soustiel et al., 2008) and which may be attributable to differences in binding sites and also the partial agonist or antagonistic effects of PK11195 (Le Fur et al., 1983). Because TSPO likely has distinct sites for ligand-binding and cholesterol binding (i.e., at the cholesterol recognition/interaction amino acid consensus [CRAC] domain; Li and Papadopoulos, 1998), the effects of ligand binding may act upstream to influence cholesterol binding and/or transport at the CRAC domain to facilitate subsequent neurosteroid production (Midzak et al., 2011). These results indicate that the therapeutic potential ascribed to synthetic ligands of TSPO (Rupprecht et al., 2010; Barron et al., 2013; Daugherty et al., 2013) may be extended to endogenous ligands such as TTN, and even possibly to ligands that modulate the function of the CRAC domain (Midzak et al., 2011; Lecanu et al., 2013), in measures to control microglia activation in chronic neuroinflammation.
How endogenous TTN-mediated signaling to TSPO results in decreases in microglial activation is incompletely understood, but likely involves, at least in part, the production of neurosteroids. TTN has been demonstrated to stimulate steroid production in endocrine cells (Berkovich et al., 1990; Papadopoulos et al., 1991) and hypothalamic steroidogenic neurons (Do-Rego et al., 1998). Recent studies have demonstrated the role of nuclear receptors, including estrogen receptor-β (ERβ) ligands in the negative regulation of microglial activation (Saijo et al., 2013). Neurosteroids such as 5-androstene-3β,7β-diol and its precursor, DHEA, have been shown to bind to ERβ in microglia and inhibit microglial activation via the suppression of AP-1-mediated transcription (Saijo et al., 2011). Therefore, TTN-mediated TSPO signaling in microglia can increase mitochondrial cholesterol flux, facilitating the production of modulatory neurosteroids that in turn repress inflammatory genes in a cell-autonomous manner. The detection of increased DHEA levels in microglia treated with TTN here supports this hypothesized mechanism. In addition, neurosteroids including allopregnanolone, DHEA, and Δ5PS have been found to modulate allosterically multiple neurotransmitter receptors, such as GABAA, NMDA, and AMPA receptors (Rupprecht and Holsboer, 1999; Belelli and Lambert, 2005; Mameli et al., 2005). Increased neurosteroid synthesis can thus induce changes in neurotransmission, consequently affecting the activation state of microglia (Lee, 2013) either via direct reception (Pocock and Kettenmann, 2007) or indirect modulation of overall neuronal activity (Fontainhas et al., 2011), thus facilitating an alternative non-cell-autonomous mechanism underlying neurosteroid effects on microglia. Endogenous TSPO signaling may also result in other physiological changes in microglia beyond neurosteroid synthesis, including regulation of calcium influx (Hong et al., 2006), mitochondrial function (Casellas et al., 2002), and apoptosis (Hirsch et al., 1998), all of which may potentially modulate aspects of microglial activation.
Macroglia-microglia interactions via TSPO signaling shape retinal inflammatory responses
Effective immune responses rely on rapid and robust microglial activation for pathogen clearance and tissue repair. However, this prompt “on” phase also needs to be followed by an appropriate and timely downregulation of activation responses and a reestablishment of the quiescent state. Activation patterns that are excessive in magnitude or prolonged in duration are detrimental to tissue homeostasis and are thought to contribute to chronic neuroinflammatory states in neurodegenerative disease (Lull and Block, 2010). Although some microglial cell-surface receptors (e.g., CX3CR1 and CD200R) serve as constitutive negative regulators of activation (Kierdorf and Prinz, 2013), dynamically expressed regulatory molecules may be required to shape the temporal features of activation and deactivation. The synchronously regulated expression of TSPO in retinal microglia and DBI in retinal macroglia provides a mechanism of coordinated macroglia-microglia interactions that drive the resolution phase shortly after an injury/inflammation response is generated. In the retina, physical microglia-macroglia interactions increase after activation as microglial processes reorient to fasciculate closely with the radial Müller cells processes (Wang et al., 2011), promoting intercellular signaling. TSPO may thus represent a prominent example of reciprocal modulatory interactions hypothesized to exist between macroglia and microglia that influence the overall beneficial-versus-detrimental outcome of inflammatory responses (Gao et al., 2013). These intercellular molecular interactions can therefore constitute novel targets for interventions that are aimed, not at completely eliminating immune function, but at modulating the temporal features of immune responses to avoid excessive or chronic activation.
It is interesting that the expression of DBI in the retina, unlike that for TSPO, is constitutively expressed by astrocytes and Müller glia, suggesting the presence of constitutive non-TSPO signaling. In the brain, DBI and its peptide products act as allosteric modulators of GABAA receptor function (Costa and Guidotti, 1991), modulating neuronal activity and synaptic plasticity in ways that influence epileptic discharge (Christian et al., 2013), learning and memory (Siiskonen et al., 2007), and behaviors including anxiety (De Mateos-Verchere et al., 1998), aggression (Kavaliers and Hirst, 1986), and feeding (Do-Rego et al., 2007). In the retina, constitutive DBI signaling under normal conditions can influence GABAA receptor-mediated visual processing (Barmack et al., 2004); how these GABAergic effects feedback onto microglia to influence their function in retinal inflammation is a current topic of study in our laboratory (Fontainhas et al., 2011).
In conclusion, we show here that TSPO and its endogenous ligand, DBI, are expressed in the retinal microglia and macroglia, respectively, and are regulated during retinal development and in different phases of the inflammatory response. TSPO signaling in microglia, which is mediated via its endogenous ligand, TTN, is capable of suppressing features of microglial activation in the retina in vitro and in vivo. Regulated microglial TSPO expression in the retina suggests its suitability for use as a biomarker in retinal diseases visualizable using high-resolution retinal imaging techniques. Coordinated TSPO and DBI signaling in retinal microglia and macroglia, respectively, constitute an endogenous and dynamic immodulatory mechanism that may be targeted in treatments to alleviate chronic neuroinflammation underlying retinal disease.
Footnotes
This study was supported by funds from the National Eye Institute Intramural Research Program, National Institutes of Health.
The authors declare no competing financial interests.
References
- Alho H, Harjuntausta T, Schultz R, Pelto-Huikko M, Bovolin P. Immunohistochemistry of diazepam binding inhibitor (DBI) in the central nervous system and peripheral organs: its possible role as an endogenous regulator of different types of benzodiazepine receptors. Neuropharmacology. 1991;30:1381–1386. doi: 10.1016/S0028-3908(11)80005-5. [DOI] [PubMed] [Google Scholar]
- Barmack NH, Bilderback TR, Liu H, Qian Z, Yakhnitsa V. Activity-dependent expression of acyl-coenzyme a-binding protein in retinal muller glial cells evoked by optokinetic stimulation. J Neurosci. 2004;24:1023–1033. doi: 10.1523/JNEUROSCI.3936-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett EM, Zhang X, Maxwell D, Chang Q, Piwnica-Worms D. Single-cell imaging of retinal ganglion cell apoptosis with a cell-penetrating, activatable peptide probe in an in vivo glaucoma model. Proc Natl Acad Sci U S A. 2009;106:9391–9396. doi: 10.1073/pnas.0812884106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron AM, Garcia-Segura LM, Caruso D, Jayaraman A, Lee JW, Melcangi RC, Pike CJ. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer's disease. J Neurosci. 2013;33:8891–8897. doi: 10.1523/JNEUROSCI.1350-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Berkovich A, McPhie P, Campagnone M, Guidotti A, Hensley P. A natural processing product of rat diazepam binding inhibitor, triakontatetraneuropeptide (diazepam binding inhibitor 17–50) contains an alpha-helix, which allows discrimination between benzodiazepine binding site subtypes. Mol Pharmacol. 1990;37:164–172. [PubMed] [Google Scholar]
- Bringmann A, Wiedemann P. Muller glial cells in retinal disease. Ophthalmologica. 2012;227:1–19. doi: 10.1159/000328979. [DOI] [PubMed] [Google Scholar]
- Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol. 2011;95:14–25. doi: 10.1016/j.pneurobio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Kassiou M, Meikle SR, Banati RB. Positron emission tomography imaging of neuroinflammation. Neurotherapeutics. 2007;4:443–452. doi: 10.1016/j.nurt.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi ME, Gordon AY, Penn JS, Jayagopal A. Molecular imaging of retinal disease. J Ocul Pharmacol Ther. 2013;29:275–286. doi: 10.1089/jop.2012.0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40:475–486. doi: 10.1016/S0197-0186(01)00118-8. [DOI] [PubMed] [Google Scholar]
- Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1–17. doi: 10.1016/j.pharmthera.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HB, Khoo C, Ryu JK, van Breemen E, Kim SU, McLarnon JG. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-alpha and [Ca2+]i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J Neurochem. 2002;83:546–555. doi: 10.1046/j.1471-4159.2002.01122.x. [DOI] [PubMed] [Google Scholar]
- Choi J, Ifuku M, Noda M, Guilarte TR. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia. 2011;59:219–230. doi: 10.1002/glia.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Herbert AG, Holt RL, Peng K, Sherwood KD, Pangratz-Fuehrer S, Rudolph U, Huguenard JR. Endogenous positive allosteric modulation of GABA(A) receptors by diazepam binding inhibitor. Neuron. 2013;78:1063–1074. doi: 10.1016/j.neuron.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinone N, Hötje HD, Carotti A. Development of a unique 3D interaction model of endogenous and synthetic peripheral benzodiazepine receptor ligands. Journal of Computer-Aided Molecular Design. 2000;14:753–768. doi: 10.1023/A:1008168127539. [DOI] [PubMed] [Google Scholar]
- Congdon N, O'Colmain B, Klaver CC, Klein R, Muñoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P Eye Diseases Prevalence Research G. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, Lee SC. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–328. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E, Guidotti A. Diazepam binding inhibitor (DBI): a peptide with multiple biological actions. Life Sci. 1991;49:325–344. doi: 10.1016/0024-3205(91)90440-M. [DOI] [PubMed] [Google Scholar]
- Cuadros MA, Navascués J. Early origin and colonization of the developing central nervous system by microglial precursors. Prog Brain Res. 2001;132:51–59. doi: 10.1016/S0079-6123(01)32065-4. [DOI] [PubMed] [Google Scholar]
- Daugherty DJ, Selvaraj V, Chechneva OV, Liu XB, Pleasure DE, Deng W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med. 2013;5:891–903. doi: 10.1002/emmm.201202124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mateos-Verchere JG, Leprince J, Tonon MC, Vaudry H, Costentin J. The octadecaneuropeptide ODN induces anxiety in rodents: possible involvement of a shorter biologically active fragment. Peptides. 1998;19:841–848. doi: 10.1016/S0196-9781(98)00037-0. [DOI] [PubMed] [Google Scholar]
- Do-Rego JC, Orta MH, Leprince J, Tonon MC, Vaudry H, Costentin J. Pharmacological characterization of the receptor mediating the anorexigenic action of the octadecaneuropeptide: evidence for an endozepinergic tone regulating food intake. Neuropsychopharmacology. 2007;32:1641–1648. doi: 10.1038/sj.npp.1301280. [DOI] [PubMed] [Google Scholar]
- Do-Rego JL, Mensah-Nyagan AG, Feuilloley M, Ferrara P, Pelletier G, Vaudry H. The endozepine triakontatetraneuropeptide diazepam-binding inhibitor [17–50] stimulates neurosteroid biosynthesis in the frog hypothalamus. Neuroscience. 1998;83:555–570. doi: 10.1016/s0306-4522(97)00362-x. [DOI] [PubMed] [Google Scholar]
- Ferrero P, Santi MR, Conti-Tronconi B, Costa E, Guidotti A. Study of an octadecaneuropeptide derived from diazepam binding inhibitor (DBI): biological activity and presence in rat brain. Proc Natl Acad Sci U S A. 1986;83:827–831. doi: 10.1073/pnas.83.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontainhas AM, Wang M, Liang KJ, Chen S, Mettu P, Damani M, Fariss RN, Li W, Wong WT. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One. 2011;6:e15973. doi: 10.1371/journal.pone.0015973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvao J, Davis BM, Cordeiro MF. In vivo imaging of retinal ganglion cell apoptosis. Curr Opin Pharmacol. 2013;13:123–127. doi: 10.1016/j.coph.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhu Q, Zhang Y, Zhao Y, Cai L, Shields CB, Cai J. Reciprocal modulation between microglia and astrocyte in reactive gliosis following the CNS injury. Mol Neurobiol. 2013;48:690–701. doi: 10.1007/s12035-013-8460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier M, Boujrad N, Oke BO, Brown AS, Riond J, Ferrara P, Shoyab M, Suarez-Quian CA, Papadopoulos V. Diazepam binding inhibitor is a paracrine/autocrine regulator of Leydig cell proliferation and steroidogenesis: action via peripheral-type benzodiazepine receptor and independent mechanisms. Endocrinology. 1993;132:444–458. doi: 10.1210/endo.132.1.8380386. [DOI] [PubMed] [Google Scholar]
- Girard C, Liu S, Cadepond F, Adams D, Lacroix C, Verleye M, Gillardin JM, Baulieu EE, Schumacher M, Schweizer-Groyer G. Etifoxine improves peripheral nerve regeneration and functional recovery. Proc Natl Acad Sci U S A. 2008;105:20505–20510. doi: 10.1073/pnas.0811201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Forchetti CM, Corda MG, Konkel D, Bennett CD, Costa E. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc Natl Acad Sci U S A. 1983;80:3531–3535. doi: 10.1073/pnas.80.11.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471. doi: 10.1016/S0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- Hirsch T, Decaudin D, Susin SA, Marchetti P, Larochette N, Resche-Rigon M, Kroemer G. PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp Cell Res. 1998;241:426–434. doi: 10.1006/excr.1998.4084. [DOI] [PubMed] [Google Scholar]
- Hong SH, Choi HB, Kim SU, McLarnon JG. Mitochondrial ligand inhibits store-operated calcium influx and COX-2 production in human microglia. J Neurosci Res. 2006;83:1293–1298. doi: 10.1002/jnr.20829. [DOI] [PubMed] [Google Scholar]
- Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, Zhang MR, Suzuki K, Ando K, Staufenbiel M, Trojanowski JQ, Lee VM, Higuchi M, Suhara T. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer's and other CNS pathologies. J Neurosci. 2008;28:12255–12267. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology. 2010;215:685–691. doi: 10.1016/j.imbio.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Kavaliers M, Hirst M. An octadecaneuropeptide (ODN) derived from diazepam binding inhibitor increases aggressive interactions in mice. Brain Res. 1986;383:343–349. doi: 10.1016/0006-8993(86)90037-5. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacapère JJ, Papadopoulos V. Peripheral-type benzodiazepine receptor: structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids. 2003;68:569–585. doi: 10.1016/S0039-128X(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Adjali O, Galéotti N, Poncet J, Jouin P, Homburger V, Bockaert J, Marin P. Proteomic analysis of astrocytic secretion in the mouse. Comparison with the cerebrospinal fluid proteome. J Biol Chem. 2003;278:24438–24448. doi: 10.1074/jbc.M211980200. [DOI] [PubMed] [Google Scholar]
- Langmann T. Microglia activation in retinal degeneration. J Leukocyte Biol. 2007;81:1345–1351. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- Lecanu L, Yao ZX, McCourty A, Sidahmed el-K, Orellana ME, Burnier MN, Papadopoulos V. Control of hypercholesterolemia and atherosclerosis using the cholesterol recognition/interaction amino acid sequence of the translocator protein TSPO. Steroids. 2013;78:137–146. doi: 10.1016/j.steroids.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. Neurotransmitters and microglial-mediated neuroinflammation. Curr Protein Pept Sci. 2013;14:21–32. doi: 10.2174/1389203711314010005. [DOI] [PubMed] [Google Scholar]
- Le Fur G, Vaucher N, Perrier ML, Flamier A, Benavides J, Renault C, Dubroeucq MC, Guérémy C, Uzan A. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5–4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 1983;33:449–457. doi: 10.1016/0024-3205(83)90794-4. [DOI] [PubMed] [Google Scholar]
- Lesouhaitier O, Feuilloley M, Vaudry H. Effect of the triakontatetraneuropeptide (TTN) on corticosteroid secretion by the frog adrenal gland. J Mol Endocrinol. 1998;20:45–53. doi: 10.1677/jme.0.0200045. [DOI] [PubMed] [Google Scholar]
- Li H, Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport–identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–4997. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- Loomis WF, Behrens MM, Williams ME, Anjard C. Pregnenolone sulfate and cortisol induce secretion of acyl-CoA-binding protein and its conversion into endozepines from astrocytes. J Biol Chem. 2010;285:21359–21365. doi: 10.1074/jbc.M110.105858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS One. 2009;4:e7945. doi: 10.1371/journal.pone.0007945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007;1157:100–111. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]
- Mameli M, Carta M, Partridge LD, Valenzuela CF. Neurosteroid-induced plasticity of immature synapses via retrograde modulation of presynaptic NMDA receptors. J Neurosci. 2005;25:2285–2294. doi: 10.1523/JNEUROSCI.3877-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masmoudi O, Gandolfo P, Tokay T, Leprince J, Ravni A, Vaudry H, Tonon MC. Somatostatin down-regulates the expression and release of endozepines from cultured rat astrocytes via distinct receptor subtypes. J Neurochem. 2005;94:561–571. doi: 10.1111/j.1471-4159.2005.03076.x. [DOI] [PubMed] [Google Scholar]
- Midzak A, Akula N, Lecanu L, Papadopoulos V. Novel androstenetriol interacts with the mitochondrial translocator protein and controls steroidogenesis. J Biol Chem. 2011;286:9875–9887. doi: 10.1074/jbc.M110.203216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothdurfter C, Baghai TC, Schüle C, Rupprecht R. Translocator protein (18 kDa) (TSPO) as a therapeutic target for anxiety and neurologic disorders. Eur Arch Psychiatry Clin Neurosci. 2012;262:S107–S112. doi: 10.1007/s00406-012-0352-5. [DOI] [PubMed] [Google Scholar]
- Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer's disease. Prog Retin Eye Res. 2011;30:217–238. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Berkovich A, Krueger KE, Costa E, Guidotti A. Diazepam binding inhibitor and its processing products stimulate mitochondrial steroid biosynthesis via an interaction with mitochondrial benzodiazepine receptors. Endocrinology. 1991;129:1481–1488. doi: 10.1210/endo-129-3-1481. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapère JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Patte C, Gandolfo P, Leprince J, Thoumas JL, Fontaine M, Vaudry H, Tonon MC. GABA inhibits endozepine release from cultured rat astrocytes. Glia. 1999;25:404–411. doi: 10.1002/(SICI)1098-1136(19990215)25:4<404::AID-GLIA9>3.0.CO%3B2-Q. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Politis M, Su P, Piccini P. Imaging of microglia in patients with neurodegenerative disorders. Front Pharmacol. 2012;3:96. doi: 10.3389/fphar.2012.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Bilderback TR, Barmack NH. Acyl coenzyme A-binding protein (ACBP) is phosphorylated and secreted by retinal Muller astrocytes following protein kinase C activation. J Neurochem. 2008;105:1287–1299. doi: 10.1111/j.1471-4159.2008.05229.x. [DOI] [PubMed] [Google Scholar]
- Rone MB, Fan J, Papadopoulos V. Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim Biophys Acta. 2009;1791:646–658. doi: 10.1016/j.bbalip.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht R, Holsboer F. Neuropsychopharmacological properties of neuroactive steroids. Steroids. 1999;64:83–91. doi: 10.1016/S0039-128X(98)00101-9. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Choi HB, McLarnon JG. Peripheral benzodiazepine receptor ligand PK11195 reduces microglial activation and neuronal death in quinolinic acid-injected rat striatum. Neurobiol Dis. 2005;20:550–561. doi: 10.1016/j.nbd.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Crotti A, Glass CK. Regulation of microglia activation and deactivation by nuclear receptors. Glia. 2013;61:104–111. doi: 10.1002/glia.22423. [DOI] [PubMed] [Google Scholar]
- Santos AM, Calvente R, Tassi M, Carrasco MC, Martín-Oliva D, Marín-Teva JL, Navascués J, Cuadros MA. Embryonic and postnatal development of microglial cells in the mouse retina. J Comp Neurol. 2008;506:224–239. doi: 10.1002/cne.21538. [DOI] [PubMed] [Google Scholar]
- Siiskonen H, Oikari S, Korhonen VP, Pitkänen A, Voikar V, Kettunen M, Hakumäki J, Wahlfors T, Pussinen R, Penttonen M, Kiehne K, Kaasinen SK, Alhonen L, Jänne J, Herzig KH. Diazepam binding inhibitor overexpression in mice causes hydrocephalus, decreases plasticity in excitatory synapses and impairs hippocampus-dependent learning. Mol Cell Neurosci. 2007;34:199–208. doi: 10.1016/j.mcn.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Sivak JM. The aging eye: common degenerative mechanisms between the Alzheimer's brain and retinal disease. Invest Ophthalmol Vis Sci. 2013;54:871–880. doi: 10.1167/iovs.12-10827. [DOI] [PubMed] [Google Scholar]
- Slobodyansky E, Guidotti A, Wambebe C, Berkovich A, Costa E. Isolation and characterization of a rat brain triakontatetraneuropeptide, a posttranslational product of diazepam binding inhibitor: specific action at the Ro 5–4864 recognition site. J Neurochem. 1989;53:1276–1284. doi: 10.1111/j.1471-4159.1989.tb07425.x. [DOI] [PubMed] [Google Scholar]
- Soustiel JF, Zaaroor M, Vlodavsky E, Veenman L, Weizman A, Gavish M. Neuroprotective effect of Ro5–4864 following brain injury. Exp Neurol. 2008;214:201–208. doi: 10.1016/j.expneurol.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15:5263–5274. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Tang Z, Zhang S, Lee C, Kumar A, Arjunan P, Li Y, Zhang F, Li X. An optic nerve crush injury murine model to study retinal ganglion cell survival. J Vis Exp. 2011;pii:2685. doi: 10.3791/2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos S. The relationship of microglial cells to dying neurons during natural neuronal cell death and axotomy-induced degeneration of the rat retina. Eur J Neurosci. 1991;3:1189–1207. doi: 10.1111/j.1460-9568.1991.tb00054.x. [DOI] [PubMed] [Google Scholar]
- Tokay T, Masmoudi O, Gandolfo P, Leprince J, Pelletier G, Vaudry H, Tonon MC. Beta-amyloid peptides stimulate endozepine biosynthesis in cultured rat astrocytes. J Neurochem. 2005;94:607–616. doi: 10.1111/j.1471-4159.2005.03102.x. [DOI] [PubMed] [Google Scholar]
- Veiga S, Azcoitia I, Garcia-Segura LM. Ro5-4864, a peripheral benzodiazepine receptor ligand, reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J Neurosci Res. 2005;80:129–137. doi: 10.1002/jnr.20430. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. Molecular imaging of microglia/macrophages in the brain. Glia. 2013;61:10–23. doi: 10.1002/glia.22357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohra R, Tsai JC, Kolko M. The role of inflammation in the pathogenesis of glaucoma. Surv Ophthalmol. 2013;58:311–320. doi: 10.1016/j.survophthal.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive Muller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation. 2011;8:173. doi: 10.1186/1742-2094-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanase H, Shimizu H, Yamada K, Iwanaga T. Cellular localization of the diazepam binding inhibitor in glial cells with special reference to its coexistence with brain-type fatty acid binding protein. Arch Histol Cytol. 2002;65:27–36. doi: 10.1679/aohc.65.27. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ma W, Fariss RN, Wong WT. Minocycline attenuates photoreceptor degeneration in a mouse model of subretinal hemorrhage microglial: inhibition as a potential therapeutic strategy. Am J Pathol. 2011;179:1265–1277. doi: 10.1016/j.ajpath.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]