Mispair-specific Recruitment of the Mlh1-Pms1 Complex Identifies Repair Substrates of the Saccharomyces cerevisiae Msh2-Msh3 Complex (original) (raw)
Background: Msh2-Msh6 and Msh2-Msh3 bind to and initiate the repair of mismatched DNA.
Results: Purified Msh2-Msh3 binds specific base:base mispairs and small to large insertion/deletions and recruits a downstream mismatch repair component, Mlh1-Pms1.
Conclusion: Msh2-Msh3 functions in the repair of specific base:base and insertion/deletion mispairs.
Significance: Our results show that mispair specificity of Mlh1-Pms1 recruitment by Msh2-Msh3 explains the specificity of Msh2-Msh3 in mismatch repair.
Keywords: DNA Mismatch Repair, DNA Recombination, DNA Repair, DNA Replication, Mutagenesis Mechanisms, Mismatch Binding, Mismatch Repair Specificity, Msh6, Replication Fidelity
Abstract
DNA mismatch repair is initiated by either the Msh2-Msh6 or the Msh2-Msh3 mispair recognition heterodimer. Here we optimized the expression and purification of Saccharomyces cerevisiae Msh2-Msh3 and performed a comparative study of Msh2-Msh3 and Msh2-Msh6 for mispair binding, sliding clamp formation, and Mlh1-Pms1 recruitment. Msh2-Msh3 formed sliding clamps and recruited Mlh1-Pms1 on +1, +2, +3, and +4 insertion/deletions and CC, AA, and possibly GG mispairs, whereas Msh2-Msh6 formed mispair-dependent sliding clamps and recruited Mlh1-Pms1 on 7 of the 8 possible base:base mispairs, the +1 insertion/deletion mispair, and to a low level on the +2 but not the +3 or +4 insertion/deletion mispairs and not on the CC mispair. The mispair specificity of sliding clamp formation and Mlh1-Pms1 recruitment but not mispair binding alone correlated best with genetic data on the mispair specificity of Msh2-Msh3- and Msh2-Msh6-dependent mismatch repair in vivo. Analysis of an Msh2-Msh6/Msh3 chimeric protein and mutant Msh2-Msh3 complexes showed that the nucleotide binding domain and communicating regions but not the mispair binding domain of Msh2-Msh3 are responsible for the extremely rapid dissociation of Msh2-Msh3 sliding clamps from DNA relative to that seen for Msh2-Msh6, and that amino acid residues predicted to stabilize Msh2-Msh3 interactions with bent, strand-separated mispair-containing DNA are more critical for the recognition of small +1 insertion/deletions than larger +4 insertion/deletions.
Introduction
DNA mismatch repair (MMR)2 is a critical pathway that repairs base:base and insertion/deletion mispairs that occur as the result of errors during replication (1–7). Because MMR increases the fidelity of DNA replication, defects in MMR result in increased mutation rates, and consequently, defects in MMR in mammals underlie the development of both inherited and sporadic cancers (8–13). MMR also acts on mispairs that occur in recombination intermediates and as a result of chemical damage to DNA (3, 4). In addition, MMR can prevent recombination between divergent DNAs and aid in the resolution of some types of recombination intermediates (14–20).
The early steps of MMR are highly conserved. In bacteria, the MutS homodimer recognizes mispaired bases in DNA and upon mispair binding recruits a critical accessory factor, the MutL homodimer (21–23). In eukaryotes, mispaired bases are recognized by two heterodimers of MutS homolog proteins, Msh2-Msh6 and Msh2-Msh3, that have partially overlapping mispair recognition specificities (3, 24–28). Msh2-Msh6 is thought to be responsible for the repair of base:base and small insertion/deletion mispairs, whereas Msh2-Msh3 is thought to be responsible for the repair of both small and large insertion/deletion mispairs as well as a limited spectrum of base:base mispairs (1, 3, 25, 26, 29). However, there are conflicting data on whether Msh2-Msh3 plays a role in repairing +1 insertion/deletion mispairs (24–26, 30–33). In addition, the specificity of Msh2-Msh6 and Msh2-Msh3-dependent MMR in vitro does not appear to correlate well with the mispair specificity of MMR in vivo or with mispair binding specificity in the case of Msh2-Msh6, where sufficient data exist for comparative purposes (32, 34). The Msh2-Msh3 complex also binds branched DNAs, which may reflect the role of Msh2-Msh3 in the resolution of some types of recombination intermediates (35, 36). On binding mispairs, the Msh2-Msh6 complex recruits a heterodimer of MutL homolog proteins, the Mlh1-Pms1 complex (Mlh1-Pms2 in mammals) (37–41). The Msh2-Msh3 complex also appears to recruit the Mlh1-Pms1 complex to mispairs (42), although this has not been characterized as extensively as the interaction between Msh2-Msh6 and Mlh1-Pms1. Eukaryotes also contain two other MutL-related complexes, Mlh1-Mlh2 (Mlh1-Pms1 in mammals) and Mlh1-Mlh3 (43, 44). Mlh1-Mlh3 plays a role in meiosis and is thought to function in a fraction of Msh2-Msh3-dependent MMR (29, 44–48). The function of Mlh1-Mlh2 is less clear (43, 46, 49), although recent studies have shown that Mlh1-Mlh2 is recruited to foci in vivo by Msh2-Msh6 in response to mispaired bases in DNA, suggesting a role as an accessory factor in MMR.3
The binding of the mispair recognition proteins to DNA has been extensively characterized by biochemical, structural, and genetics-linked structure-function approaches, particularly in the case of MutS and Msh2-Msh6. X-ray crystallography of mispair-bound bacterial MutS demonstrated that MutS forms a ring around the DNA and that mispair binding involves bending of the DNA, flipping of the mispaired base out of the DNA helix, and stabilization of this base by π-stacking interactions with a Phe in the mispair-binding domain as well as contacts between other amino acid residues and the DNA backbone (50, 51). The structure of human Msh2-Msh6 is very similar, and the DNA bending at the mispair and stabilization of an extrahelical base by a stacking Phe is conserved (52, 53). In the absence of a mispair, MutS and Msh2-Msh6 bind more weakly and directly dissociate from DNA when ATP binds the ATPase domains (21, 38). In contrast, ATP causes mispair-bound MutS and Msh2-Msh6 to adopt a conformation called a sliding clamp, which is trapped on the DNA and slides along the DNA until it dissociates from the ends (38, 54). As a consequence of this property, these sliding clamps can be trapped on the DNA by end blocks (38, 54). A sliding clamp conformation for Msh2-Msh3 has been inferred from the ATP sensitivity and the ATP hydrolysis properties of mispair-bound Msh2-Msh3 but has not been directly demonstrated using end blocked DNA substrates (55). Interestingly, deuterium exchange mass spectrometry has demonstrated that MutS and Saccharomyces cerevisiae Msh2-Msh6 likely form a ring on DNA lacking a mispair, with the stacking Phe contacting the DNA; the presence of a mispair stabilizes this ring through yet other contacts between MutS or Msh6 and the DNA backbone (56). More recently, molecular modeling, genetics-based structure-function studies, and x-ray crystallography have demonstrated that although the overall structure of the mispair-bound Msh2-Msh3 is similar to Msh2-Msh6, the interactions with the mispair are different and involve bending and strand separation of the DNA by the core mispair-contacting residues and stabilization by additional contacts with the Msh3 mispair binding domain (57, 58). This importance of these additional contacts to stabilize the bound and bent DNA appears to be reduced on larger insertion/deletions (57).
MutS and Msh2-Msh6 have been extensively characterized with regard to their interactions with DNA, nucleotides, and with MutL and Mlh1-Pms1, respectively. In contrast, analysis of the Msh2-Msh3 complex has lagged behind that of MutS and Msh2-Msh6. One reason for this is the expectation that the properties of Msh2-Msh3 would resemble those of MutS and Msh2-Msh6. Furthermore, in S. cerevisiae, where it is possible to perform detailed functional studies in vivo to correlate with in vitro biochemical studies, the availability of purified Msh2-Msh3 has been limited. Previous studies have utilized an _S. cerevisiae_-based Msh2-Msh3 expression system in which translation of Msh3 was driven from a start codon upstream of the correct start codon (36, 59), yielding Msh2-Msh3 preparations that appeared to contain multiple Msh3 species (59) or were of undocumented purity (36) or utilized a _S. cerevisiae_-based Msh2-Msh3 expression system in which translation of Msh3 was driven from the correct start codon but which yielded very low amounts of Msh2-Msh3 (29). To overcome these problems, in the present study, we have optimized the expression and purification of S. cerevisiae Msh2-Msh3 to obtain the amounts of purified Msh2-Msh3 required for biochemical studies. Using this protein, we have extensively characterized Msh2-Msh3 for mispair binding, sliding clamp formation, and Mlh1-Pms1 recruitment and performed structure-function studies of mispair binding and dissociation. From this, we show that Msh2-Msh3 forms an ATP-dependent sliding clamp that has a much more rapid dissociation rate from DNA than the Msh2-Msh6 sliding clamp. We also show that the mispair specificity of MMR in vivo determined by genetics studies correlates best with the mispair specificity of Msh2-Msh3 sliding clamp formation and recruitment of Mlh1-Pms1.
EXPERIMENTAL PROCEDURES
Media, Strains, and Plasmids
S. cerevisiae strains were grown in standard media, either 10 g of yeast extract, 20 g of peptone, and 20 g of dextrose per liter (YPD) or complete supplement mixture (U. S. Biological, Salem, MA) containing 2% glucose and lacking specific amino acids to select for plasmid markers and 2.4% agar if required. Escherichia coli strains were grown in either Luria-Bertani broth (10 g of Tryptone, 5 g of yeast extract, 5 g of sodium chloride, and 50 mg of thymine per liter) or Terrific Broth (12 g of peptone, 2.31 g of monopotassium phosphate, 12.54 g of dipotassium phosphate, 24 g of yeast extract, and 4 ml of glycerol per liter) with antibiotics to select for plasmid markers and 1.5% agar if required. Plasmids were typically maintained and propagated in the E. coli strain TOP 10F′ (Invitrogen). Overproduction of proteins in S. cerevisiae was performed using the strains RDKY1293 (_MAT_α ura3-52 trp1 leu2_Δ_1 his3_Δ_200 pep4::HIS3 prb1_Δ_1.6R GAL can1) and RDKY2418 (RDKY1293 msh2::hisG msh6::hisG). Overproduction of the Msh2-Msh6 complex in E. coli was performed using the strain BL21 CodonPlus (DE3) RIL (Stratagene, La Jolla, CA), and overproduction of LacI was performed using E. coli BLIM (ompT hsdSBa [_rBa_−, _mBa gal dcm lac_]) containing the plasmid pLS1 Ampr LacI (a gift from Dr. Kathleen Matthews) (60–62). The plasmid used for overexpression of the Msh2-Msh6 complex was pET11a-MSH2-MSH6 (63). The plasmids used for overexpression of the wild-type Msh2-Msh3 complex were pRDK354 (_2_μ URA3 P_GAL1–10_-MSH2) and pRDK1596 (_2_μ LEU2 P_GAL1–10_-MSH3-FLAG) (29). The plasmids used for overexpression of the Msh2-Msh3 mutant complexes were pRDK354 with pRDK1769 and pRDK1768 for MSH3-K158E-FLAG and MSH3-R195D-FLAG, respectively, which were constructed from the plasmid pRDK1596 using the GeneArt® site-directed mutagenesis system (Invitrogen) to introduce the nucleotide changes A472G and AGG583_585GAT, respectively. The plasmids used for overexpression of the Mlh1-Pms1 complex were pRDK573 (_2_μ PGAL1–10-MLH1 TRP1) and pRDK1099 (_2_μ PGAL1–10-PMS1-FLAG LEU2) (64, 65). All plasmids were periodically verified by DNA sequencing to ensure the absence of unwanted mutations.
Proteins
Purification of Msh2-Msh3
The protease-defective S. cerevisiae strains RDKY2418 or RDKY1293 were transformed with the plasmids pRDK354 and pRDK1596 for purification of the wild-type Msh2-Msh3 complex, pRDK354 and pRDK1769 for purification of the Msh2-Msh3-K158E complex, or pRDK354 and pRDK1768 for purification of the Msh2-Msh3-R195D complex. Liquid starter cultures were grown overnight at 30 °C in SCGL medium lacking specific amino acids and containing 0.1% glucose (w/v), 3% glycerol (v/v), and 2% lactic acid (v/v; 24.7 ml of 80% lactic acid per liter) (pH adjusted to 5–6 using concentrated NaOH) (66). The starter culture was then used to inoculate 1 or 1.5 liters of the same medium, and the culture was grown at 30 °C until it reached an _A_600 = 1. Protein overexpression was induced by addition of an equal volume of YPGL medium consisting of 10 g of yeast extract and 20 g of peptone per liter, 0.2% (w/v) glucose, 3% (v/v) glycerol, and 2% (v/v) lactic acid (pH adjusted to 5–6 using concentrated NaOH) followed by the addition of galactose to a final concentration of 2% (w/v) (66). 6–18 h after induction, the cells were harvested by centrifugation, and the pellets were either snap-frozen at −80 °C and later thawed or directly used for protein purification as follows. The cells were resuspended in Buffer A (50 mm Tris-Cl, pH 8, 1 mm EDTA, pH 8, 10% glycerol (v/v), protease inhibitor mixture PIC-D (to obtain final concentrations of 1 mm PMSF, 1 μg/liter chymostatin, and 1 μg/liter pepstatin A), and protease inhibitor mixture PIC-W (to obtain final concentrations of 1 mm benzamidine, 0.5 μg/liter bestatin, 1 μg/liter aprotinin, and 1 μg/liter leupeptin)) containing 1 mm DTT and 500 mm NaCl and lysed by 7 passes through a microfluidizer (Microfluidics, Westwood, MA). The lysate was clarified by centrifugation at 16,000 rpm in a Sorvall SA600 Rotor at 4 °C for 1 h. The supernatant was diluted with an equal volume of Buffer A containing 1 mm DTT and no NaCl and loaded onto two 5-ml HiTrap heparin HP columns (GE Healthcare) connected in series that were equilibrated with Buffer A containing 200 mm NaCl and 2 mm β-mercaptoethanol using an AKTA FPLC system (GE Healthcare). The columns were washed with 50 ml of Buffer A containing 2 mm β-mercaptoethanol and 200 mm NaCl and eluted in a 50-ml gradient of 200 mm to 1 m NaCl in Buffer A containing 2 mm β-mercaptoethanol. The peak fractions containing Msh2-Msh3 were pooled and diluted in an equal volume of Buffer A (no salt) containing 2 mm β-mercaptoethanol instead of DTT to reduce the salt concentration to ∼200 mm NaCl. The sample was loaded onto a 4-ml Anti-FLAG M2 affinity gel (Sigma) column, which was washed with 21 ml of Buffer A containing 2 mm β-mercaptoethanol and 200 mm NaCl. The bound proteins were eluted using 15 ml of 200 μg/ml FLAG peptide in the same buffer. The peak fractions were pooled and diluted in an equal volume of Buffer A containing 1 mm DTT and no NaCl, to lower the salt concentration to 100 mm. The sample was loaded on a 1-ml HiTrap Q-Sepharose FF column (GE Healthcare), which was washed with 5 ml of Buffer A containing 1 mm DTT and 100 mm NaCl. The bound proteins were eluted using a 10-ml gradient of 100 mm to 1 m NaCl in Buffer A containing 1 mm DTT. The fractions containing Msh2-Msh3 or the mutant Msh2-Msh3 complexes were pooled and stored in small aliquots at −80 °C. All purification steps were performed at 4 °C.
Purification of Mlh1-Pms1
S. cerevisiae Mlh1-Pms1 was overproduced in the S. cerevisiae strain RDKY1293 using the plasmids pRDK573 and pRDK1099 and purified to >98% purity as previously described (64, 65).
Purification of LacI
LacI was purified exactly as previously described (60–62) with the following three modifications: 1) the DNase step was omitted, 2) the viscosity of the cell lysate was reduced by sonication prior to the first chromatography step, and 3) the phosphocellulose column was run using an AKTA FPLC system.
Msh2-Msh6 and Msh2-Msh6(3-MBD)
Msh2-Msh6 and Msh2-Msh6(3-MBD) were from previously published studies from our laboratory and were purified to >98% purity (64, 67).
Surface Plasmon Resonance Analysis
Protein-DNA and protein-protein-DNA interactions were monitored using a Biacore T100 instrument (GE Healthcare) using the buffers and protein flow schedules exactly as described previously (38, 64). The DNA substrates used were 236 bp in length with biotin conjugated at one end, the lacO sequence at the other end, and a centrally located base-base or insertion/deletion mispair or a GC base pair in the homoduplex control that were constructed as previously described (38, 40, 64, 67, 68). Approximately 20 ng (100 ± 5 resonance units (RUs)) of different DNA substrates were conjugated to 3 flow cells of streptavidin-coated Biacore SA chips (GE Healthcare), and the 4th flow cell was used as an unmodified reference surface in each experiment. The experiments investigating DNA binding and sliding clamp formation were performed using 50 nm Msh2-Msh6, Msh2-Msh3, or Msh2-Msh6(3-MBD) unless otherwise indicated in specific experiments. The experiments investigating recruitment of Mlh1-Pms1 were performed using 20 nm Msh2-Msh6 or Msh2-Msh3 and 40 nm Mlh1-Pms1. In experiments with end blocked DNA substrates, the DNA ends were blocked by including 30 nm LacI in the reactions, and the bound LacI was subsequently released by challenge with 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) when indicated. ATP or ADP was included in the reaction mixtures at a final concentration of 250 μm as indicated. All experiments were performed at 25 °C at a flow rate of 20 μl/min, and data were collected at a frequency of 10 Hz. The data were analyzed using the BiaEvaluation v3.1 (GE Healthcare) and Prism 6 (GraphPad Software, Inc., La Jolla, CA) software.
RESULTS
Purification of the S. cerevisiae Msh2-Msh3 Complex
To improve the purification of Msh2-Msh3, we expressed Msh2-Msh3 in protease-defective S. cerevisiae strains containing two plasmids encoding _GAL_-promoter driven MSH2 and MSH3 in which MSH3 contains a C-terminal FLAG tag that had no effect on MSH3 function (29, 69). We optimized protein expression using a glycerol-lactate media to galactose media growth protocol for inducing Msh2-Msh3 expression and performed rapid purification by sequential chromatography on HiTrap heparin, anti-FLAG antibody and HiTrap Q-Sepharose columns. We were able to obtain 1–1.5 mg of Msh2-Msh3 complex per liter of S. cerevisiae culture that was >99% pure and had a 1:1 ratio of Msh2 to Msh3 subunits (Fig. 1). As part of these studies, we also expressed Msh2-Msh3 and codon-optimized Msh2-Msh3 in E. coli using T7 promoter expression vectors, but we were unsuccessful in purifying Msh2-Msh3 from these E. coli expression strains.
FIGURE 1.
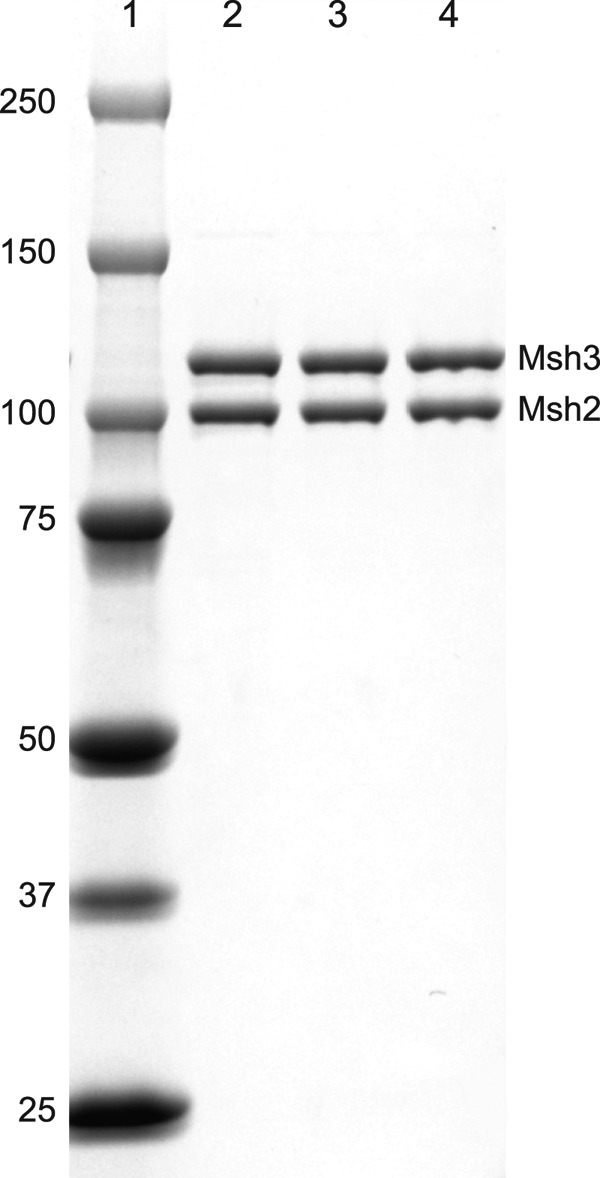
SDS-PAGE analysis of purified wild-type and mutant Msh2-Msh3 complexes. The wild-type Msh2-Msh3 complex and Msh2-Msh3-K158E and Msh2-Msh3-R195D mutant complexes were purified as described under “Experimental Procedures” and analyzed by SDS-PAGE followed by staining the gel with Coomassie Blue. Lane 1 contains the molecular mass standards (kDa) indicated on the left. Lanes 2–4 contain 0.5 μg each of the purified Msh2-Msh3, Msh2-Msh3-K158E, and Msh2-Msh3-R195D complexes, respectively. The bands corresponding to the Msh2 and Msh3 subunits are indicated.
Msh2-Msh3 Preferentially Binds to Insertion/Deletions and Specific Base-Base Mispairs
To determine the specificity of Msh2-Msh3 for binding different mispaired bases, we systematically monitored the binding of Msh2-Msh3 to various insertion/deletion mispairs and base-base mispairs using Surface Plasmon Resonance (SPR) and compared the binding observed to the binding of Msh2-Msh6 performed in parallel (Figs. 2 and 3). Each of the DNA substrates, including the fully base-paired control DNA (referred to as GC), was 236 bp long with a biotin at one end and a lacO site at the other end and, with the exception of the centrally located mispair, had the same sequence (38, 67, 68). Mispair binding was evaluated under three different conditions: 1) in the absence of added nucleotide, where the MutS family proteins bind mispaired bases in DNA and then directly dissociate from the DNA, 2) in the presence of ADP, where the MutS-family proteins bind mispaired bases but do not form sliding clamps and directly dissociate from the DNA, 3) in the presence of ATP, where the MutS family proteins do not bind mispaired bases in DNA until their bound ATP is hydrolyzed to ADP and then dissociate from DNA by forming a sliding clamp and sliding off the end of DNA when they exchange the bound ADP for ATP (38, 54, 55, 70, 71). Under these different conditions, the level of binding to mispaired DNA relative to base paired DNA reflects the mispair binding specificity of the protein.
FIGURE 2.

Msh2-Msh3 binds preferentially to insertion/deletion mispairs and specific base-base mispairs. Binding of Msh2-Msh3 (red) or Msh2-Msh6 (black) to homoduplex DNA (dashed lines) and various mispair-containing DNA substrates (solid lines) was monitored by SPR as described under “Experimental Procedures.” In all cases the background binding to a flow cell lacking bound DNA run in the same experiment was subtracted. The presence of no nucleotide (−NT), ADP (+ADP), or ATP (+ATP) in the binding reactions is indicated at the top of each column of sensorgrams. The mispair substrate analyzed is indicated at the right of each row of sensorgrams. The x axis indicates the time of association in seconds, the y axis indicates the extent of binding in RUs, and the black arrows indicate the time at which the injection of protein was initiated.
FIGURE 3.
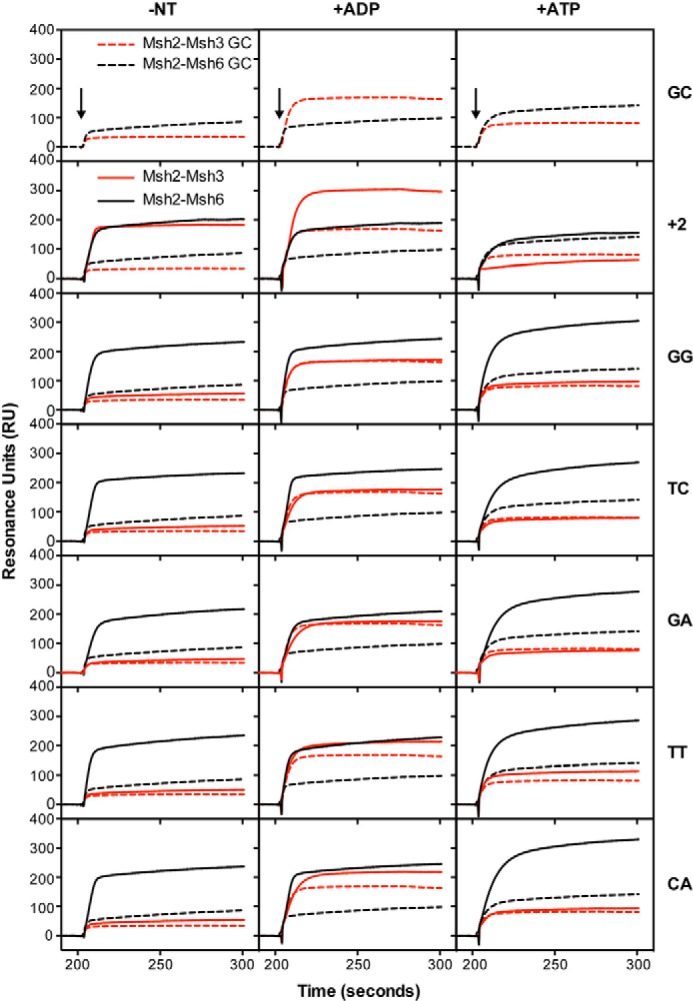
Msh2-Msh3 binds preferentially to insertion/deletion mispairs and specific base-base mispairs. See the legend to Fig. 2 for experimental details and a key for abbreviations used.
In the absence of added nucleotide, Msh2-Msh3 showed increased binding to substrates containing +1, +2, +3, and +4 insertion/deletion mispairs as well as to the CC and AA base:base mispairs relative to binding to the base paired control, whereas only very minor binding to the TG, GG, TC, GA, TT, and CA mispairs was observed. The relative level of mispair binding observed for Msh2-Msh3 was +4, +2, +3, +1 ≫ AA > CC ≫ GG, TC > CA, TT, TG, GA (Table 1). The observation of Msh2-Msh3 binding to AA and CC mispairs is consistent with the results of a previous genetic study indicating that Msh2-Msh3-dependent MMR can repair some base:base mispairs (29). In contrast, Msh2-Msh6 bound to substrates containing +1 and +2, to a lesser extent to the +3 and +4 insertion/deletion mispairs, and all eight of the base:base mispairs tested. The relative level of mispair binding observed for Msh2-Msh6 was GG, TC, TG > CA, +1, TT, AA, GA > +2, CC ≫ +3 > +4 (Table 1). The addition of ADP to the binding reactions had relatively small effects on the relative mispair binding specificity of Msh2-Msh3 (relative binding +1 > +2, +4, +3 ≫ CC, AA ≫ CA, TC, TT, GG > TG, GA) or Msh2-Msh6 (relative binding TC, TG, GG, CA > +1, TT, AA > GA, +2 > CC > +3 > +4), with the exception that the overall level of binding of Msh2-Msh3 to both base paired and mispaired DNA was somewhat higher in the presence of ADP than in the absence of nucleotide. Strikingly, the addition of ATP to the binding reactions eliminated the preferential binding of Msh2-Msh3 to mispaired versus base-paired DNA. In contrast, the addition of ATP to the Msh2-Msh6 binding reactions had complex effects resulting in modestly reduced binding to the +1 insertion/deletion, almost complete elimination of binding to the +2, +3, and +4 insertion/deletions, enhancement of binding to the TG and CA mispairs, modestly reduced binding to the AA and CC mispairs, and little if any effect on binding to the other four mispairs (relative binding TG ≫ CA, GG > TC, TT > GA > +1 ≫ AA > +3, CC, +4, +2). Overall, the observations that Msh2-Msh3 and Msh2-Msh6 bind relatively equally to the +1 insertion/deletion mispair, that Msh2-Msh3 binds to the +2, +3, and +4 insertion/deletions to an increasingly greater extent than Msh2-Msh6 as the size of the insertion/deletion increases, and that Msh2-Msh6 binds to the different base:base mispairs to a greater extent than Msh2-Msh3 generally parallel the results of genetic studies on the mispair specificity of Msh2-Msh3- and Msh2-Msh6-mediated MMR with the exception that the binding of Msh2-Msh6 to +2, +3, and +4 insertion/deletion mispairs is greater than predicted from genetic studies (25, 26, 29, 30, 33, 72, 73).
TABLE 1.
Binding of Msh2-Msh6 and Msh2-Msh3 to different mispairs determined by SPR
The binding of Msh2-Msh6 or Msh2-Msh3 to the indicated mispairs was determined by SPR as described under “Experimental Procedures” in the absence of nucleotide (−NT), the presence of ADP (+ADP), or the presence of ATP (+ATP) as indicated. The values reported are the RUs bound to the mispair at 100 s after the start of the protein injection minus the RU value obtained for the base pair (GC) control run in each experiment. The average value from up to five independent experiments is reported. The S.E. was typically in the range of 3–10% except for +2 (Msh2-Msh6 -NT, +ADP), CC (Msh2-Msh6 +ADP, +ATP), GG (Msh2-Msh3 +ADP), and TC (Msh2-Msh3 +ADP), where the S.E. was in the range of 12–20%. NS, not significantly above the binding to the GC control DNA.
| Mispair | Msh2-Msh6 | Msh2-Msh3 | ||||
|---|---|---|---|---|---|---|
| −NT | +ADP | +ATP | −NT | +ADP | +ATP | |
| +1 | 137 | 123 | 98 | 130 | 150 | NS |
| +2 | 97 | 91 | NS | 142 | 130 | NS |
| +3 | 67 | 51 | 25 | 133 | 123 | NS |
| +4 | 29 | 33 | NS | 144 | 129 | NS |
| CC | 91 | 67 | 24 | 57 | 80 | NS |
| TG | 153 | 141 | 258 | NS | NS | NS |
| GG | 163 | 139 | 165 | 26 | 22 | NS |
| TC | 163 | 141 | 130 | 22 | 24 | NS |
| AA | 135 | 106 | 50 | 83 | 76 | NS |
| GA | 124 | 95 | 108 | NS | NS | NS |
| TT | 136 | 119 | 129 | NS | 24 | NS |
| CA | 139 | 137 | 173 | NS | 29 | NS |
Msh2-Msh3 Forms ATP-dependent Sliding Clamps on Substrates That Contain Insertion/deletion Mispairs and Specific Base-Base Mispairs
Previous studies have shown that in the presence of ATP, the mispair-bound but not base pair-bound Msh2-Msh6 complex forms a clamp that slides away from the mispair and dissociates from the ends of the mispaired base-containing DNA (38, 54, 70, 74, 75). Because much less is known about the formation and properties of Msh2-Msh3 sliding clamps, we tested whether the Msh2-Msh3 complex forms a sliding clamp on different mispair-containing DNA substrates (Figs. 4 and 5). These experiments used a previously described reversible _lac_O-LacI-based end blocking system in which LacI bound to the free end of the substrate DNA traps the sliding clamp on the DNA, and addition of IPTG to release the LacI end block results in diagnostic rapid dissociation of the trapped sliding clamp (38). The binding of Msh2-Msh3 to the GC DNA was not enhanced by the end block in the presence of ATP. In contrast, the binding of Msh2-Msh3 to the +1, +2, +3, and +4 insertion/deletions and the AA, CC and to a much lesser extent to the TG mispair-containing substrates but not the other five base:base mispairs was significantly higher in the presence of the end block and ATP. Removal of the end block by the addition of IPTG caused the rapid dissociation of the Msh2-Msh3 complex. These results indicate that the Msh2-Msh3 complex efficiently forms an ATP-dependent sliding clamp on its preferred mispaired substrates.
FIGURE 4.

Msh2-Msh3 forms ATP-dependent sliding clamps on substrates containing insertion/deletion mispairs and specific base-base mispairs. The ability of Msh2-Msh3 and Msh2-Msh6 to form sliding clamps on homoduplex DNA or various mispair-containing substrates was monitored using SPR analysis as described under “Experimental Procedures” in the presence of ADP (+ADP) or ATP (+ATP), as indicated. The sensorgrams show the binding to substrates with free ends (dashed red lines) or ends blocked by bound LacI (solid red) followed by release of the end block (green) by the addition of IPTG. The mispair substrate analyzed is indicated at the right of each row of sensorgrams. The x axis indicates the time of association in seconds, the y axis indicates the extent of binding in RUs after reference subtraction, and the black arrows indicate the time at which the injection of protein was initiated.
FIGURE 5.

Msh2-Msh3 forms ATP-dependent sliding clamps on substrates containing insertion/deletion mispairs and specific base-base mispairs. See the legend to Fig. 4 for experimental details and a key for abbreviations used.
Consistent with previous results (38), the end block increased the binding of Msh2-Msh6 to the +1 insertion/deletion and TG mispair-containing substrates relative to the non-end blocked substrates in the presence of ATP, and the addition of IPTG resulted in rapid dissociation of Msh2-Msh6 from these two substrates consistent with the formation of ATP-dependent sliding clamps. We also found that the end block increased Msh2-Msh6 binding to the +2 insertion/deletion and the AA, GG, TC, TT, GA, and CA mispair-containing substrates relative to the non-end blocked substrates in the presence of ATP and that the addition of IPTG resulted in rapid dissociation of Msh2-Msh6, consistent with the formation of ATP-dependent sliding clamps. Strikingly, the end block did not increase Msh2-Msh6 binding to the +3 and +4 insertion/deletions in the presence of ATP and the addition of IPTG did not result in dissociation of Msh2-Msh6, consistent with the lack of formation of ATP-dependent sliding clamps on these two substrates. The end block also resulted in very little increased binding of Msh2-Msh6 to the CC mispair in the presence of ATP, although the addition of IPTG did result in dissociation of Msh2-Msh6 consistent with a much lower level of sliding clamp formation than seen on the other base:base mispairs.
The observation that Msh2-Msh3 did not show any mispair-specific binding in the presence of ATP on DNA substrates lacking an end block was in striking contrast to the behavior of Msh2-Msh6. This suggests that Msh2-Msh3 may have different kinetics of mispair association and/or dissociation in the presence of ATP compared with Msh2-Msh6. To investigate this, we determined the half times of association and IPTG-induced dissociation for Msh2-Msh3 and Msh2-Msh6 in the presence of ATP for the +1 insertion/deletion substrate, because Msh2-Msh3 and Msh2-Msh6 show relatively equal binding to this substrate. Under these conditions, Msh2-Msh3 and Msh2-Msh6 had similar half times of association (12.0 ± 1.2 s versus 15.7 ± 3.0 s, respectively), whereas Msh2-Msh3 had a more rapid half time of dissociation compared with that of Msh2-Msh6 (0.67 ± 0.08 s versus 2.61 ± 0.42 s, respectively) consistent with the lack of a steady state level of mispair binding in the presence of ATP on DNA substrates lacking an end block observed for Msh2-Msh3. This rapid dissociation seen with Msh2-Msh3 could reflect altered mispair and/or ATP binding compared with Msh2-Msh6. To gain insight into this, we compared Msh2-Msh3, Msh2-Msh6, and a chimeric derivative of Msh2-Msh6 containing the Msh3 mispair binding domain (called Msh2-Msh6(3-MBD)) that confers Msh3 mispair binding specificity onto Msh2-Msh6 (67) for their ability to bind to the +1 insertion/deletion substrate lacking an end block in the presence of ATP (Fig. 6). High steady state mispair binding was observed with Msh2-Msh6 and Msh2-Msh6(3-MBD) but not with Msh2-Msh3, supporting the idea that the rapid ATP-induced dissociation of Msh2-Msh3 from mispairs is a property of the Msh3 ATP binding domain or regions that communicate with the mispair binding domain but not the mispair binding domain itself and that the observed rapid dissociation may reflect the ATP binding properties of Msh2-Msh3. The slower end-dependent dissociation of Msh2-Msh6 could also reflect a combination of a major, rapidly dissociating component and a smaller fraction of sliding clamps that become trapped on the mispaired substrate and dissociate more slowly. However, this seems unlikely because our previous SPR study showed that most of the Msh2-Msh6 bound to an unblocked TG substrate dissociated rapidly when challenged with ATP and that the levels of ATP-resistant Msh2-Msh6 binding to the TG substrate were equal to the levels of ATP-resistant nonspecific binding to control DNA lacking a mispair (40).
FIGURE 6.
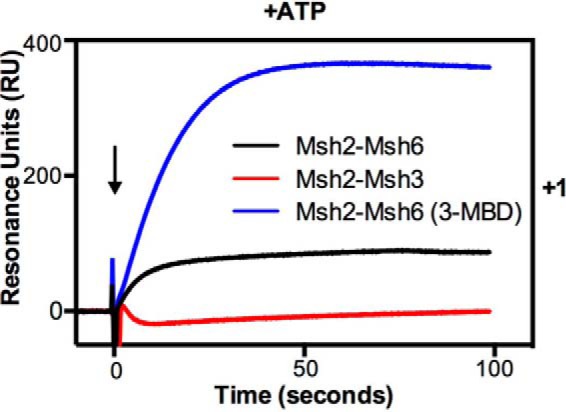
The low steady-state binding in the presence of ATP is not dependent on the Msh2-Msh3 mispair-binding domain. The binding of Msh2-Msh3 (red), Msh2-Msh6 (black), and the chimeric protein Msh2-Msh6(3-MBD) (blue) to the +1 insertion/deletion mispair and the homoduplex substrate was monitored using SPR analysis as described under “Experimental Procedures” in the presence of ATP (+ATP). The sensorgram shows the binding to +1 after subtraction of the RUs observed on the homoduplex substrate. The x axis indicates the time of association in seconds, the y axis indicates the extent of binding in RUs, and the black arrow indicates the time at which the injection of protein was initiated.
The msh3 K158E and R195D Mutations Cause Differential Effects on the Binding of the Msh2-Msh3 Complex to Small and Large Insertion/Deletion Mispairs
Previous experiments have suggested that the interaction of Msh2-Msh3 with small (+1) insertion/deletion mispairs involves additional contacts with the DNA compared with the interaction with larger (+4) insertion/deletion mispairs (57). To investigate this model, we tested the effect of two msh3 mutations on binding of Msh2-Msh3 to the +1 and +4 insertion/deletion mispairs: the msh3-K158E mutation, which causes as high an increase in mutation rate in mono-nucleotide and tetra-nucleotide frameshift reporter assays as an _msh3_Δ mutation, and the msh3-R195D mutation, which causes ∼80 and ∼10% increases in mutation rate in the mono-nucleotide and tetra-nucleotide frameshift reporter assays, respectively, relative to an _msh3_Δ mutation (57). Initially, mispair binding by the mutant complexes was evaluated using DNA substrates without an end block in the absence of added nucleotide (Fig. 7A). Under these conditions, the Msh2-Msh3-K158E protein showed essentially no binding, and the Msh2-Msh3-R195D protein showed reduced but still significant binding to the +1 insertion/deletion compared with Msh2-Msh3, whereas both mutant proteins showed reduced but still significant binding to the +4 insertion/deletion compared with Msh2-Msh3. Next, we evaluated mispair binding under a range of protein concentrations using DNA substrates with an end block in the presence of ATP followed by the addition of IPTG to determine whether sliding clamps were formed (Fig. 7, B and C). Under these conditions the Msh2-Msh3-K158E protein showed little or no binding to the +1 insertion/deletion at any protein concentration and showed reduced binding to the +4 insertion/deletion that was ∼80% of the level seen with Msh2-Msh3. The Msh2-Msh3-K158E protein formed sliding clamps on the +4 insertion/deletion as the bound protein dissociated rapidly on addition of IPTG. In contrast, the Msh2-Msh3-R195D protein showed markedly reduced but still significant binding to the +1 insertion/deletion and showed only slightly reduced binding to the +4 insertion/deletion that was ∼40 and ∼90% of the levels seen with Msh2-Msh3, respectively. The Msh2-Msh3-R195D protein formed sliding clamps on the +1 and +4 insertion/deletions because the bound protein dissociated rapidly on the addition of IPTG. These results show that the Msh2-Msh3-K158E protein is more generally defective in mispair binding than the Msh2-Msh3-R195D protein and that the Msh2-Msh3-R195D protein has little or no defect in binding +4 insertion/deletions consistent with the results of previous genetic experiments (57).
FIGURE 7.
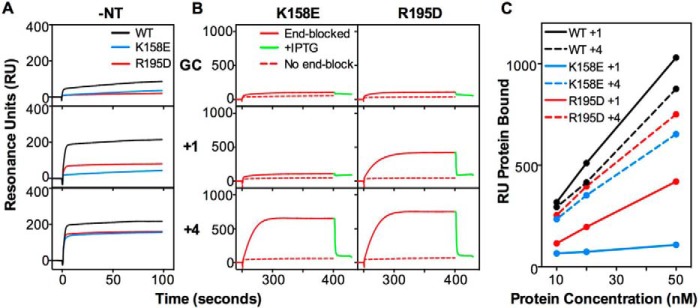
The Msh3 K158E and R195D mutations differentially affect binding to small versus large insertion-containing mispairs. A, the binding of wild-type Msh2-Msh3 (black) and the Msh2-Msh3-K158E (blue) and Msh2-Msh3-R195D (red) mutant complexes (50 nm) to homoduplex DNA and substrates containing a +1 or +4 insertion/deletion mispairs was monitored using SPR as described under “Experimental Procedures” in the absence of added nucleotide (−NT). B, sliding clamp formation by the Msh2-Msh3-K158E and Msh2-Msh3-R195D mutant complexes (50 nm) on the indicated substrates was monitored in the presence of ATP by SPR. The sensorgrams show the binding to substrates with free ends (dashed red lines) or ends blocked by bound LacI (solid red) followed by release of the end block (green) by the addition of IPTG. C, comparison of the binding of wild-type Msh2-Msh3 (black) and the K158E (blue) and R195D (red) mutant complexes to the +1 (solid lines) and +4 (dashed lines) end blocked substrates at different protein concentrations. The RUs observed immediately before IPTG addition are plotted.
Msh2-Msh3 Recruits Mlh1-Pms1 to Substrates That Contain Insertion/Deletion Mispairs and Specific Base-Base Mispairs
The ATP- and mispair-dependent recruitment of Mlh1-Pms1 by Msh2-Msh6 and Msh2-Msh3 is thought to be a critical step in MMR (1–3, 6, 76). Here we used end-blocked substrates to preclude the binding of Mlh1-Pms1 to the DNA ends (38) to evaluate the ability of Msh2-Msh3 to recruit Mlh1-Pms1 on different mispairs in the presence of ATP. Parallel control experiments were performed with Msh2-Msh6 for comparative purposes and to evaluate the recruitment of Mlh1-Pms1 by Msh2-Msh6 using a broader set of mispaired substrates than has been previously studied. In these experiments Msh2-Msh3 or Msh2-Msh6 was first flowed over the DNA substrate to allow mispair binding, and then a mixture of Mlh1-Pms1 and either Msh2-Msh3 or Msh2-Msh6 (to allow for recruitment of Mlh1-Pms1) or Msh2-Msh3 or Msh2-Msh6 alone (as a control) was flowed over the DNA substrate (Fig. 8).
FIGURE 8.

Msh2-Msh3 recruits Mlh1-Pms1 preferentially to insertion/deletion mispairs and specific base-base mispairs. The recruitment of Mlh1-Pms1 by Msh2-Msh6 (left) and Msh2-Msh3 (right) to different mispaired substrate DNAs containing ends blocked by bound LacI was monitored SPR in the presence of ATP as described under “Experimental Procedures.” The binding of Msh2-Msh6 or Msh2-Msh3 alone (black lines) followed by the addition of Msh2-Msh6 or Msh2-Msh3 plus Mlh1-Pms1 (purple lines) or Msh2-Msh6 or Msh2-Msh3 minus Mlh1-Pms1 (dashed black lines) are shown. The mispair substrate analyzed is indicated at the right of each row of sensorgrams. The x axis indicates the time of association in seconds, and the y axis indicates the extent of binding in RUs after subtraction of the no DNA reference binding and the binding observed with Mlh1-Pms1 alone (in the absence of Msh2-Msh3 or Msh2-Msh6).
We observed time-dependent binding of Mlh1-Pms1 to homoduplex DNA in the presence of Msh2-Msh3 but not in the absence of Msh2-Msh3; this mispair-independent recruitment of Mlh1-Pms1 by Msh2-Msh3 was greater than observed with Msh2-Msh6. In contrast, we observed much greater levels of recruitment of Mlh1-Pms1 by Msh2-Msh3 on the +1, +2, +3, and +4 insertion/deletion- and CC and AA mispair-containing substrates, which are the preferred mispaired substrates for Msh2-Msh3, as well as possibly a low level of recruitment of Mlh1-Pms1 on the GG mispair substrate. The recruitment of Mlh1-Pms1 by Msh2-Msh3 to the TG, TC, TT, GA, and CA substrates did not exceed the level of recruitment to the homoduplex control DNA substrate. Consistent with previous studies (37, 38, 41), we detected the recruitment of Mlh1-Pms1 by Msh2-Msh6 to the +1 insertion/deletion and TG mispair substrates. We also detected the recruitment of Mlh1-Pms1 by Msh2-Msh6 to the +2 insertion/deletion and CC, AA, GG, TC, TT, GA, and CA mispair substrates with the level of recruitment to the CC, AA, and GA mispairs being somewhat lower than seen with the other mispaired substrates. In contrast, there did not appear to be any significant recruitment of Mlh1-Pms1 by Msh2-Msh6 to the +3 and +4 insertion/deletion mispair substrates above that seen with the homoduplex control DNA substrate. Overall, the mispair substrate dependence for Mlh1-Pms1 recruitment by both Msh2-Msh3 and Msh2-Msh6 correlated well with the mispair substrate dependence for the formation of sliding clamps.
DISCUSSION
In the present study we optimized the expression and purification of the S. cerevisiae Msh2-Msh3 complex. Previous biochemical studies used Msh2-Msh3 purified from the wrong Msh3 start codon or used protocols that generated very limited amounts of purified protein (29, 36, 59). Here, the use of a plasmid-based expression system greatly facilitated the production of mutant proteins, and the availability of larger amounts of highly purified Msh2-Msh3 facilitated analysis using biophysical methods such as SPR. We performed a comparative study of the ability of the Msh2-Msh3 and Msh2-Msh6 complexes to bind to four different insertion/deletion mispairs and eight different base:base mispairs in the same sequence context, form ATP-induced sliding clamps, and recruit Mlh1-Pms1. This analysis showed that the Msh2-Msh3 complex formed functional mispair-bound complexes as evidenced by sliding clamp formation and Mlh1-Pms1 recruitment on the +1, +2, +3, and +4 insertion/deletions and CC, AA, and to a lesser extent on the GG mispairs but did not functionally interact with the other five base:base mispairs. In contrast, the Msh2-Msh6 complex formed functional mispair-bound complexes on the +1 and +2 insertion/deletions and seven of the eight base:base mispairs but did not functionally interact with the +3 or +4 insertion/deletion mispairs and showed very little functional binding of the CC mispair. Compared with Msh2-Msh6, the Msh2-Msh3 complex exhibited a much faster sliding clamp mode of dissociation, a property that appears to be independent of the mispair binding domain of Msh3. Analysis of two different mutant Msh2-Msh3 complexes containing the Msh3-K158E or R195D amino acid substitutions provided support for the view from genetic and structural studies that binding of smaller mispairs requires a greater number of protein-DNA contacts than binding of larger mispairs (57, 58). Together, the results from these experiments provide a base-line characterization of the interaction between Msh2-Msh3 and the major mispairs thought to be repaired by MMR, an extension of the available mispair interaction data for Msh2-Msh6, and biochemical evidence for the functional differences between Msh2-Msh3 and Msh2-Msh6 predicted from genetic studies.
Previous studies have suggested that the recognition of mispairs by the Msh2-Msh3 and Msh2-Msh6 complexes occurs by related but different mechanisms. This was initially suggested by the fact that the key mispair-contacting residue is different in Msh6 (Phe-337 in S. cerevisiae) and Msh3 (Lys-158 in S. cerevisiae), and the fact that Msh2 domain 1 that contains DNA backbone-contacting residues is only required for MMR by Msh2-Msh3 but not Msh2-Msh6 (29, 53, 57, 77). It was subsequently proposed that the recognition of smaller insertion/deletion mispairs by Msh2-Msh3 requires mispair binding-induced bending and strand separation of the DNA by the core mispair-contacting residues and stabilization by additional contacts with the Msh3 mispair-binding domain (57, 58). In contrast, Msh2-Msh3-mediated recognition of DNAs that contain larger insertion/deletion mispairs, which are intrinsically bent and strand-separated, was suggested to only require the core mispair-contacting residues and be less dependent on the stabilization by additional contacts with Msh3 (57, 58). Consistent with this, the msh3-K158E mutation affecting the core mispair-contacting residue (57) caused strong defects in the binding of Msh2-Msh3 to both +1 and +4 insertion/deletion mispairs and in sliding clamp formation. In contrast, the msh3-R195D mutation that affects one of the stabilization contacts (57) caused no defect in Msh2-Msh3 binding or sliding clamp formation with the +4 insertion/deletion and caused a stronger but still modest defect in Msh2-Msh3 binding with the +1 insertion/deletion. It should be noted that the msh3-R195D mutation, like the other mutations predicted to affect stabilizing contacts, does not cause complete loss of repair of a one-base insertion/deletions in vivo (57). These results support the model that the stabilization contacts are more important for the recognition of mispaired DNAs that are less intrinsically bent than for recognition of mispairs that are more intrinsically bent but suggest that stabilization of less intrinsically bent mispaired DNAs requires contributions from many DNA contacting residues, and hence, no one such residue is absolutely required (57, 58). It is not readily apparent how Msh2-Msh3 recognizes some base:base mispairs (CC, AA, GG) but not others, although all of the recognized base:base mispairs appear to have increased flexibility and helix destabilization, as shown by NMR spectroscopy studies (78–82). We also observed that Msh2-Msh3 shows extremely rapid ATP-induced sliding-mediated dissociation from mispairs compared with Msh2-Msh6 and demonstrated that this property is conferred by the nucleotide binding domains of Msh2-Msh3; however, additional studies are required to elucidate how ATP binding induces this difference in the kinetics of dissociation.
Analysis of MMR in S. cerevisiae using frameshift reversion and forward mutation assays has led to the hypothesis that the Msh2-Msh6 and Msh2-Msh3 complexes play distinct but partially overlapping roles in MMR. Msh2-Msh6 was initially suggested to be responsible for repair of base:base mispairs as well as functioning in the repair of +1 and to a lesser extent +2 insertion/deletion mispairs with Msh2-Msh3 being equally responsible for the repair of +1 insertion/deletions and playing an increasingly dominant role in the repair of larger insertion/deletions with insertion/deletions as large as +4 being solely repaired by Msh2-Msh3 (1, 3, 25, 26, 76). However, recent analysis of mutation spectra for different MMR defective mutants has suggested that Msh2-Msh3 can also repair selected base:base mispairs (29). The mispair specificity of human and mouse MMR, although being based on less extensive genetic experiments, generally agrees with the S. cerevisiae data on mispair specificity with the exception of conflicting data in regard to whether Msh2-Msh3 can repair +1 insertion/deletions (24, 30–33). The data presented here clearly show that Msh2-Msh3 can bind and form sliding clamps on and recruit Mlh1-Pms1 to +1, +2, +3, and +4 insertion/deletion substrates as well as at a lower level to the CC, AA, and GG base:base mispair substrates that were previously implicated as potential substrates for Msh2-Msh3 in vivo. The analysis presented also showed that Msh2-Msh6 can bind, form sliding clamps on, and recruit Mlh1-Pms1 to seven of the eight base:base mispairs and a +1 insertion/deletion; this analysis only detected a weak interaction with the +2 insertion/deletion, no functional interaction with the +3 and +4 insertion/deletions, and little functional interaction with the CC mispair. By incorporating analysis of sliding clamp formation and Mlh1-Pms1 recruitment into our studies of Msh2-Msh3 and Msh2-Msh6, both of which have generally not been performed on an extended series of mispaired substrates, we were able to clearly distinguish simple mispair binding from more functionally significant interactions. Overall, the results presented support the view that Msh2-Msh3 can act in the repair of some base:base mispairs, that Msh2-Msh6 can act in the repair of +1 and +2 insertion/deletions, and that Msh2-Msh3 is likely exclusively responsible for the repair of insertion/deletion mispairs larger than +2. In addition, the observation that Msh2-Msh3 forms sliding clamps and recruits Mlh1-Pms1 on a CC mispair to a greater extent than Msh2-Msh6 raises the possibility that CC mispairs, which are generally thought to be poorly repaired mispairs (83–85), are repaired to a greater extent by Msh2-Msh3-dependent MMR than Msh2-Msh6-dependent MMR.
Interestingly, the specificity of Msh2-Msh3 and Msh2-Msh6 for repair of mispairs in MMR reactions in vitro does not correlate with mispair specificity for in vitro sliding clamp formation and Mlh1-Pms1 recruitment or in vivo MMR but does correlate to a greater extent with mispair binding in the absence of added nucleotide in vitro (32, 34). There are several possible explanations for this. First, the rate-limiting step for in vitro MMR, which is potentially the mispair- and Msh2-Msh6-dependent stimulation of exonuclease 1 (86), may depend only on mispair binding by Msh2-Msh6 or Msh2-Msh3. Second, in vitro MMR may not require Msh2-Msh6 or Msh2-Msh3 sliding clamp formation or Mlh1-Pms1 recruitment. This is likely true in the case of 5′ repair that does not require Mlh1-Pms1/Pms2 in vitro and could also be the case for the reconstituted 3′ repair using human proteins, which requires the Mlh1-Pms2 endonuclease, especially if the recruitment and activation of Mlh1-Pms2 are primarily dependent on proliferating cell nuclear antigen and Replication Factor C (34, 86–91). Finally, in vitro MMR may not reflect all of the steps of in vivo MMR such as coupling to replication (1–7, 69, 76, 92). The differences between MMR in vivo, the biochemical properties of MMR proteins, and in vitro MMR argue for continued efforts to develop MMR reaction conditions in vitro that allow these reactions to better mimic the in vivo pathways.
Acknowledgments
We thank the members of the Kolodner laboratory for helpful discussions, Drs. Victoria Hargreaves and Scarlet Shell for samples of Msh2-Msh6 and Msh2-Msh6(3-MBD), respectively, and Drs. Eva Goellner, Christopher Putnam, and Catherine Smith for comments on the manuscript.
*
This work was supported, in whole or in part, by National Institutes of Health Grant GM50006 (to R. D. K.).
3
C. S. Campbell, H. Hombauer, A. Srivatsan, N. Bowen, A. Desai, C. D. Putnam, and R. D. Kolodner, personal communication.
2
The abbreviations used are:
MMR
DNA mismatch repair
SPR
surface plasmon resonance
RU
resonance units
SD
standard deviation
IPTG
isopropyl β-d-1-thiogalactopyranoside
Msh2
MutS homolog 2
Msh3
MutS homolog 3
Msh6
MutS homolog 6
Pms1
post meiotic segregation 1
Mlh1
MutL homolog 1
Mlh2
MutL homolog 2
Mlh3
MutL homolog 3.
REFERENCES
- 1.Harfe B. D., Jinks-Robertson S. (2000) DNA mismatch repair and genetic instability. Annu. Rev. Genet. 34, 359–399 [DOI] [PubMed] [Google Scholar]
- 2.Iyer R. R., Pluciennik A., Burdett V., Modrich P. L. (2006) DNA mismatch repair. Functions and mechanisms. Chem. Rev. 106, 302–323 [DOI] [PubMed] [Google Scholar]
- 3.Kolodner R. D., Marsischky G. T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9, 89–96 [DOI] [PubMed] [Google Scholar]
- 4.Modrich P. (1991) Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 25, 229–253 [DOI] [PubMed] [Google Scholar]
- 5.Jiricny J. (2013) Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 5, a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel T. A., Erie D. A. (2005) DNA mismatch repair. Annu. Rev. Biochem. 74, 681–710 [DOI] [PubMed] [Google Scholar]
- 7.Modrich P., Lahue R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65, 101–133 [DOI] [PubMed] [Google Scholar]
- 8.de la Chapelle A. (2004) Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 4, 769–780 [DOI] [PubMed] [Google Scholar]
- 9.Peltomäki P., Vasen H. F. (1997) Mutations predisposing to hereditary nonpolyposis colorectal cancer. Database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 113, 1146–1158 [DOI] [PubMed] [Google Scholar]
- 10.BØrresen A. L., Lothe R. A., Meling G. I., Lystad S., Morrison P., Lipford J., Kane M. F., Rognum T. O., Kolodner R. D. (1995) Somatic mutations in the hMSH2 gene in microsatellite unstable colorectal carcinomas. Hum. Mol. Genet. 4, 2065–2072 [DOI] [PubMed] [Google Scholar]
- 11.Kane M. F., Loda M., Gaida G. M., Lipman J., Mishra R., Goldman H., Jessup J. M., Kolodner R. (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 57, 808–811 [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research Network, Kandoth C., Schultz N., Cherniack A. D., Akbani R., Liu Y., Shen H., Robertson A. G., Pashtan I., Shen R., Benz C. C., Yau C., Laird P. W., Ding L., Zhang W., Mills G. B., Kucherlapati R., Mardis E. R., Levine D. A. (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peltomäki P. (2003) Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 21, 1174–1179 [DOI] [PubMed] [Google Scholar]
- 14.Matic I., Rayssiguier C., Radman M. (1995) Interspecies gene exchange in bacteria. The role of SOS and mismatch repair systems in evolution of species. Cell 80, 507–515 [DOI] [PubMed] [Google Scholar]
- 15.Sugawara N., Goldfarb T., Studamire B., Alani E., Haber J. E. (2004) Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. U.S.A. 101, 9315–9320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datta A., Adjiri A., New L., Crouse G. F., Jinks Robertson S. (1996) Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harfe B. D., Jinks-Robertson S. (2000) Mismatch repair proteins and mitotic genome stability. Mutat. Res. 451, 151–167 [DOI] [PubMed] [Google Scholar]
- 18.Selva E. M., New L., Crouse G. F., Lahue R. S. (1995) Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. Genetics 139, 1175–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stambuk S., Radman M. (1998) Mechanism and control of interspecies recombination in Escherichia coli. I. Mismatch repair, methylation, recombination and replication functions. Genetics 150, 533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Putnam C. D., Hayes T. K., Kolodner R. D. (2009) Specific pathways prevent duplication-mediated genome rearrangements. Nature 460, 984–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acharya S., Foster P. L., Brooks P., Fishel R. (2003) The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell 12, 233–246 [DOI] [PubMed] [Google Scholar]
- 22.Grilley M., Welsh K. M., Su S. S., Modrich P. (1989) Isolation and characterization of the Escherichia coli mutL gene product. J. Biol. Chem. 264, 1000–1004 [PubMed] [Google Scholar]
- 23.Su S. S., Modrich P. (1986) Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc. Natl. Acad. Sci. U.S.A. 83, 5057–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acharya S., Wilson T., Gradia S., Kane M. F., Guerrette S., Marsischky G. T., Kolodner R., Fishel R. (1996) hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. U.S.A. 93, 13629–13634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsischky G. T., Filosi N., Kane M. F., Kolodner R. (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 10, 407–420 [DOI] [PubMed] [Google Scholar]
- 26.Sia E. A., Kokoska R. J., Dominska M., Greenwell P., Petes T. D. (1997) Microsatellite instability in yeast. Dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol. 17, 2851–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drummond J. T., Li G. M., Longley M. J., Modrich P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 268, 1909–1912 [DOI] [PubMed] [Google Scholar]
- 28.Palombo F., Gallinari P., Iaccarino I., Lettieri T., Hughes M., D'Arrigo A., Truong O., Hsuan J. J., Jiricny J. (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 268, 1912–1914 [DOI] [PubMed] [Google Scholar]
- 29.Harrington J. M., Kolodner R. D. (2007) Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol. Cell. Biol. 27, 6546–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Risinger J. I., Umar A., Boyd J., Berchuck A., Kunkel T. A., Barrett J. C. (1996) Mutation of MSH3 in endometrial cancer and evidence for its functional role in heteroduplex repair. Nat. Genet. 14, 102–105 [DOI] [PubMed] [Google Scholar]
- 31.Palombo F., Iaccarino I., Nakajima E., Ikejima M., Shimada T., Jiricny J. (1996) hMutSβ, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr. Biol. 6, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 32.Genschel J., Littman S. J., Drummond J. T., Modrich P. (1998) Isolation of MutSβ from human cells and comparison of the mismatch repair specificities of MutSβ and MutSα. J. Biol. Chem. 273, 19895–19901 [DOI] [PubMed] [Google Scholar]
- 33.Edelmann W., Umar A., Yang K., Heyer J., Kucherlapati M., Lia M., Kneitz B., Avdievich E., Fan K., Wong E., Crouse G., Kunkel T., Lipkin M., Kolodner R. D., Kucherlapati R. (2000) The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 60, 803–807 [PubMed] [Google Scholar]
- 34.Bowen N., Smith C. E., Srivatsan A., Willcox S., Griffith J. D., Kolodner R. D. (2013) Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 18472–18477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugawara N., Pâques F., Colaiácovo M., Haber J. E. (1997) Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc. Natl. Acad. Sci. U.S.A. 94, 9214–9219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surtees J. A., Alani E. (2006) Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J. Mol. Biol. 360, 523–536 [DOI] [PubMed] [Google Scholar]
- 37.Habraken Y., Sung P., Prakash L., Prakash S. (1998) ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2-MSH6 and MLH1-PMS1 protein complexes. J. Biol. Chem. 273, 9837–9841 [DOI] [PubMed] [Google Scholar]
- 38.Mendillo M. L., Mazur D. J., Kolodner R. D. (2005) Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end blocking system. J. Biol. Chem. 280, 22245–22257 [DOI] [PubMed] [Google Scholar]
- 39.Prolla T. A., Pang Q., Alani E., Kolodner R. D., Liskay R. M. (1994) MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science 265, 1091–1093 [DOI] [PubMed] [Google Scholar]
- 40.Mendillo M. L., Hargreaves V. V., Jamison J. W., Mo A. O., Li S., Putnam C. D., Woods V. L., Jr., Kolodner R. D. (2009) A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc. Natl. Acad. Sci. U.S.A. 106, 22223–22228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blackwell L. J., Wang S., Modrich P. (2001) DNA chain length dependence of formation and dynamics of hMutSα·hMutLα·heteroduplex complexes. J. Biol. Chem. 276, 33233–33240 [DOI] [PubMed] [Google Scholar]
- 42.Habraken Y., Sung P., Prakash L., Prakash S. (1997) Enhancement of MSH2-MSH3-mediated mismatch recognition by the yeast MLH1-PMS1 complex. Curr. Biol. 7, 790–793 [DOI] [PubMed] [Google Scholar]
- 43.Wang T. F., Kleckner N., Hunter N. (1999) Functional specificity of MutL homologs in yeast. Evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc. Natl. Acad. Sci. U.S.A. 96, 13914–13919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flores-Rozas H., Kolodner R. D. (1998) The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. U.S.A. 95, 12404–12409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charbonneau N., Amunugama R., Schmutte C., Yoder K., Fishel R. (2009) Evidence that hMLH3 functions primarily in meiosis and in hMSH2-hMSH3 mismatch repair. Cancer Biol. Ther. 8, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harfe B. D., Minesinger B. K., Jinks-Robertson S. (2000) Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 10, 145–148 [DOI] [PubMed] [Google Scholar]
- 47.Nishant K. T., Plys A. J., Alani E. (2008) A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics 179, 747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zakharyevich K., Tang S., Ma Y., Hunter N. (2012) Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 149, 334–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abdullah M. F., Hoffmann E. R., Cotton V. E., Borts R. H. (2004) A role for the MutL homologue MLH2 in controlling heteroduplex formation and in regulating between two different crossover pathways in budding yeast. Cytogenet. Genome Res. 107, 180–190 [DOI] [PubMed] [Google Scholar]
- 50.Lamers M. H., Perrakis A., Enzlin J. H., Winterwerp H. H., de Wind N., Sixma T. K. (2000) The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature 407, 711–717 [DOI] [PubMed] [Google Scholar]
- 51.Obmolova G., Ban C., Hsieh P., Yang W. (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407, 703–710 [DOI] [PubMed] [Google Scholar]
- 52.Warren J. J., Pohlhaus T. J., Changela A., Iyer R. R., Modrich P. L., Beese L. S. (2007) Structure of the human MutSα DNA lesion recognition complex. Mol. Cell 26, 579–592 [DOI] [PubMed] [Google Scholar]
- 53.Bowers J., Sokolsky T., Quach T., Alani E. (1999) A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2-MSH6 complex disrupts mismatch recognition. J. Biol. Chem. 274, 16115–16125 [DOI] [PubMed] [Google Scholar]
- 54.Gradia S., Subramanian D., Wilson T., Acharya S., Makhov A., Griffith J., Fishel R. (1999) hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol. Cell 3, 255–261 [DOI] [PubMed] [Google Scholar]
- 55.Wilson T., Guerrette S., Fishel R. (1999) Dissociation of mismatch recognition and ATPase activity by hMSH2-hMSH3. J. Biol. Chem. 274, 21659–21664 [DOI] [PubMed] [Google Scholar]
- 56.Mendillo M. L., Putnam C. D., Mo A. O., Jamison J. W., Li S., Woods V. L., Jr., Kolodner R. D. (2010) Probing DNA- and ATP-mediated conformational changes in the MutS family of mispair recognition proteins using deuterium exchange mass spectrometry. J. Biol. Chem. 285, 13170–13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dowen J. M., Putnam C. D., Kolodner R. D. (2010) Functional studies and homology modeling of Msh2-Msh3 predict that mispair recognition involves DNA bending and strand separation. Mol. Cell. Biol. 30, 3321–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta S., Gellert M., Yang W. (2012) Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 19, 72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Habraken Y., Sung P., Prakash L., Prakash S. (1996) Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr. Biol. 6, 1185–1187 [DOI] [PubMed] [Google Scholar]
- 60.Swint-Kruse L., Elam C. R., Lin J. W., Wycuff D. R., Shive Matthews K. (2001) Plasticity of quaternary structure. Twenty-two ways to form a LacI dimer. Protein Sci. 10, 262–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wycuff D. R., Matthews K. S. (2000) Generation of an AraC-araBAD promoter-regulated T7 expression system. Anal. Biochem. 277, 67–73 [DOI] [PubMed] [Google Scholar]
- 62.Zhan H., Swint-Kruse L., Matthews K. S. (2006) Extrinsic interactions dominate helical propensity in coupled binding and folding of the lactose repressor protein hinge helix. Biochemistry 45, 5896–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Antony E., Hingorani M. M. (2003) Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry 42, 7682–7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hargreaves V. V., Shell S. S., Mazur D. J., Hess M. T., Kolodner R. D. (2010) Interaction between the Msh2 and Msh6 nucleotide-binding sites in the Saccharomyces cerevisiae Msh2-Msh6 complex. J. Biol. Chem. 285, 9301–9310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith C. E., Mendillo M. L., Bowen N., Hombauer H., Campbell C. S., Desai A., Putnam C. D., Kolodner R. D. (2013) Dominant mutations in S. cerevisiae PMS1 identify the Mlh1-Pms1 endonuclease active site and an exonuclease 1-independent mismatch repair pathway. PLoS Genet. 9, e1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerik K. J., Gary S. L., Burgers P. M. (1997) Overproduction and affinity purification of Saccharomyces cerevisiae replication factor C. J. Biol. Chem. 272, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 67.Shell S. S., Putnam C. D., Kolodner R. D. (2007) Chimeric Saccharomyces cerevisiae Msh6 protein with an Msh3 mispair-binding domain combines properties of both proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 10956–10961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin Y. L., Shivji M. K., Chen C., Kolodner R., Wood R. D., Dutta A. (1998) The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J. Biol. Chem. 273, 1453–1461 [DOI] [PubMed] [Google Scholar]
- 69.Hombauer H., Campbell C. S., Smith C. E., Desai A., Kolodner R. D. (2011) Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147, 1040–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gradia S., Acharya S., Fishel R. (1997) The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell 91, 995–1005 [DOI] [PubMed] [Google Scholar]
- 71.Mazur D. J., Mendillo M. L., Kolodner R. D. (2006) Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol. Cell 22, 39–49 [DOI] [PubMed] [Google Scholar]
- 72.Harfe B. D., Jinks-Robertson S. (1999) Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 4766–4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lühr B., Scheller J., Meyer P., Kramer W. (1998) Analysis of in vivo correction of defined mismatches in the DNA mismatch repair mutants msh2, msh3, and msh6 of Saccharomyces cerevisiae. Mol. Gen. Genet. 257, 362–367 [DOI] [PubMed] [Google Scholar]
- 74.Blackwell L. J., Martik D., Bjornson K. P., Bjornson E. S., Modrich P. (1998) Nucleotide-promoted release of hMutSα from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. J. Biol. Chem. 273, 32055–32062 [DOI] [PubMed] [Google Scholar]
- 75.Jiang J., Bai L., Surtees J. A., Gemici Z., Wang M. D., Alani E. (2005) Detection of high-affinity and sliding clamp modes for MSH2-MSH6 by single-molecule unzipping force analysis. Mol. Cell 20, 771–781 [DOI] [PubMed] [Google Scholar]
- 76.Modrich P. (2006) Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281, 30305–30309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee S. D., Surtees J. A., Alani E. (2007) Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J. Mol. Biol. 366, 53–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arnold F. H., Wolk S., Cruz P., Tinoco I., Jr. (1987) Structure, dynamics, and thermodynamics of mismatched DNA oligonucleotide duplexes d(CCCAGGG)2 and d(CCCTGGG)2. Biochemistry 26, 4068–4075 [DOI] [PubMed] [Google Scholar]
- 79.Borden K. L., Jenkins T. C., Skelly J. V., Brown T., Lane A. N. (1992) Conformational properties of the G.G mismatch in d(CGCGAATTGGCG)2 determined by NMR. Biochemistry 31, 5411–5422 [DOI] [PubMed] [Google Scholar]
- 80.Lane A. N., Jenkins T. C., Brown D. J., Brown T. (1991) NMR determination of the solution conformation and dynamics of the A.G mismatch in the d(CGCAAATTGGCG)2 dodecamer. Biochem. J. 279, 269–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kouchakdjian M., Li B. F., Swann P. F., Patel D. J. (1988) Pyrimidine·pyrimidine base-pair mismatches in DNA. A nuclear magnetic resonance study of T.T pairing at neutral pH and C·C pairing at acidic pH in dodecanucleotide duplexes. J. Mol. Biol. 202, 139–155 [DOI] [PubMed] [Google Scholar]
- 82.Lane A. N., Peck B. (1995) Conformational flexibility in DNA duplexes containing single G·G mismatches. Eur. J. Biochem. 230, 1073–1087 [DOI] [PubMed] [Google Scholar]
- 83.Su S. S., Lahue R. S., Au K. G., Modrich P. (1988) Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem. 263, 6829–6835 [PubMed] [Google Scholar]
- 84.Bishop D. K., Andersen J., Kolodner R. D. (1989) Specificity of mismatch repair following transformation of Saccharomyces cerevisiae with heteroduplex plasmid DNA. Proc. Natl. Acad. Sci. U.S.A. 86, 3713–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kramer B., Kramer W., Fritz H. J. (1984) Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell 38, 879–887 [DOI] [PubMed] [Google Scholar]
- 86.Genschel J., Bazemore L. R., Modrich P. (2002) Human exonuclease I is required for 5′ and 3′ mismatch repair. J. Biol. Chem. 277, 13302–13311 [DOI] [PubMed] [Google Scholar]
- 87.Dzantiev L., Constantin N., Genschel J., Iyer R. R., Burgers P. M., Modrich P. (2004) A defined human system that supports bidirectional mismatch-provoked excision. Mol. Cell 15, 31–41 [DOI] [PubMed] [Google Scholar]
- 88.Zhang Y., Yuan F., Presnell S. R., Tian K., Gao Y., Tomkinson A. E., Gu L., Li G. M. (2005) Reconstitution of 5′-directed human mismatch repair in a purified system. Cell 122, 693–705 [DOI] [PubMed] [Google Scholar]
- 89.Pluciennik A., Dzantiev L., Iyer R. R., Constantin N., Kadyrov F. A., Modrich P. (2010) PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 107, 16066–16071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kadyrov F. A., Dzantiev L., Constantin N., Modrich P. (2006) Endonucleolytic function of MutLα in human mismatch repair. Cell 126, 297–308 [DOI] [PubMed] [Google Scholar]
- 91.Kadyrov F. A., Holmes S. F., Arana M. E., Lukianova O. A., O'Donnell M., Kunkel T. A., Modrich P. (2007) Saccharomyces cerevisiae MutLα is a mismatch repair endonuclease. J. Biol. Chem. 282, 37181–37190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hombauer H., Srivatsan A., Putnam C. D., Kolodner R. D. (2011) Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334, 1713–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]