Phosphorylation of Myosin II-interacting Guanine Nucleotide Exchange Factor (MyoGEF) at Threonine 544 by Aurora B Kinase Promotes the Binding of Polo-like Kinase 1 to MyoGEF (original) (raw)
Background: Aurora B kinase (aurora B) and polo-like kinase 1 (Plk1) are critical regulators of cytokinesis.
Results: Phosphorylation of MyoGEF at Thr-544 by aurora B promotes the binding of Plk1 to MyoGEF.
Conclusion: Phosphorylation of MyoGEF by aurora B creates a docking site for Plk1.
Significance: Coordinated regulation of MyoGEF localization and activation by aurora B and Plk1 contributes to the regulation of cytokinesis.
Keywords: Cytokinesis, Guanine Nucleotide Exchange Factor (GEF), Mitosis, Phosphorylation, Rho GTPases, MyoGEF, Aurora B Kinase, Central Spindle, Plk1
Abstract
We previously reported that phosphorylation of myosin II-interacting guanine nucleotide exchange factor (MyoGEF) by polo-like kinase 1 (Plk1) promotes the localization of MyoGEF to the central spindle and increases MyoGEF activity toward RhoA during mitosis. In this study we report that aurora B-mediated phosphorylation of MyoGEF at Thr-544 creates a docking site for Plk1, leading to the localization and activation of MyoGEF at the central spindle. In vitro kinase assays show that aurora B can phosphorylate MyoGEF. T544A mutation drastically decreases aurora B-mediated phosphorylation of MyoGEF in vitro and in transfected HeLa cells. Coimmunoprecipitation and in vitro pulldown assays reveal that phosphorylation of MyoGEF at Thr-544 enhances the binding of Plk1 to MyoGEF. Immunofluorescence analysis shows that aurora B colocalizes with MyoGEF at the central spindle and midbody during cytokinesis. Suppression of aurora B activity by an aurora B inhibitor disrupts the localization of MyoGEF to the central spindle. In addition, T544A mutation interferes with the localization of MyoGEF to the cleavage furrow and decreases MyoGEF activity toward RhoA during mitosis. Taken together, our results suggest that aurora B coordinates with Plk1 to regulate MyoGEF activation and localization, thus contributing to the regulation of cytokinesis.
Introduction
Polo-like kinase 1 (Plk1)2 plays a central role in controlling multiple events during cell cycle progression, such as centrosome amplification and maturation, mitotic exit, mitotic spindle assembly, spindle pole maintenance, and cytokinesis (1). It has been demonstrated that Plk1 associates with its substrates by binding to a docking site (S-pS or S-pT) that is created by the phosphorylation mediated by priming kinases (2, 3). Plk1 docking sites often overlap with Cdk1 consensus sites ((pS/pT)P_X_(K/R)), and Cdk1 can act as a priming kinase to generate Plk1 docking sites on a number of proteins, such as INCENP, vimentin, GRASP65, Cep55, Nir2, Bub1, MYPT1, and PICH (4–11). However, Cdk1 is inactivated at anaphase onset (12). Therefore, other priming kinases are likely to be responsible for creating docking sites that allow Plk1 to bind to its substrates at the central spindle during anaphase (13).
Aurora kinase also belongs to a class of serine/threonine kinases. There are three members of the aurora kinase family (aurora A, B, and C) in mammalian cells (14). Aurora B kinase (aurora B) plays a critical role in the regulation of chromosome segregation and cytokinesis (15). Perturbation of aurora B function leads to defects in chromosome alignment, segregation, and cytokinesis (16–18). Plk1 and aurora B continuously remains active at the central spindle during anaphase (13, 19–22); both Plk1 and aurora B can phosphorylate a number of cytokinesis regulators that localize to the central spindle (18, 19, 23–26).
RhoA is a key regulator of actin-myosin contractility (27–30). It can activate Rho kinase that, in turn, inhibits myosin phosphatase and directly phosphorylates myosin light chain, thus leading to an increase in myosin contractile activity (31). Activation of RhoA and an increase in myosin contractile activity are required for the initiation and progression of furrow ingression (32–34). It is generally thought that Ect2, a guanine nucleotide exchange factor (GEF) for RhoA, is a key activator of RhoA at the cleavage furrow during cytokinesis (20, 34–38). However, it has also been shown that GEF-H1 (a RhoGEF) contributes to RhoA activation at the cleavage furrow (39). More recently, a RhoGEF termed leukemia-associated RhoGEF (LARG) is involved in the late stages of cytokinesis (40). Furthermore, our previous findings have shown that another RhoGEF, MyoGEF, is implicated in equatorial RhoA activation during cytokinesis (41, 42). In addition, Plk1 can phosphorylate MyoGEF at Thr-574 and promote the activation and localization of MyoGEF at the central spindle (43). In this study we provide evidence demonstrating that aurora B can phosphorylate MyoGEF at Thr-544, creating a docking site for Plk1.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa cells (ATCC) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. A double thymidine block was used to synchronize cells at mitosis as described previously (43). Briefly, the transfected HeLa cells were treated with 2 mm thymidine for 24 h, cultured in thymidine-free medium for 10 h, and treated with 2 mm thymidine for 16 h followed by “release” to progress through the cell cycle in thymidine-free medium for 12 h. Lipofectamine was used for transfection with plasmids or siRNA according to the manufacturer's instructions (Invitrogen). For transfection with plasmids, the transfected cells were analyzed ∼24 h after transfection. For transfection with siRNA, the transfected cells were analyzed ∼48–72 h after transfection. The siRNA for human aurora B (5′-CGCGGCACUUC ACAAUUGATT-3′) (44) was purchased from Dharmacon. To disrupt aurora B activation, HeLa cells were treated with vehicle (DMSO; Sigma) or ZM447439 (10 μm; Tocris Bioscience) for 30–60 min.
Plasmid Constructs
Plasmids encoding Myc- and GFP-tagged MyoGEF were generated as described previously (41, 43). To generate plasmids encoding His- and GST-tagged MyoGEF, the human MyoGEF cDNA was cloned into pDEST26 and pDEST27 vectors, respectively, according to the manufacturer's instructions (Invitrogen). The MyoGEF mutant T544A was generated by site-directed mutagenesis according to the manufacturer's instructions (Agilent Technologies) and confirmed by DNA sequencing (Eurofins MWG Operon). A plasmid encoding GFP-RhoA was obtained from Addgene (45).
Expression and Purification of Recombinant Polypeptides
A bacterial expression system was used to express GST- and His-tagged recombinant polypeptides. BL21 bacterial cells expressing GST-tagged recombinant polypeptides were resuspended in PBS and homogenized by sonication. Triton X-100 was added to a final concentration of 1%. The bacterial lysates were then incubated by shaking at 23 °C for 1 h. A glutathione-conjugated agarose (Sigma) column was used to purify GST-tagged recombinant polypeptides. The proteins were eluted with 100 mm Tris-HCl (pH 8.0), 10 mm glutathione and dialyzed against 50 mm Tris-HCl (pH 7.5), 50 mm NaCl. BL21 bacterial cells expressing His-tagged recombinant polypeptides were resuspended in 50 mm sodium phosphate (pH 8.0), 300 mm NaCl and homogenized by sonication. A nickel-nitrilotriacetic acid-agarose (Qiagen) column was used to purify His-tagged recombinant polypeptides. The proteins were eluted with 50 mm sodium phosphate (pH 8.0), 300 mm NaCl, 250 mm imidazole and dialyzed against 50 mm Tris-HCl (pH 7.5), 50 mm NaCl.
GST Pulldown Assays
GST pulldown assays were done as described previously (42). Briefly, the immobilized GST-MyoGEF polypeptides were incubated with _in vitro_-translated Myc-aurora B overnight at 4 °C. After washing four times with binding buffer (50 mm Tris-HCl (pH 7.4), 100 mm NaCl, 0.05% Triton X-100, 10% glycerol, 0.2 mm EDTA, and 1 mm DTT), the beads were resuspended in SDS loading buffer to elute the bound proteins. In vitro translated Myc-aurora B was synthesized using the TNT SP6 quick-coupled transcription/translation system (Promega, Madison, WI) according to the manufacturer's instructions.
Coimmunoprecipitation Assays
Coimmunoprecipitation assays were carried out as described previously (46, 47). Briefly, transfected HeLa cells were lysed in radioimmune precipitation lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 2.5% sodium deoxycholate, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm Na3VO4, 1 mm NaF) for 10 min on ice. Cell extracts were collected and precleared with protein A/G-agarose beads. The precleared lysate was incubated with agarose-conjugated anti-Myc antibody overnight at 4 °C. After 3 washes with radioimmune precipitation lysis buffer, the bound proteins were eluted with SDS loading buffer.
Immunoblot Analysis
Cell lysates or immunoprecipitates were separated on 10% or 4–12% SDS-PAGE gels (Bio-Rad), transferred to an Immobilon-P transfer membrane (Millipore), blocked in 5% nonfat milk, and incubated with primary antibodies as indicated. The following primary antibodies were used: mouse anti-Myc (1:1000, 9E10, catalogue number sc-40, Santa Cruz), mouse anti-Plk1 (1:1000, 3F8, catalogue number, sc-53751, Santa Cruz), rabbit anti-aurora B (1:2000, catalogue number, sc-25426, Santa Cruz), rabbit anti-phosphohistone 3 (1:1000, catalogue number, 06-570, Millipore), rabbit anti-phosphothreonine (1:500, catalogue number 71-8200, Invitrogen), rabbit anti-GFP (1:1000, catalogue number sc-8334, Santa Cruz), rabbit anti-β-tubulin (1:2000, catalogue number sc-9104, Santa Cruz), and rabbit anti-MyoGEF (1:250) (41). After 3 washes, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (1:5000, Santa Cruz) for 1 h at 23 °C and visualized by SuperSignal West Pico Luminol/Enhancer solution (Pierce).
Immunofluorescence Analysis
HeLa cells grown on coverslips were fixed with methanol for 12 min at −20 °C. After blocking with 1% bovine serum albumin for 1 h at 23 °C, the fixed HeLa cells were incubated with primary antibodies as indicated for 3 h at 23 °C or overnight at 4 °C followed by incubation with secondary antibodies for 40 min at 23 °C. The primary antibodies used for immunofluorescence analysis were mouse monoclonal anti-MyoGEF antibody (medium of antibody-producing hybridoma cells; without dilution) and rabbit polyclonal aurora B antibody (1:5000, catalogue number sc-25426; Santa Cruz). The mouse monoclonal anti-MyoGEF antibody was generated using the C-terminal 290 amino acid residues of human MyoGEF as antigen. The hybridoma cells for anti-MyoGEF antibody were produced by Promab Biotechnologies, Inc. (Richmond, CA). The secondary antibodies Alexa Fluor 594 goat anti-mouse IgG (1:500) and Alexa Fluor 488 goat anti-rabbit IgG (1:500) were purchased from Invitrogen. Nuclei were visualized by 4′,6-diamidino-2-phenylindole (DAPI; Sigma). The coverslips were mounted using a Prolong antifade kit (Invitrogen). Images were collected using the Nikon TiE Perfect Focus Digital Fluorescence Imaging System (Morrell Instrument Company, Inc.) with an Andor Zyla sCMOS 2560 × 2160 camera.
Biotinylated Peptide Pulldown Assays and Far Western Blot Analysis
Biotinylated peptides (biotin-SPSTRPS(pT)PSLEGSQ and biotin-SPSTRPS(T)PSLEGSQ; both peptides contain amino acid residues 537–551 from MyoGEF) were purchased from Sigma. The peptide pulldown assay was done as described previously (48). Briefly, 20 μg of biotinylated peptides were incubated with 10 μg of GST-Plk1-PBD overnight in peptide binding buffer (50 mm Tris-HCl at pH 7.5, 150 mm NaCl, 1 mm EDTA, 2 mm dithiothreitol, 0.05% Nonidet P-40). Twenty-five microliters of streptavidin-agarose beads (Vector Laboratories) were added and incubated by shaking at 23 °C for 2 h. The beads were washed 5 times with 500 μl of peptide binding buffer on a rotator for 1 min per each washing. Twenty microliters of SDS-PAGE gel loading buffer were added to streptavidin-agarose beads and boiled for 5 min. The samples were separated on 4–12% SDS-PAGE gels (Bio-Rad) and subjected to immunoblot analysis with an antibody specific for Plk1 (1:1000; Santa Cruz). Far Western blot analysis was done as described previously (23).
In Vitro Kinase Assays
For the aurora B in vitro kinase assay, 5 μg of purified GST-tagged MyoGEF polypeptides were incubated with 1 μg of recombinant His-aurora B (Invitrogen) in kinase buffer (5 mm MOPS (pH 7.2), 2.5 mm β-glycerophosphate, 1 mm EGTA, 4 mm MgCl2, 0.05 mm DTT, 250 μm ATP) and 1 μCi of [γ-32P]ATP. The reaction mixtures (50 μl) were incubated at 30 °C for 30 min and resolved on SDS-PAGE gels. The gels were dried and subjected to autoradiography.
In Vitro Guanine Nucleotide Exchange Analysis
The GEF exchange assay was conducted using a fluorescence-based spectrometry analysis, which measured the incorporation of a fluorescence analog of GTP (_N_-methylanthraniloyl (mant)-GTP) onto the small GTPase RhoA. Briefly, HeLa cells transfected with an empty vector or plasmids encoding GST-MyoGEF-WT or GST-MyoGEF-T544A were enriched in mitosis by release from double thymidine block. Glutathione-conjugated agarose beads were used to precipitate GST-tagged recombinant proteins from the transfected HeLa cells. The beads were washed 4 times with lysis buffer (50 mm Tris-HC1 (pH 7.5), 1 mm dithiothreitol, 100 mm NaCl, 0.5% Triton X-100). 10 μg of His-tagged RhoA (Cytoskeleton, Inc.) were equilibrated in exchange buffer containing 40 mm Tris-HCl (pH 7.5), 100 mm NaCl, 20 mm MgCl2, 1 mm dithiothreitol, 100 μg/ml BSA, and 800 nm mant-GTP (Invitrogen). After taking a few measurements of steady readings at 20 °C using a SpectraMax® i3 Multi-Mode Microplate reader (Molecular Devices), 20 μl of empty vector, GST-MyoGEF-WT, or GST-MyoGEF-T544A beads were added, and relative fluorescence of mant-GTP (excitation λ, 360 nm; emission λ, 440 nm) was monitored.
Rhotekin Pulldown Assay
The rhotekin pulldown assay was carried out as described previously (41).
RESULTS
Phosphorylation of MyoGEF at Thr-544 by Aurora B
Our previous studies have shown that Plk1 can phosphorylate MyoGEF at Thr-574 (43). Plk1 contains an N-terminal kinase domain and a C-terminal polo-box domain (PBD). The PBD can recognize and bind to a docking site on the substrates. In turn, the kinase domain catalyzes the phosphorylation of the target proteins (2, 3). Consensus Plk1 docking sites contain a phosphothreonine or phosphoserine that is preceded by a serine (S(pS/pT)) (3). To identify Plk1 docking sites on MyoGEF, we used NetPhos 2.0 Server to analyze the predicted phosphorylation sites in the MyoGEF sequence to identify any potential phosphothreonines or phosphoserines that are preceded by a serine. We found that there are four potential Plk1 docking sites on MyoGEF, i.e. Thr-544, Ser-553, Ser-697, and Ser-712.
It is well established that aurora B is a mitotic kinase that remains active throughout anaphase and plays a pivotal role in the regulation of cytokinesis regulators (19). Thus, we carried out in vitro kinase assays to examine whether aurora B could phosphorylate MyoGEF. As shown in Fig. 1, aurora B could phosphorylate MyoGEF fragments MyoGEF-392–565-WT and MyoGEF-392–780-WT (Fig. 1B, lanes 1 and 3 in the upper panels). Importantly, T544A mutation almost completely abrogated the phosphorylation of MyoGEF fragments by aurora B (Fig. 1B; compare lane 1 with lane 2 or lane 3 with lane 4 in the upper panels), indicating that aurora B can predominantly phosphorylate Thr-544, a potential Plk1 docking site. Therefore, we further analyzed the phosphorylation of MyoGEF at Thr-544 by aurora B and determined whether Thr-544 could serve as a docking site for Plk1.
FIGURE 1.
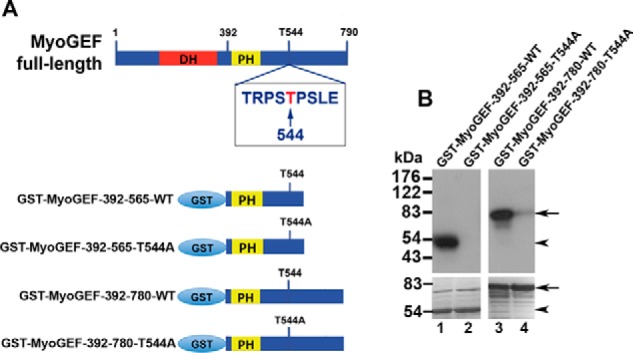
In vitro phosphorylation of MyoGEF at Thr-544 by aurora B. A, schematic diagram of GST-MyoGEF polypeptides. The numbers indicate the amino acids. T544, threonine 544. DH, Dbl-homology domain. B, in vitro phosphorylation of MyoGEF by aurora B. GST-tagged MyoGEF polypeptides were incubated with or without purified aurora B in the presence of [γ-32P]ATP. The proteins were separated by SDS-PAGE and visualized by autoradiography (upper panel) or Coomassie Blue staining (lower panel).
To confirm the phosphorylation of Thr-544 by aurora B, a MyoGEF fragment (His-MyoGEF-501–790) was phosphorylated in vitro by aurora B and subjected to mass spectrometry analysis. Phosphorylation of Thr-544 by aurora B was confirmed by mass spectrometry analysis (supplemental Fig. S1). This analysis also revealed several additional aurora B sites: Thr-540, Ser-543, Ser-546, Thr-620, Ser-645, and Ser-697 (Supplemental Fig. S1). It should be noted that Thr-540, Thr-544, and Ser-697 are all preceded by a serine, indicating that they can potentially act as Plk1 docking sites. However, our result that T544A mutation almost completely abrogated the phosphorylation (Fig. 1B) indicates Thr-544 as the major phosphorylation site on MyoGEF by aurora B. In addition, dual phosphorylation at Ser-543/Thr-544 or at Thr-544/Ser-546 was not revealed by mass spectrometry analysis.
We then examined whether Thr-544 could be phosphorylated during mitosis. HeLa cells transfected with plasmids encoding Myc-MyoGEF-WT or Myc-MyoGEF-T544A were enriched in mitosis as described under “Experimental Procedures.” Coimmunoprecipitation assays were carried out to examine whether the T544A mutation decreased the phosphorylation of MyoGEF in the transfected HeLa cells. As shown in Fig. 2A, phosphorylation of Myc-MyoGEF-WT in the transfected HeLa cells could be detected by immunoblot analysis with a phospho-Thr antibody (anti-p-Thr; Fig. 2A; lane 3 in the upper panel). However, T544A mutation drastically decreased the Thr phosphorylation of MyoGEF in the transfected HeLa cells (Fig. 2A; lane 6 in the upper panel), suggesting that Thr-544 is phosphorylated during mitosis. Next, we asked whether aurora B could phosphorylate MyoGEF during mitosis. HeLa cells were transfected with a plasmid encoding Myc-MyoGEF-WT together with control or aurora B siRNA. The transfected HeLa cells were enriched in mitosis and subjected to coimmunoprecipitation assays with anti-Myc antibody. Fig. 2B shows that depletion of aurora B by RNA interference (RNAi) drastically decreased the Thr phosphorylation of Myc-MyoGEF-WT (Fig. 2B; compare lane 3 with lane 6 in the upper panel). Therefore, our results indicate that Thr-544 can be phosphorylated by aurora B during mitosis. The arrest of HeLa cells in mitosis was confirmed by immunoblot analysis with an antibody specific for phosphorylated histone 3 as shown in Fig. 2C. Depletion of aurora B by RNAi was confirmed by immunoblot analysis with an antibody specific for aurora B (Fig. 2D). As shown in Fig. 2D, ∼80% of aurora B were depleted by RNAi.
FIGURE 2.
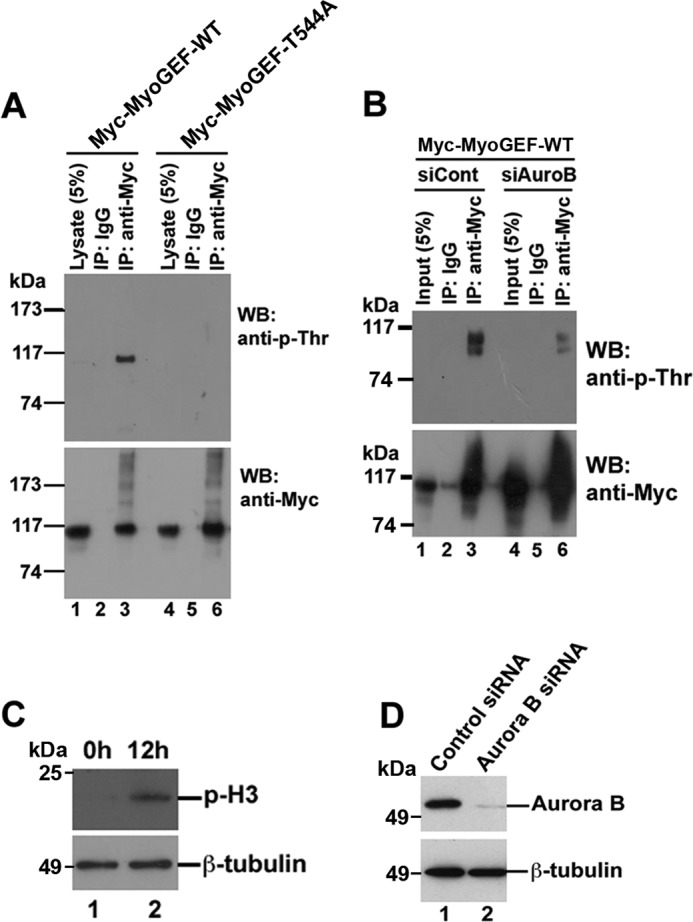
Phosphorylation of MyoGEF at Thr-544 in transfected HeLa cells. A, HeLa cells were transfected with plasmids encoding Myc-MyoGEF-WT or Myc-MyoGEF-T544A and enriched in mitosis as described under “Experimental Procedures.” Anti-Myc-conjugated agarose was used to precipitate Myc-tagged proteins from transfected cell lysates followed by immunoblot (WB) analysis with anti-phosphothreonine antibody (anti-p-Thr). IP, immunoprecipitation. B, depletion of aurora B decreases threonine-phosphorylation of MyoGEF in transfected HeLa cells. HeLa cells were transfected with a plasmid encoding Myc-MyoGEF-WT and control siRNA (siCont; lanes 1–3) or aurora B siRNA (siAuroB; lanes 4–6). The transfected HeLa cells were enriched in mitosis as described under “Experimental Procedures” and subjected to coimmunoprecipitation assays with normal control IgG or anti-Myc antibody followed by immunoblot analysis with anti-phosphothreonine antibody (anti-p-Thr). C, arrest of HeLa cells in mitosis. At 12 h after release from double thymidine block, HeLa cells were subjected to immunoblot analysis with antibodies specific for phosphohistone 3 (p-H3; upper panel) or β-tubulin (lower panel). D, depletion of aurora B by RNAi. HeLa cells transfected with aurora B siRNA were enriched in mitosis and subjected to immunoblot analysis with antibodies specific for aurora B (upper panel) or β-tubulin (lower panel).
It should be noted that Cdk1/cyclin B often acts as a primase that can phosphorylate Plk1 substrates and create docking sites for Plk1 (3). Therefore, we also examined whether Cdk1/cyclin B could phosphorylate MyoGEF at Thr-544. In vitro kinase assays revealed that Cdk1/Cyclin B could phosphorylate MyoGEF-392–780. However, T544A or S697A mutation did not substantially interfere with Cdk1 phosphorylation of the MyoGEF fragment (data not shown), indicating that Thr-544 and Ser-697 are unlikely the major sites for Cdk1 on MyoGEF. However, it remains to be determined whether two other potential Plk1 docking sites (Ser-553 and Ser-712) can be phosphorylated by Cdk1.
Phosphorylation of Thr-544 Increases the Binding of Plk1 to MyoGEF
Our previous studies have shown that Plk1 can bind to MyoGEF and phosphorylate MyoGEF at Thr-574 (43). Our findings in this study demonstrate that aurora B can phosphorylate MyoGEF at Thr-544, a potential Plk1 docking site (Figs. 1 and 2). Therefore, we asked whether phosphorylation of MyoGEF at Thr-544 by aurora B could promote the binding of Plk1 to MyoGEF. To this end, we carried out a pulldown assay to examine whether the PBD of Plk1 could bind specifically to the phosphorylated Thr-544 motif on MyoGEF. The PBD was used for the binding assay because it has been shown that this domain can recognize and bind to a docking site on Plk1 substrates (2). Bacterially expressed GST-Plk1-PBD was purified using a glutathione-affinity column (Fig. 3A). Two biotin-labeled peptides that contain phosphorylated or non-phosphorylated Thr-544 as well as the surrounding amino acid residues (biotin-SPSTRPS(pT)PSLEGSQ and biotin-SPSTRPS(T)PSLEGSQ; both peptides contain amino acid residues 537–551 from MyoGEF) were incubated with an equal amount of GST-Plk1-PBD. Streptavidin-conjugated beads were used to pull down biotin-labeled peptides. As shown in Fig. 3C, the biotin-labeled peptide containing phospho-Thr-544 (biotin-SPSTRPS(pT)PSLEGSQ) co-precipitated with more GST-Plk1-PBD as compared with the biotin-labeled peptide containing non-phosphorylated Thr-544 (biotin-SPSTRPS(T)PSLEGSQ) (Fig. 3C; compare lane 3 with lane 4). Consistent with this finding, Far Western blot analysis also revealed that aurora B-mediated phosphorylation of His-MyoGEF-501–790 increased its interaction with GST-Plk1-PBD (Fig. 3E; compare lane 1 with lane 2 in the upper panel).
FIGURE 3.
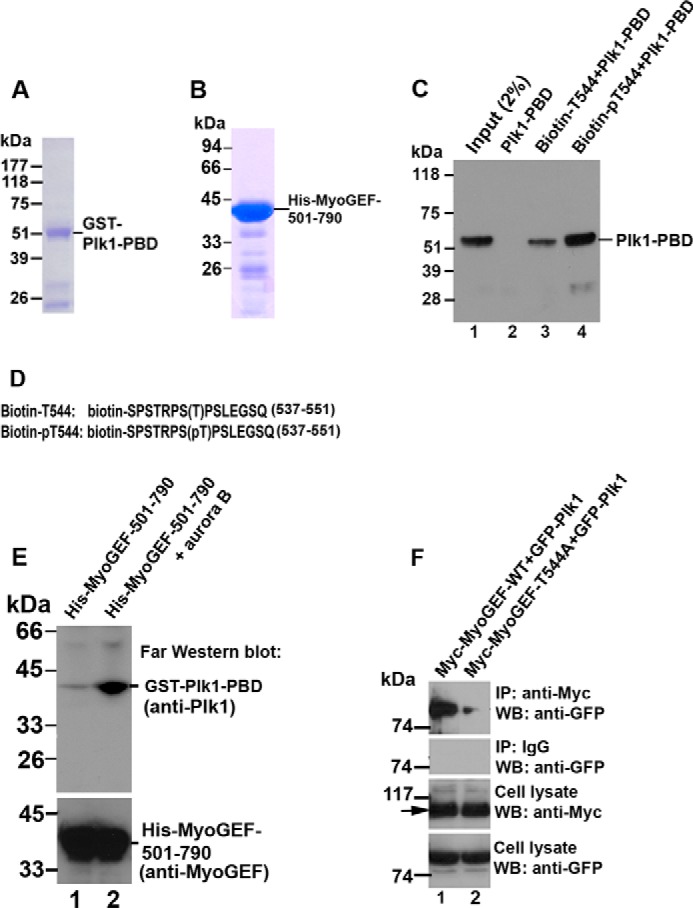
Phosphorylation of Thr-544 increases the binding of Plk1 to MyoGEF. A, purified GST-tagged Plk1-PBD that was used in the peptide binding assay in C and in the Far Western blot assay in E. B, purified His-tagged MyoGEF fragment (His-MyoGEF-501–790) that was used in the Far Western blot assay in E. C, biotinylated peptides containing phosphorylated threonine (biotin-pT544; lane 4) or non-phosphorylated threonine (biotin-T544; lane 3) were incubated with GST-Plk1-PBD and subjected to pulldown assays with streptavidin-conjugated beads followed by immunoblot analysis with an antibody specific for Plk1. D, the amino acid sequences of the biotinylated-peptides (biotin-T544 and biotin-pT544) that were used in the peptide-binding assay in C. The numbers 537–551 indicate the amino acid residues from MyoGEF. E, Far Western blot analysis of the Plk1-MyoGEF interaction. In vitro phosphorylation of His-MyoGEF-501–790 was carried out in the absence (lane 1) or presence (lane 2) of aurora B. The treated polypeptides were separated by SDS-PAGE, transferred to a PVDF membrane, incubated with Plk1-PBD, and blotted with anti-Plk1 antibody. F, the non-phosphorylatable mutation T544A decreases the binding of Plk1 to MyoGEF. HeLa cells were transfected with plasmids encoding GFP-Plk1 and Myc-MyoGEF-WT or Myc-MyoGEF-T544A. The transfected cells were enriched in mitosis as described under “Experimental Procedures.” Anti-Myc-conjugated agarose was used to precipitate Myc-MyoGEF-WT or Myc-MyoGEF-T544A from the transfected cell lysate followed by immunoblot (WB) analysis with an antibody specific for GFP. Arrows indicate Myc-MyoGEF-WT (lane 1) or Myc-MyoGEF-T544A (lane 2). IP, immunoprecipitation.
We then asked whether phosphorylation of MyoGEF at Thr-544 increased the interaction between MyoGEF and Plk1 during mitosis. To this end we examined whether T544A mutation interfered with the interaction between MyoGEF and Plk1 in transfected HeLa cells. HeLa cells transfected with plasmids encoding GFP-Plk1 and Myc-MyoGEF-WT or Myc-MyoGEF-T544A were enriched in mitosis and subjected to coimmunoprecipitation assays with an antibody specific for Myc. As shown in Fig. 3F, GFP-Plk1 co-precipitated with Myc-MyoGEF-WT (Fig. 3F; lane 1 in the top panel). However, decreased amounts of GFP-Plk1 co-precipitated with Myc-MyoGEF-T544A (Fig. 3F; compare lane 1 with lane 2 in the top panel). Therefore, our results demonstrate that Thr-544 phosphorylation increases the binding of Plk1 to MyoGEF, suggesting that phosphorylation of MyoGEF at Thr-544 by aurora B creates a docking site for Plk1.
Aurora B Promotes MyoGEF Localization to the Central Spindle
It is known that aurora B localizes to the central spindle and plays a pivotal role in regulating cytokinesis (15). Our previous studies have shown that MyoGEF also localizes to the central spindle and cleavage furrow (41–43). Thus, we examined whether aurora B and MyoGEF colocalized to the central spindle. HeLa cells were fixed with methanol and subjected to immunofluorescence analysis with antibodies specific for aurora B and MyoGEF. As shown in Fig. 4, aurora B and MyoGEF colocalized to the central spindle and midbody (Fig. 4; arrowheads in panels a–l). This finding also led us to examine whether aurora B interacts with MyoGEF. HeLa cells transfected with plasmids encoding GFP-MyoGEF and Myc-aurora B were enriched in mitosis and subjected to coimmunoprecipitation assays with an antibody specific for Myc. As shown in Fig. 5A, GFP-MyoGEF co-precipitated with Myc-aurora B (Fig. 5A; lane 3 in the upper panel), suggesting that aurora B can interact with MyoGEF. GST pulldown assays were also carried out to examine the direct interaction between MyoGEF and aurora B. Three GST-MyoGEF fragments that were used in the GST pulldown assay are as follows; MyoGEF-71–388 contains the Dbl homology domain (amino acid residues 154–351), and MyoGEF-392–565 contains the pleckstrin homology (PH) domain (amino acid residues 410–500), whereas MyoGEF-392–780 contains the PH domain as well as most of the C-terminal region (Fig. 5, B and C). As shown in Fig. 5D, GST-MyoGEF-392–565 and GST-MyoGEF-392–780 (but not GST-MyoGEF-71–388) strongly interacted with _in vitro_-translated Myc-aurora B (Fig. 5D; compare lanes 3 and 5 with lane 4), suggesting that aurora B can bind to a region corresponding to amino acid residues 392–565 of MyoGEF. However, it is still unclear whether the PH domain (amino acid residues 410–500) of MyoGEF is responsible for MyoGEF interaction with aurora B.
FIGURE 4.
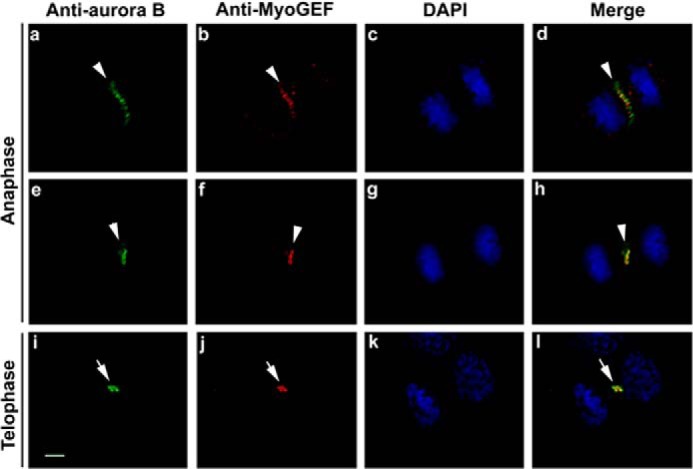
Colocalization of MyoGEF and aurora B at the central spindle and midbody. HeLa cells were fixed with methanol and subjected to immunofluorescence analysis with antibodies specific for MyoGEF (arrowheads in panels b, f, and j; red) and aurora B (arrowheads in panels a, e, and i; green). The nuclei were stained with DAPI (panels c, g, and k; blue). Bar, 5 μm.
FIGURE 5.
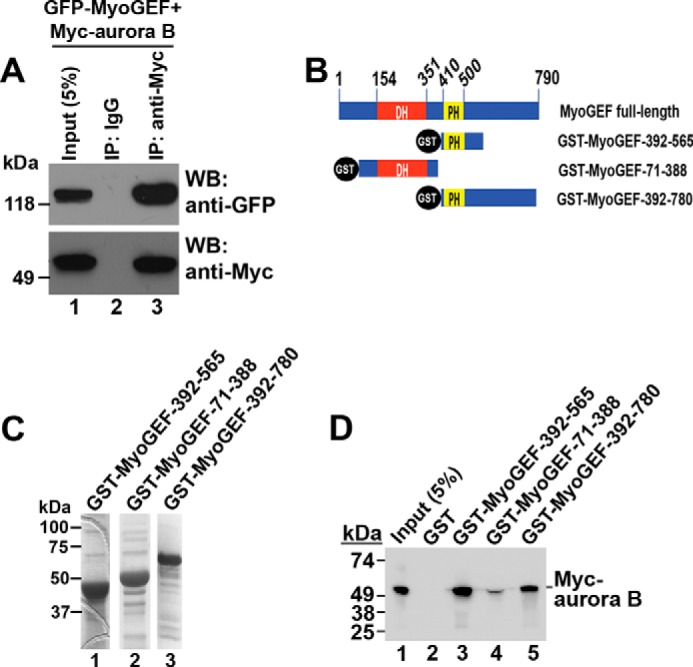
Binding of aurora B to MyoGEF. A, coimmunoprecipitation (IP) of Myc-aurora B and GFP-MyoGEF from transfected HeLa cells. HeLa cells transfected with plasmids encoding Myc-aurora B and GFP-MyoGEF were subjected to coimmunoprecipitation assays with normal IgG (lane 2) or anti-Myc antibody (lane 3). WB, Western blot. B, schematic diagram of GST-MyoGEF polypeptides. The numbers indicate the amino acids. DH, Dbl-homology domain. C, purified GST-tagged MyoGEF fragments that were used in GST pulldown assays in D. D, GST pulldown assays. GST-tagged MyoGEF fragments were used to precipitate _in vitro_-translated Myc-aurora B.
Our previous studies have demonstrated that Plk1 promotes the localization of MyoGEF to the central spindle (43). Our results in this study further indicate that phosphorylation of Thr-544 by aurora B creates a docking site for Plk1 (Fig. 3). Therefore, we asked whether phosphorylation of MyoGEF at Thr-544 by aurora B increased the localization of MyoGEF to the central spindle. The aurora B inhibitor ZM447439 has been successfully used to block the biological function of aurora B (16, 50, 51). To assess the importance of aurora B-mediated phosphorylation of MyoGEF in localizing MyoGEF to the central spindle, HeLa cells treated with DMSO or ZM447439 were fixed and stained for aurora B, MyoGEF, and nuclei (DAPI). As shown in Fig. 6, in HeLa cells treated with DMSO, MyoGEF colocalized with aurora B to the central spindle (Fig. 6; arrowheads in panels a–d). Conversely, in HeLa cells treated with ZM447439 (ZM), neither aurora B nor MyoGEF localized to the central spindle (Fig. 6; arrowheads in panels e–h). This finding suggests that aurora B plays a critical role in localizing MyoGEF to the central spindle.
FIGURE 6.
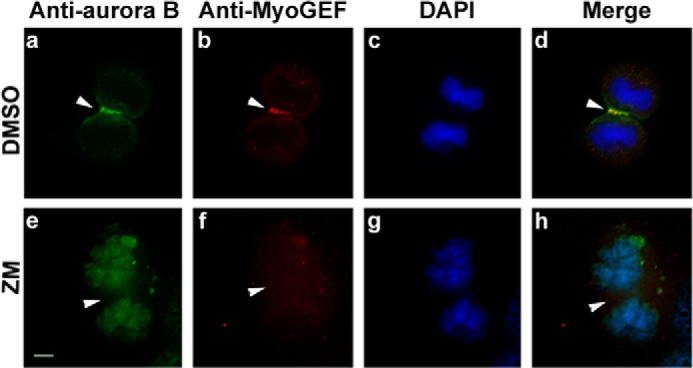
Aurora B is required for MyoGEF localization to the central spindle. HeLa cells were treated with DMSO (a–d) or the aurora B inhibitor ZM447439 (e–h) and subjected to immunofluorescence analysis with antibodies specific for aurora B (arrowheads in panels a and e) and MyoGEF (arrowheads in panels b and f). The nuclei were stained with DAPI (panels c and g). The merged images are shown in panels d and h. Bar, 5 μm. ZM, ZM447439.
It has been demonstrated that aurora B also plays a central role in the assembly of the central spindle. In turn, the formation of the central spindle leads to the localization of numerous cytokinesis regulators to the central spindle (15). Therefore, it was possible that inhibition of aurora B by ZM447439 interfered with the formation of the central spindle and thus indirectly disrupted MyoGEF localization. To confirm that Thr-544 phosphorylation was implicated in promoting the localization of MyoGEF to the central spindle, we examined the localization of GFP-MyoGEF-T544A during cytokinesis. HeLa cells transfected with plasmids encoding GFP-MyoGEF-WT or GFP-MyoGEF-T544A were fixed and stained for nuclei (DAPI). As shown in Fig. 7, GFP-MyoGEF-WT concentrated at the cleavage furrow. Arrowheads in Fig. 7, panel a, indicate the localization of GFP-MyoGEF-WT at the cleavage furrow. However, GFP-MyoGEF-T544A was diffusely localized to the cleavage furrow and the pole regions during anaphase (Fig. 7, panel b). Therefore, our results suggest that phosphorylation of MyoGEF at Thr-544 by aurora B can promote the localization of MyoGEF to the central spindle and cleavage furrow.
FIGURE 7.
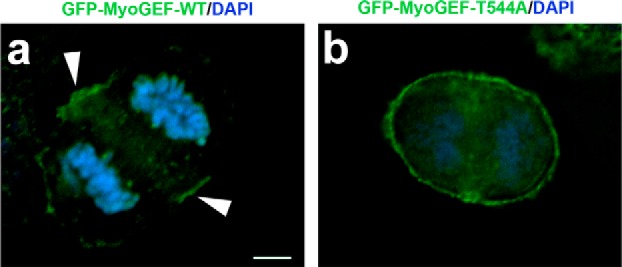
Non-phosphorylatable mutation T544A decreases the localization of MyoGEF to the cleavage furrow. HeLa cells transfected with a plasmid encoding GFP-MyoGEF-WT or GFP-MyoGEF-T544A were fixed with 3.7% of paraformaldehyde and subjected to immunofluorescence analysis. The nuclei were stained with DAPI. Unlike the generally cortical localization of GFP-MyoGEF-T544A (panel b), the localization of GFP-MyoGEF-WT at the cleavage furrow is evident (arrowheads in panel a). Bar, 5 μm.
Phosphorylation of Thr-544 Increases MyoGEF Activity toward RhoA
Our previous studies have shown that phosphorylation of MyoGEF at Thr-574 by Plk1 enhances MyoGEF activity toward RhoA (43). Our results in this study indicate that aurora B can phosphorylate MyoGEF at Thr-544 and that this phosphorylation increases the binding of Plk1 to MyoGEF (Fig. 3). Therefore, we asked whether phosphorylation of Thr-544 by aurora B had an impact on MyoGEF activity toward RhoA. A plasmid encoding GFP-RhoA was co-transfected into HeLa cells with an empty vector or with plasmids encoding His-MyoGEF-WT or His-MyoGEF-T544A. The transfected HeLa cells were enriched in mitosis and subjected to rhotekin pulldown assays to measure the levels of activated GFP-RhoA. Consistent with our previous findings (43, 52), exogenous expression of His-MyoGEF-WT in HeLa cells increased the levels of activated GFP-RhoA (Fig. 8A; compare lane 1 with lane 2 in the top panel). Conversely, HeLa cells transfected with plasmids encoding GFP-RhoA and His-MyoGEF-T544A expressed a lower level of activated GFP-RhoA (Fig. 8A; compare lane 2 with lane 3 in the top panel). This finding suggests that T544A mutation decreases the ability of MyoGEF to activate RhoA during mitosis.
FIGURE 8.
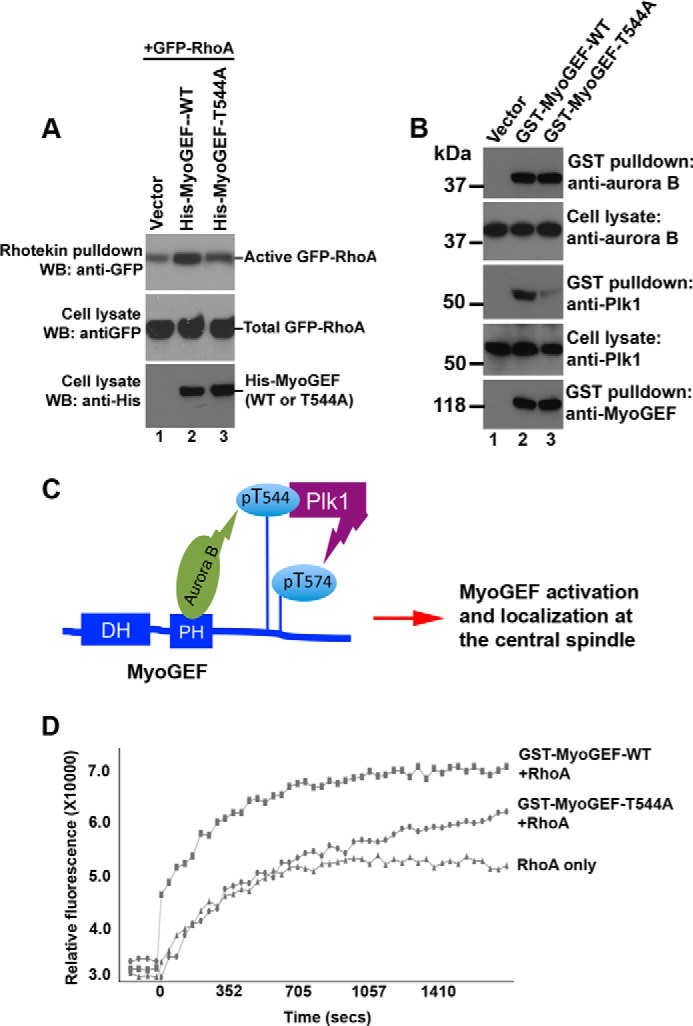
Phosphorylation of Thr-544 increases MyoGEF activity toward RhoA. A, Rhotekin pulldown assays were carried out to precipitate activated RhoA from HeLa cells transfected with a plasmid encoding GFP-RhoA plus an empty vector (lane 1) or a plasmid encoding His-MyoGEF-WT (lane 2) or His-MyoGEF-T544A (lane 3). WB, Western blot. B, glutathione-conjugated beads were used to precipitate GST-tagged proteins from HeLa cells transfected with an empty vector (lane 1) or a plasmid encoding GST-MyoGEF-WT (lane 2) or GST-MyoGEF-T544A (lane 3). Cell lysates and GST pulldown proteins were separated by SDS-PAGE followed by immunoblot analysis with antibodies as indicated. C, a schematic diagram showing that sequential phosphorylation of MyoGEF by aurora B and Plk1 leads to the activation and localization of MyoGEF at the central spindle. DH, Dbl-homology domain. D, the glutathione-conjugated beads in B were used in an in vitro GEF exchange assay (as described under “Experimental Procedures”) to assess the impact of T544A mutation on MyoGEF activity toward RhoA.
To further confirm the role of Thr-544 phosphorylation in promoting Plk1-MyoGEF interactions and MyoGEF activity toward RhoA during mitosis, we also examined whether aurora B and Plk1 co-precipitated with GST-MyoGEF-WT or GST-MyoGEF-T544A from transfected HeLa cells. The precipitated GST-MyoGEF-WT and GST-MyoGEF-T544A were then used in an in vitro GEF exchange assay to assess the impact of T544A mutation on MyoGEF-mediated activation of RhoA. HeLa cells were transfected with plasmids encoding GST-MyoGEF-WT or GST-MyoGEF-T544A and enriched in mitosis. Glutathione-conjugated beads were used to precipitate GST-MyoGEF-WT and GST-MyoGEF-T544A from the transfected HeLa cells (Fig. 8B; lanes 2 and 3 in the bottom panel). As shown in Fig. 8B, both aurora B and Plk1 co-precipitated with GST-MyoGEF-WT (Fig. 8B; lane 2 in top four panels). In addition, aurora B also co-precipitated with GST-MyoGEF-T544A (Fig. 8B; lane 3 in the top two panels). However, only small amounts of Plk1 co-precipitated with GST-MyoGEF-T544A (Fig. 8B; lane 3 in the second and third panels from the bottom). These findings further confirm that aurora B can bind to MyoGEF and phosphorylate MyoGEF at Thr-544, creating a docking site for Plk1. In turn, Plk1 binds to the docking site (i.e. phospho-Thr-544) and phosphorylates Thr-574 (Fig. 8C) (43).
The glutathione-conjugated beads that associated with GST-MyoGEF-WT or GST-MyoGEF-T544A were then used in an in vitro GEF exchange assay (as described under “Experimental Procedures”). In this GEF exchange assay, an activated RhoGEF should enhance the incorporation of a fluorescence analog of GTP (_N_-methylanthraniloyl (mant)-GTP) onto RhoA, thus increasing the fluorophore emission intensity. As shown in Fig. 8D, GST-MyoGEF-WT (but not GST-MyoGEF-T544A) beads substantially increased the fluorescence intensity, suggesting that T544A mutation decreases MyoGEF activity toward RhoA during mitosis. Taken together, our results suggest that phosphorylation of MyoGEF at Thr-544 contributes to RhoA activation during mitosis.
DISCUSSION
In this study we have demonstrated that aurora B can phosphorylate MyoGEF at Thr-544. In addition, phosphorylation of MyoGEF at Thr-544 increases the binding of Plk1 to MyoGEF, promotes the localization of MyoGEF to the central spindle, and enhances MyoGEF activity toward RhoA. Our results suggest that aurora B coordinates with Plk1 to regulate MyoGEF activation and localization during cytokinesis (Fig. 8C).
Plk1 has been shown to play a central role in controlling the progression and completion of cytokinesis (1). The timely localization of Plk1 to the central spindle during anaphase leads to the recruitments of RhoGEFs to the central spindle, resulting in the activation of RhoA and the assembly of the actomyosin contractile ring (20, 22, 32–34, 36, 43). Localization of myosin at the cleavage furrow and the binding of myosin to actin are required for furrow formation and ingression (54). It has been shown that Plk1 can bind and phosphorylate Ect2 (36). Inhibition of Plk1 activity by small-molecule inhibitors prevents localization of Ect2 to the central spindle and abolishes equatorial RhoA localization and contractile ring formation (20, 22). We have also shown previously that Plk1 can phosphorylate MyoGEF at Thr-574 and promotes MyoGEF activation and localization at the central spindle (43). Plk1 associates with its substrates by binding to a docking site on the substrates (2, 3). The main goal of this study was to identify the Plk1 docking site on MyoGEF. We found that phosphorylation of MyoGEF at Thr-544 increased the binding of Plk1 to MyoGEF as well as promoted MyoGEF activity toward RhoA and the localization of MyoGEF to the central spindle (Figs. 3, 6, 7, and 8). These results are consistent with our previous findings that inhibition of Plk1 activity by a small molecule inhibitor or depletion of Plk1 by RNAi disrupts the localization of MyoGEF to the central spindle and decreases MyoGEF activity toward RhoA (43). Therefore, we propose that phosphorylation of MyoGEF at Thr-544 by aurora B promotes the binding of Plk1 to MyoGEF. In turn, Plk1 phosphorylates MyoGEF at Thr-574, thus leading to MyoGEF activation and localization at the central spindle (Fig. 8C).
Aurora B has been shown to positively regulate RhoA activation during cytokinesis. A critical event during cytokinesis is that the centralspindlin complex, which is composed of MgcRacGAP and Mklp1, localizes to the central spindle. In turn, the centralspindlin complex recruits Ect2 to the central spindle, leading to cortical RhoA activation and the formation of myosin contractile ring at the cleavage furrow (18). Importantly, localization of the centralspindlin complex to the central spindle requires the correct localization and the activity of aurora B (18). In this study we demonstrate that aurora B-mediated phosphorylation of MyoGEF at Thr-544 promotes the localization and activation of MyoGEF at the central spindle, suggesting a role for the aurora B-MyoGEF pathway in contributing to the regulation of cytokinesis. Therefore, it is likely that aurora B-mediated signals can recruit multiple RhoGEFs including Ect2 and MyoGEF to the central spindle and cleavage furrow, thus contributing to the regulation of equatorial RhoA activation and contractile ring formation during cytokinesis.
Ect2 is a conserved RhoGEF that is implicated in cytokinesis in multiple organisms and cell lines (20, 34–38). However, studies in different types of cells and tissues reveal that redundant and/or tissue-specific mechanisms may exist for the regulation of cytokinesis (55, 56). Citron kinase is a downstream effector for RhoA and is important for cytokinesis in cultured cells (57–59). However, mice deficient for citron kinase show cytokinesis defects only in some cell populations of the central nervous system and during spermatogenesis, suggesting that citron kinase regulates cytokinesis in a tissue/cell-specific manner (53, 60). Furthermore, expression of a dominant negative mutant of Ect2 or treatment with Y-27632 (an inhibitor of Rho kinase) inhibits cytokinesis in HeLa cells but not in Rat1A cells (30). Additionally, RhoA can be activated at the cleavage furrow via an Ect2-independent pathway in HT1080 cells (49). These data suggest that there are redundant mechanisms and/or cell type-specific pathways to regulate RhoA activation at the cleavage furrow in mammals. Thus, it is possible that additional RhoGEFs (MyoGEF, GEF-H1, and LARG (leukemia-associated RhoGEF)) that contribute to equatorial RhoA activation during cytokinesis may be implicated in these redundant or cell type-specific regulatory mechanisms for cytokinesis.
Supplementary Material
Supplemental Data
Acknowledgments
We thank Dr. Robert S. Adelstein and Dr. Mary Anne Conti for critical reading and comments on the manuscript.
*
This work was supported, in whole or in part, by National Institutes of Health Grant R15GM097702 (to Q. W.).
2
The abbreviations used are:
Plk
Polo-like kinase 1
GEF
guanine nucleotide exchange factor
PBD
polo-box domain
PH
pleckstrin homology domain
MyoGEF
myosin II-interacting guanine nucleotide exchange factor.
REFERENCES
- 1.Barr F. A., Silljé H. H., Nigg E. A. (2004) Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5, 429–440 [DOI] [PubMed] [Google Scholar]
- 2.Elia A. E., Rellos P., Haire L. F., Chao J. W., Ivins F. J., Hoepker K., Mohammad D., Cantley L. C., Smerdon S. J., Yaffe M. B. (2003) The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115, 83–95 [DOI] [PubMed] [Google Scholar]
- 3.Elia A. E., Cantley L. C., Yaffe M. B. (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299, 1228–1231 [DOI] [PubMed] [Google Scholar]
- 4.Baumann C., Körner R., Hofmann K., Nigg E. A. (2007) PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 128, 101–114 [DOI] [PubMed] [Google Scholar]
- 5.Qi W., Tang Z., Yu H. (2006) Phosphorylation- and polo-box-dependent binding of Plk1 to Bub1 is required for the kinetochore localization of Plk1. Mol. Biol. Cell 17, 3705–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goto H., Kiyono T., Tomono Y., Kawajiri A., Urano T., Furukawa K., Nigg E. A., Inagaki M. (2006) Complex formation of Plk1 and INCENP required for metaphase-anaphase transition. Nat. Cell Biol. 8, 180–187 [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi T., Goto H., Yokoyama T., Silljé H., Hanisch A., Uldschmid A., Takai Y., Oguri T., Nigg E. A., Inagaki M. (2005) Phosphorylation by Cdk1 induces Plk1-mediated vimentin phosphorylation during mitosis. J. Cell Biol. 171, 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preisinger C., Körner R., Wind M., Lehmann W. D., Kopajtich R., Barr F. A. (2005) Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 24, 753–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabbro M., Zhou B. B., Takahashi M., Sarcevic B., Lal P., Graham M. E., Gabrielli B. G., Robinson P. J., Nigg E. A., Ono Y., Khanna K. K. (2005) Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev. Cell 9, 477–488 [DOI] [PubMed] [Google Scholar]
- 10.Litvak V., Argov R., Dahan N., Ramachandran S., Amarilio R., Shainskaya A., Lev S. (2004) Mitotic phosphorylation of the peripheral Golgi protein Nir2 by Cdk1 provides a docking mechanism for Plk1 and affects cytokinesis completion. Mol. Cell 14, 319–330 [DOI] [PubMed] [Google Scholar]
- 11.Yamashiro S., Yamakita Y., Totsukawa G., Goto H., Kaibuchi K., Ito M., Hartshorne D. J., Matsumura F. (2008) Myosin phosphatase-targeting subunit 1 regulates mitosis by antagonizing polo-like kinase 1. Dev. Cell 14, 787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King R. W., Deshaies R. J., Peters J. M., Kirschner M. W. (1996) How proteolysis drives the cell cycle. Science 274, 1652–1659 [DOI] [PubMed] [Google Scholar]
- 13.Neef R., Gruneberg U., Kopajtich R., Li X., Nigg E. A., Sillje H., Barr F. A. (2007) Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat. Cell Biol. 9, 436–444 [DOI] [PubMed] [Google Scholar]
- 14.Nigg E. A. (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2, 21–32 [DOI] [PubMed] [Google Scholar]
- 15.Carmena M., Wheelock M., Funabiki H., Earnshaw W. C. (2012) The chromosomal passenger complex (CPC). From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 13, 789–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ditchfield C., Johnson V. L., Tighe A., Ellston R., Haworth C., Johnson T., Mortlock A., Keen N., Taylor S. S. (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161, 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauf S., Cole R. W., LaTerra S., Zimmer C., Schnapp G., Walter R., Heckel A., van Meel J., Rieder C. L., Peters J. M. (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guse A., Mishima M., Glotzer M. (2005) Phosphorylation of ZEN-4/MKLP1 by aurora B regulates completion of cytokinesis. Curr. Biol. 15, 778–786 [DOI] [PubMed] [Google Scholar]
- 19.Fuller B. G., Lampson M. A., Foley E. A., Rosasco-Nitcher S., Le K. V., Tobelmann P., Brautigan D. L., Stukenberg P. T., Kapoor T. M. (2008) Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature 453, 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petronczki M., Glotzer M., Kraut N., Peters J. M. (2007) Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev. Cell 12, 713–725 [DOI] [PubMed] [Google Scholar]
- 21.Santamaria A., Neef R., Eberspächer U., Eis K., Husemann M., Mumberg D., Prechtl S., Schulze V., Siemeister G., Wortmann L., Barr F. A., Nigg E. A. (2007) Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis. Mol. Biol. Cell 18, 4024–4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burkard M. E., Randall C. L., Larochelle S., Zhang C., Shokat K. M., Fisher R. P., Jallepalli P. V. (2007) Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc. Natl. Acad. Sci. U.S.A. 104, 4383–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neef R., Preisinger C., Sutcliffe J., Kopajtich R., Nigg E. A., Mayer T. U., Barr F. A. (2003) Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J. Cell Biol. 162, 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou T., Aumais J. P., Liu X., Yu-Lee L. Y., Erikson R. L. (2003) A role for Plk1 phosphorylation of NudC in cytokinesis. Dev. Cell 5, 127–138 [DOI] [PubMed] [Google Scholar]
- 25.Lowery D. M., Clauser K. R., Hjerrild M., Lim D., Alexander J., Kishi K., Ong S. E., Gammeltoft S., Carr S. A., Yaffe M. B. (2007) Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. 26, 2262–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfe B. A., Takaki T., Petronczki M., Glotzer M. (2009) Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol. 7, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumura F., Ono S., Yamakita Y., Totsukawa G., Yamashiro S. (1998) Specific localization of serine 19 phosphorylated myosin II during cell locomotion and mitosis of cultured cells. J. Cell Biol. 140, 119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamakita Y., Yamashiro S., Matsumura F. (1994) In vivo phosphorylation of regulatory light chain of myosin II during mitosis of cultured cells. J. Cell Biol. 124, 129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshizaki H., Ohba Y., Kurokawa K., Itoh R. E., Nakamura T., Mochizuki N., Nagashima K., Matsuda M. (2003) Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J. Cell Biol. 162, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshizaki H., Ohba Y., Parrini M. C., Dulyaninova N. G., Bresnick A. R., Mochizuki N., Matsuda M. (2004) Cell type-specific regulation of RhoA activity during cytokinesis. J. Biol. Chem. 279, 44756–44762 [DOI] [PubMed] [Google Scholar]
- 31.Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 32.Bement W. M., Benink H. A., von Dassow G. (2005) A microtubule-dependent zone of active RhoA during cleavage plane specification. J. Cell Biol. 170, 91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosako H., Yoshida T., Matsumura F., Ishizaki T., Narumiya S., Inagaki M. (2000) Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene 19, 6059–6064 [DOI] [PubMed] [Google Scholar]
- 34.Kamijo K., Ohara N., Abe M., Uchimura T., Hosoya H., Lee J. S., Miki T. (2006) Dissecting the role of Rho-mediated signaling in contractile ring formation. Mol. Biol. Cell 17, 43–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yüce O., Piekny A., Glotzer M. (2005) An ECT2-centralspindlin complex regulates the localization and function of RhoA. J. Cell Biol. 170, 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niiya F., Tatsumoto T., Lee K. S., Miki T. (2006) Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene 25, 827–837 [DOI] [PubMed] [Google Scholar]
- 37.Nishimura Y., Yonemura S. (2006) Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J. Cell Sci. 119, 104–114 [DOI] [PubMed] [Google Scholar]
- 38.Cook D. R., Solski P. A., Bultman S. J., Kauselmann G., Schoor M., Kuehn R., Friedman L. S., Cowley D. O., Van Dyke T., Yeh J. J., Johnson L., Der C. J. (2011) The ect2 rho guanine nucleotide exchange factor is essential for early mouse development and normal cell cytokinesis and migration. Genes Cancer 2, 932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birkenfeld J., Nalbant P., Bohl B. P., Pertz O., Hahn K. M., Bokoch G. M. (2007) GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev. Cell 12, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martz M. K., Grabocka E., Beeharry N., Yen T. J., Wedegaertner P. B. (2013) Leukemia-associated RhoGEF (LARG) is a novel RhoGEF in cytokinesis and required for the proper completion of abscission. Mol. Biol. Cell 24, 2785–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu D., Asiedu M., Adelstein R. S., Wei Q. (2006) A novel guanine nucleotide exchange factor MyoGEF is required for cytokinesis. Cell Cycle 5, 1234–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asiedu M., Wu D., Matsumura F., Wei Q. (2009) Centrosome/spindle pole-associated protein regulates cytokinesis via promoting the recruitment of MyoGEF to the central spindle. Mol. Biol. Cell 20, 1428–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asiedu M., Wu D., Matsumura F., Wei Q. (2008) Phosphorylation of MyoGEF on Thr-574 by Plk1 promotes MyoGEF localization to the central spindle. J. Biol. Chem. 283, 28392–28400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Z., Sun Y., Harley S. E., Zou H., Yu H. (2004) Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc. Natl. Acad. Sci. U.S.A. 101, 18012–18017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts P. J., Mitin N., Keller P. J., Chenette E. J., Madigan J. P., Currin R. O., Cox A. D., Wilson O., Kirschmeier P., Der C. J. (2008) Rho Family GTPase modification and dependence on CAA_X_ motif-signaled posttranslational modification. J. Biol. Chem. 283, 25150–25163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei Q. (2005) Pitx2a binds to human Papillomavirus type 18 E6 protein and inhibits E6-mediated P53 degradation in HeLa cells. J. Biol. Chem. 280, 37790–37797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pal D., Wu D., Haruta A., Matsumura F., Wei Q. (2010) Role of a novel coiled-coil domain-containing protein CCDC69 in regulating central spindle assembly. Cell Cycle 9, 4117–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee Y. H., Bedford M. T., Stallcup M. R. (2011) Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev. 25, 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanada M., Nagasaki A., Uyeda T. Q. (2008) Novel functions of Ect2 in polar lamellipodia formation and polarity maintenance during “contractile ring-independent” cytokinesis in adherent cells. Mol. Biol. Cell 19, 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cimini D., Wan X., Hirel C. B., Salmon E. D. (2006) Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 16, 1711–1718 [DOI] [PubMed] [Google Scholar]
- 51.Gadea B. B., Ruderman J. V. (2005) Aurora kinase inhibitor ZM447439 blocks chromosome-induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol. Biol. Cell 16, 1305–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu D., Asiedu M., Wei Q. (2009) Myosin-interacting guanine exchange factor (MyoGEF) regulates the invasion activity of MDA-MB-231 breast cancer cells through activation of RhoA and RhoC. Oncogene 28, 2219–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cunto F. D., Imarisio S., Camera P., Boitani C., Altruda F., Silengo L. (2002) Essential role of citron kinase in cytokinesis of spermatogenic precursors. J. Cell Sci. 115, 4819–4826 [DOI] [PubMed] [Google Scholar]
- 54.Ma X., Kovács M., Conti M. A., Wang A., Zhang Y., Sellers J. R., Adelstein R. S. (2012) Nonmuscle myosin II exerts tension but does not translocate actin in vertebrate cytokinesis. Proc. Natl. Acad. Sci. U.S.A. 109, 4509–4514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uyeda T. Q., Nagasaki A., Yumura S. (2004) Multiple parallelisms in animal cytokinesis. Int. Rev. Cytol. 240, 377–432 [DOI] [PubMed] [Google Scholar]
- 56.Piekny A., Werner M., Glotzer M. (2005) Cytokinesis. Welcome to the Rho zone. Trends Cell Biol. 15, 651–658 [DOI] [PubMed] [Google Scholar]
- 57.Yamashiro S., Totsukawa G., Yamakita Y., Sasaki Y., Madaule P., Ishizaki T., Narumiya S., Matsumura F. (2003) Citron kinase, a Rho-dependent kinase, induces di-phosphorylation of regulatory light chain of myosin II. Mol. Biol. Cell 14, 1745–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madaule P., Furuyashiki T., Eda M., Bito H., Ishizaki T., Narumiya S. (2000) Citron, a Rho target that affects contractility during cytokinesis. Microsc Res. Tech. 49, 123–126 [DOI] [PubMed] [Google Scholar]
- 59.Madaule P., Eda M., Watanabe N., Fujisawa K., Matsuoka T., Bito H., Ishizaki T., Narumiya S. (1998) Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature 394, 491–494 [DOI] [PubMed] [Google Scholar]
- 60.Di Cunto F., Imarisio S., Hirsch E., Broccoli V., Bulfone A., Migheli A., Atzori C., Turco E., Triolo R., Dotto G. P., Silengo L., Altruda F. (2000) Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 28, 115–127 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data