A New Role for Escherichia coli DsbC Protein in Protection against Oxidative Stress (original) (raw)
Background: DsbC is a protein-disulfide isomerase present in the periplasm of Gram-negative bacteria.
Results: We discovered that DsbC also regulates the redox state of the single cysteine residue of the l-arabinose-binding protein AraF.
Conclusion: DsbC is involved in the protection of single cysteine residues against oxidative stress.
Significance: This finding reveals a new link between oxidative stress protection and oxidative protein folding.
Keywords: Disulfide, Escherichia coli, Oxidative Stress, Protein Folding, Redox Regulation, Thioredoxin, DsbC, Sulfenic Acid
Abstract
We report a new function for Escherichia coli DsbC, a protein best known for disulfide bond isomerization in the periplasm. We found that DsbC regulates the redox state of the single cysteine of the l-arabinose-binding protein AraF. This cysteine, which can be oxidized to a sulfenic acid, mediates the formation of a disulfide-linked homodimer under oxidative stress conditions, preventing l-arabinose binding. DsbC, unlike the homologous protein DsbG, reduces the intermolecular disulfide, restoring AraF binding properties. Thus, our results reveal a new link between oxidative protein folding and the defense mechanisms against oxidative stress.
Introduction
The cell envelope of Gram-negative bacteria is characterized by the presence of two membranes, the outer membrane and the inner membrane, which are separated by the periplasm, a viscous compartment that contains a thin layer of peptidoglycan and represents 10–20% of the total cell volume (1). More than 300 proteins are present in the periplasm where they perform a large variety of functions, such as uptake and transport of nutrients and detoxification of harmful substances.
In contrast to their cytoplasmic counterparts, many periplasmic proteins are stabilized by the formation of one or more disulfide bonds that are essential for conformational stability and biological activity (2). Although the majority of the cysteine residues present in periplasmic proteins are involved in disulfide bonds (3), a small but significant fraction of secreted proteins possess cysteine residues that remain in the reduced thiol (-SH) state. These reduced cysteine residues are therefore potential targets for the reactive oxygen species (ROS)2 formed as by-products of aerobic metabolism or generated by the host defenses to kill the invading bacteria.
The first oxidation product of a cysteine residue exposed to ROS is the sulfenic acid derivative (-SOH), a highly reactive intermediate that, unless stabilized within a protein microenvironment, either reacts with another thiol present in the vicinity to form a disulfide or is irreversibly oxidized to sulfinic (-SO2H) and sulfonic (-SO3H) acids (4). As these latter modifications can lead to the irreversible inactivation of the damaged proteins, cells have developed protective mechanisms to prevent sulfenic acid oxidation (5).
Recently, we discovered that Escherichia coli DsbG, a periplasmic oxidoreductase from the thioredoxin superfamily, is involved in the protection of a family of enzymes that cross-link the major outer membrane lipoprotein Lpp to the peptidoglycan (6). We found that these enzymes, known as “l,d-transpeptidases,” form a sulfenic acid on a cysteine residue essential for activity and that DsbG controls the level of sulfenylation of this residue. Our results also suggested that DsbC, an oxidoreductase homologous to DsbG, that is best known as a periplasmic protein-disulfide isomerase (7, 8), functions as a backup for DsbG in the control of sulfenylation in the periplasm (6). However, the role, if any, played by DsbC in the defense mechanisms against oxidative stress remained elusive.
We decided to investigate whether DsbC specifically protects certain periplasmic proteins from oxidative damage. Our initial strategy to address this question was to investigate whether DsbC interacts with proteins presenting a single cysteine residue, as these proteins, which do not form nonnative disulfide bonds, cannot use the disulfide isomerase activity of DsbC. By using a well established technique to trap the substrates of proteins with a thioredoxin fold (9), we found that an abundant substrate of DsbC is the arabinose-binding protein AraF, a soluble protein with a single cysteine residue. The function of AraF is to bind l-arabinose in the periplasm and to deliver it to the inner membrane protein complex AraH-AraG. We also found that AraF forms a disulfide-linked dimer under oxidative stress conditions, which prevents l-arabinose binding. Remarkably, DsbC, unlike DsbG, is able to reduce the dimer, highlighting the specificity of the interaction. Altogether, our results show that the protein-disulfide isomerase DsbC plays a role in the protection of free cysteine residues against ROS, connecting oxidative protein folding to the defense mechanisms against oxidative stress.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Bacterial strains are E. coli MC4100 derivatives (10). All alleles were moved by P1 transduction using standard procedures (11). Unless indicated, bacteria were grown aerobically at 37 °C in LB medium or in M63 minimal medium, supplemented with 0.2% glycerol, 0.2% casamino acids, vitamins (10 μg/ml thiamin, 1 μg/ml biotin, 10 μg/ml riboflavin, 10 μg/ml nicotinamide), and 1 mm MgSO4. When necessary, growth media were supplemented with chloramphenicol (25 μg/μl).
TABLE 1.
Strains and plasmids used in this study
| Plasmids | Characteristics | References |
|---|---|---|
| pBAD33a | l-Arabinose inducible, CmR selection | (35) |
| pMD35 | pBAD33a::DsbCCXXS-His6 | (31) |
| pJFC355 | pBAD33a::DsbC | Received from J. Bardwell |
| pJFC369 | pBAD33a::DsbCSXXS | Received from J. Bardwell |
| pMD41 | pBAD33a::DsbGCXXA-His6 | (6) |
| pET28a | IPTG-inducible, KanR selection | Novagen |
| pKD1 | pET28a AraF-His6 | This study |
| pKD2 | pET28a AraF | This study |
| Strains | Genotype and characteristics | References |
|---|---|---|
| MC4100 | F-, _ara_D139,Δ (arg F-lac)U169, _pts_F25, _rel_A1, _flb_5301, _rps_L 150.λ- | (10) |
| JAS27 | MC4100 Δ_ara_BAD | Received from T. Silhavy |
| MD238 | JAS27 dsbC | This study |
| KD44 | JAS27 dsbC::kan | This study |
| KD78 | JAS27 dsbG::kan | This study |
| KD80 | KD44 carrying pMD35 | This study |
| KD192 | KD78 carrying pMD41 | This study |
| KD215 | MD238 carrying pBAD33a | This study |
| KD216 | JAS27 carrying pBAD33a | This study |
| KD217 | MD238 carrying pJFC355 | This study |
Plasmid Construction
The AraF expression vector was constructed as follows. The region encoding the mature AraF protein (without the signal sequence) was amplified from the chromosome using primers AraF_ Fw (5′-AAACCATGGAGAACCTGAAGCTCGGTTTTC-3′) and AraF_ Rv (5′-CCCCTCGAGCTTACCGCCTAAACCTTTTTTC-3′) and cloned into a pET28a vector, generating plasmid pKD1. The stop codon at the 3′ end of the araF gene was added by site-directed mutagenesis using primers AraFSTOP_ Fw (5′-GGTTTAGGCGGTAAGTGAGAGCACCACCAC-3′) and AraFSTOP_ Rv (5′-GTGGTGGTGCTCTCACTTACCGCCTAAACC-3′), generating plasmid pKD2.
Trapping and Purification of the AraF-DsbCCXXS Complex
Strain KD80 was grown in LB medium at 37 °C to reach an _A_600 of 0.6. Expression of DsbCC_XX_S was induced for 5 h by the addition of 0.2% l-arabinose. After precipitation with 10% TCA, the proteins were resuspended in 100 mm NaPi, pH 8, 300 mm NaCl, 0.3% SDS, 8 m urea, and 100 mm iodoacetamide (IAM) to prevent disulfide bond rearrangement. The lysate was centrifuged at 23,000 × g for 45 min. Ni-NTA resin (Qiagen) was then added to the supernatant and the mixture incubated overnight at room temperature. The Ni-NTA resin was packed in a 1-ml column and washed thoroughly using 100 mm NaPi, pH 8, 300 mm NaCl, and 0.3% SDS. DsbCC_XX_S and the proteins bound to it were eluted with a step gradient from 30 mm to 300 mm imidazole. Only one fraction eluted from the column. This fraction was concentrated 10-fold and analyzed by SDS-PAGE with or without dithiothreitol (DTT). After electrophoresis, the SDS-polyacrylamide gel was stained with Coomassie Blue.
Identification of AraF as a DsbC Substrate by LC-MS/MS
The bands of interest were cut out from the gel and digested with trypsin. The peptides were analyzed by capillary LC-MS/MS in a LTQ XL ion trap mass spectrometer (Thermo Scientific, San Jose, CA), fitted with a microelectrospray probe. The scan routine was a top five experiment consisting of a full MS scan (400–2000 m/z) followed by a MS/MS scan for the five most abundant ions. Dynamic exclusion allowed fragmentation of co-eluting peptides. The data were analyzed with the ProteomeDiscoverer software (Thermo Scientific, version 1.4.0.288), and the proteins were identified with SEQUEST against a target decoy nonredundant E. coli protein database obtained from UniProtKB. The false discovery rate was set below 5%.
Specificity of the Interaction between DsbC and AraF
The proteins DsbCC_XX_S and DsbGC_XX_A were expressed and purified from KD80 and KD192 strains, respectively, using the protocol described above. Then, proteins were loaded on a SDS-polyacrylamide gel with or without DTT and transferred to a nitrocellulose membrane. After transfer, proteins were detected using an anti-AraF antibody (1/8000), an anti-DsbC antibody (1/10,000), or an anti-DsbG antibody (1/10,000). All of the antibodies were produced from rabbits immunized with the purified protein (Eurogentec, Liège, Belgium). Anti-rabbit IgG (Sigma) was used as the secondary antibody at a dilution of 1/5000. Thermo Scientific Pierce ECL Western blotting substrate and Fuji films were used to visualize the protein bands.
Identification of the Dimedone-modified Peptide by LC-MS/MS
Recombinant AraF (10 μg) was incubated in the presence of 5 mm dimedone with 1 mm H2O2 or 2 mm HOCl for 10 min at 37 °C. The protein was then precipitated with 10% TCA and the pellet resuspended in 50 μl of 100 mm NH4HCO3, pH 8.0, for overnight digestion at 30 °C with 0.5 μg of sequencing grade trypsin (Promega). The peptides were analyzed by LC-MS/MS as described above except that the mass spectrometer was operated in the data-dependent mode and switched automatically between MS, Zoom Scan for charge state determination and MS/MS for the three most abundant ions. Peptides were identified by Proteome Discoverer considering dynamic modifications on cysteine residues of 138.0 Da for sulfenic dimedone, 32.0 Da for sulfinic, and 48.0 Da for sulfonic. Gas-phase fragmentation and chromatographic retention times of the Cys-containing AraF peptide (GFVICTPDPK) were characterized for all oxidation states to determine the best daughter ion to be monitored for single reaction monitoring (SRM transitions) assay by LC-MS/MS (see below).
Identification of the Sulfenylated Modified Cysteine of AraF by SRM
The KD19 strain was grown in LB medium at 37 °C until reaching an _A_600 of 0.6. Expression was induced for 1 h 30 min by the addition of 0.2% l-arabinose. The culture was then harvested by centrifugation at 3000 × g for 15 min at 4 °C. Cultures were standardized to the same _A_600 before centrifugation to obtain an equal amount of bacteria. The pellet was then resuspended in 6 ml of TSE buffer (100 mm Tris-HCl, pH 8, 20% sucrose, 1 mm EDTA) containing 10 mm dimedone and 50 mm IAM. After 15 min of incubation at 4 °C with gentle shaking, the sample was centrifuged at 12,000 × g for 15 min at 4 °C. The pellet was then resuspended in buffer containing 5 mm cold MgSO4 as well as 10 mm dimedone and 50 mm IAM. After 30 min of incubation at 4 °C with gentle shaking, the sample was centrifuged for 25 min at 12,000 × g at 4 °C. An amount of 100 μg of total proteins was precipitated and digested overnight with trypsin in 50 mm NH4HCO3. Peptides (20 μg) were then analyzed by LC-MS/MS using the predefined five SRM transitions targeting the different oxidation states of the single-cysteine-containing peptide of AraF.
Monitoring AraF Dimer Formation in Vivo
KD215, KD216, and KD217 strains were grown in M63 medium at 37 °C until an _A_600 of 0.6. AraF expression was induced by the addition of 0.2% l-arabinose. After 5 h of induction, the cultures were precipitated with 10% TCA. Cultures were standardized according to _A_600 before TCA precipitation. Then, proteins were resuspended in 100 mm NaPi, pH 8, 300 mm NaCl, 0.3% SDS, 8 m urea, and 100 mm IAM to prevent any further thiol rearrangement. The lysates were incubated at room temperature for 20 min and centrifuged at 21,000 × g for 15 min. Finally, samples were loaded on a SDS-polyacrylamide gel and transferred to a nitrocellulose membrane, and proteins were detected by Western blotting as described above.
Oxidation of AraF by HOCl
KD215 and KD216 strains were grown in M63 medium at 37 °C until an _A_600 of 0.6. AraF expression was induced by the addition of 0.2% l-arabinose. After 1 h and 30 min of induction, HOCl (3 mm) was added to the cultures. After 20 min of HOCl incubation, proteins were precipitated with 10% TCA. Cultures were standardized according to _A_600.
AraF Dimer Reduction in Vitro
DsbC and DsbG were reduced first with 10 mm DTT for 1 h at 37 °C. The proteins were then loaded on a gel filtration column (NAP-5; GE Healthcare) equilibrated with 50 mm NaPi, pH 8, 100 mm NaCl and eluted with the same buffer. AraF (40 μm) was incubated either with 5 mm DTT or with 120 μm reduced DsbC or DsbG at 37 °C for 1 h. Samples were then analyzed by SDS-polyacrylamide gel stained with Coomassie Brilliant Blue.
l-[1-14C]Arabinose Binding Assays
l-Arabinose binding was assayed by equilibrium dialysis in Slide-A-Lyzer MINI Dialysis Devices (Thermo Scientific). l-[1-14C]Arabinose was purchased from Moravek Biochemicals (54 mCi/mmol). Equilibrium binding constants (Kd) were determined using purified AraF, reversibly denatured by dialysis against a solution of 2 m guanidine, 50 mm Tris, pH 7.6, 1 mm MgCl2, and 5 mm DTT to remove residual amounts of l-arabinose. Samples of 6 μm AraF were dialyzed against the assay buffer consisting of 50 mm Tris, pH 7.6, 1 mm MgCl2, 5 mm DTT and containing increasing concentrations of l-[1-14C]arabinose (from 0.02 to 0.7 μm). After overnight incubation at 4 °C, samples of the external dialysate and the dialyzed material were mixed with scintillation fluid, and counted for 2 min in a PerkinElmer Life Sciences TriCarb 2800Tr scintillation counter. The counts observed in the sample of the dialyzed material in excess of those observed in the external dialysate are considered to represent l-arabinose bound to the AraF protein.
RESULTS
AraF Is a New DsbC Substrate
To investigate the possible role of DsbC in the protection mechanisms against oxidative stress, we sought to determine whether DsbC interacts with periplasmic proteins presenting a single cysteine residue. Indeed, these proteins cannot form a nonnative disulfide bond and therefore do not use the disulfide isomerase activity of DsbC. The catalytic domain of DsbC adopts a thioredoxin fold and possesses a conserved C_XX_C catalytic motif which is maintained reduced by the inner membrane protein DsbD (7, 12). In proteins belonging to the thioredoxin superfamily, the first cysteine of the C_XX_C motif performs a nucleophilic attack on an oxidized substrate, leading to the formation of a mixed-disulfide intermediate (9). The role of the second cysteine of the C_XX_C motif is to resolve this mixed-disulfide to release the substrate. Thus, to trap DsbC in complex with its substrates, we prepared a DsbC variant in which the C_XX_C motif was replaced by C_XX_S (DsbCC_XX_S), a mutation that prevents the dissociation of the DsbCC_XX_S-substrate complexes. This approach has been used previously to trap the substrates of proteins with a thioredoxin fold (6, 13–15). After expression of DsbCC_XX_S in the periplasm of a Δ_dsbC_ mutant strain, proteins were precipitated, and free cysteine residues were alkylated with IAM to avoid any further disulfide bond rearrangement. DsbCC_XX_S was then purified under nonreducing denaturing conditions by taking advantage of the His tag at the C terminus of the protein. Only one peak eluted from the affinity column when an imidazole gradient was applied. The purified sample was then analyzed by SDS-PAGE with or without DTT, a reducing agent. As shown in Fig. 1A, several bands appeared after reducing treatment, suggesting that they correspond to proteins that were released from DsbC under the reducing conditions. These bands were cut out of the gel, digested with trypsin, and analyzed by LC-MS/MS. Remarkably, we found that the most abundant protein released upon treatment with DTT corresponds to AraF, a periplasmic protein with a single cysteine residue. AraF belongs to a family of periplasmic binding proteins that function as high affinity receptors for multiple low molecular weight compounds in the bacterial envelope. AraF mediates the uptake of l-arabinose in the periplasm by binding to this sugar with a high affinity (0.3 μm) (16, 17), transporting it across the periplasm and delivering it to the AraH-AraG IM protein complex (18). To confirm that DsbC interacts with AraF in vivo, we expressed DsbC in cells deleted for the araBAD genes. This strain cannot metabolize l-arabinose, allowing high expression levels of AraF upon l-arabinose addition. One h after adding l-arabinose to cells in exponential phase, proteins were precipitated with TCA, resuspended in a denaturing nonreducing buffer, and analyzed by Western blotting using a specific anti-AraF antibody. As shown in Fig. 1B, we observed the formation of a band migrating at the size expected for a DsbC-AraF complex, supporting that DsbC and AraF interact in living cells.
FIGURE 1.
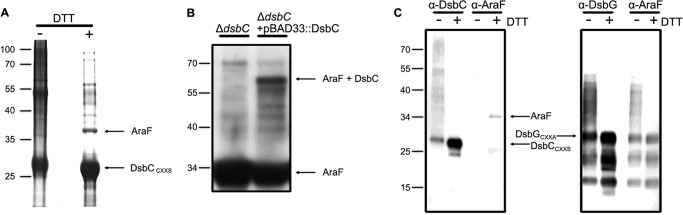
Trapping of DsbCC_XX_S substrates. A, to trap proteins linked to DsbC, DsbCC_XX_S was expressed in a Δ_dsbC_ mutant. The mixed-disulfide complexes were then purified by affinity chromatography using Ni-NTA resin. After concentration, the elution fraction was analyzed by SDS-PAGE without (−) or with DTT (+). The band migrating at ∼37 kDa was identified by MS/MS as the l-arabinose-binding protein AraF. The molecular mass markers (in kDa) are indicated on the left. B, DsbC was expressed from a plasmid in cells expressing AraF. Proteins were precipitated with TCA, resuspended in a denaturing, nonreducing buffer, and analyzed by Western blotting using a specific anti-AraF antibody. The band migrating at the size expected for a DsbC-AraF complex (∼60 kDa) is indicated. C, DsbCC_XX_S and DsbGC_XX_A were expressed in a Δ_dsbC_ mutant and a Δ_dsbG_ mutant, respectively. The mixed-disulfide complexes involving DsbC and DsbG were then purified by affinity chromatography using Ni-NTA resin. The elution fractions were analyzed by SDS-PAGE without (−) or with DTT (+). DsbC, DsbG, and AraF were then detected using specific antibodies. Treatment with DTT leads to the release of AraF from DsbC but not from DsbG. This indicates that AraF was purified in a disulfide-linked complex with DsbC but not with DsbG.
The Single Cysteine Residue of AraF Is Conserved
The overall fold of the AraF protein, illustrated in Fig. 2A, is described as a “kidney bean” and is characterized by the presence of two globular domains and a connecting hinge. These two domains enclose a deep and narrow cleft in which the binding site for l-arabinose is located (Fig. 2A) (16, 19, 20). Interestingly, the single cysteine residue of AraF is localized within this cleft, near the sugar binding site (21), and is widely conserved among the homologous proteins from α-, β-, and γ-proteobacteria (Fig. 2C). It does not, however, participate directly in l-arabinose binding (16).
FIGURE 2.

Location of the conserved cysteine residue in the structure of AraF. A, in the closed conformation with l-arabinose in the active site, the sulfur of the cysteine is directed toward l-arabinose and is not surface-exposed. The surface and the ribbon diagram of AraF (Protein Data Bank code 1ABE) are shown. l-Arabinose (ARA), present in two different orientations (orange and blue), and the cysteine (C64) are shown in sticks. B, the two globular domains of AraF are connected by a flexible hinge, allowing the l-arabinose-free protein to adopt an open conformation. This figure shows the surface and the ribbon diagram of an AraF mutant designed to bind serotonin (34) and which was crystallized in an open conformation (Protein Data Bank code 2WRZ). In the structure, Ser-64, which replaces Cys-64, is surface-exposed. Importantly, the backbone movement observed in the structure of the mutant is restricted to the hinge region (34), highlighting the high flexibility of the hinge. The side chain of Ser-64 is present in two orientations and is shown in sticks. The figures were generated using Open-Source PyMOL 1.6.x. C, multiple sequence alignment of E. coli AraF with homologous proteins is shown. The sequence of E. coli AraF (UniProtKB/TrEMBL accession no. P02924) was aligned with the sequences of homologous proteins from Yersinia pestis (Q7CIK2), Ralstonia solanacearum (M4UHU7), Pseudomonas fluorescens (G8Q167), Rhizobium etli (S5S127), Aeromonas salmonicida (A4SNH0), and Burkholderia mallei (Q62GY8). The conserved cysteine is outlined by a black rectangle. Dark gray and light gray shading indicate identical amino acids and amino acids from the same group (nonpolar, polar, acidic, or basic), respectively.
The Single Cysteine of AraF Can Be Oxidized to a Sulfenic Acid
The fact that we trapped AraF in a disulfide-linked complex with DsbC implies that the single cysteine of AraF can be oxidized. A likely hypothesis is that the cysteine of AraF can be modified to a sulfenic acid by the oxidants present in the periplasm, as observed previously for YbiS, one of the three DsbG substrates (6). Because of their high reactivity, sulfenic acids are difficult to identify. Therefore, their identification often requires the covalent modification of the sulfenic acid intermediate with specific reagents such as dimedone (5,5-dimethyl-1,3-cyclohexadione) (4). To test whether AraF is indeed able to form a stable sulfenic acid, we incubated the purified AraF protein with dimedone. The protein was then digested with trypsin and the peptide mixture analyzed by LC-MS/MS. As shown in Fig. 3, the dimedone-modified peptide, corresponding to the sulfenic acid form of AraF, was detected, indicating that the single cysteine of AraF can be oxidized to the sulfenic state. To confirm the formation of the sulfenic acid in vivo, we induced the expression of the chromosomal copy of araF by adding l-arabinose to a growing culture of the Δ_araBAD_ mutant strain. Then, in late exponential phase, dimedone was added to the culture, a periplasmic extract was prepared, treated with trypsin, and analyzed by a targeted LC-MS/MS SRM assay. The sensitivity and selectivity of this type of assay allow detection of low abundant peptide species within complex samples such as periplasmic extracts. The dimedone-modified peptide, as well as the sulfinic and sulfonic acid-modified peptides, were detected using SRM analysis, indicating that the AraF cysteine can also be sulfenylated in vivo.
FIGURE 3.

Identification of dimedone on Cys-64 of AraF. The MS/MS spectrum of the peptide containing Cys-64 of AraF (m/z = 607.7 for a doubly charged ion) shows that Cys-64 can be modified by dimedone, indicating that it can be oxidized to a sulfenic acid. The mass difference between _y_6 and _y_5 (+241 Da) indicates indeed that Cys-64 (+103 Da) is modified by dimedone (+138 Da). A major neutral loss of HS-DMD is observed resulting from a C-S bond cleavage in the side chain of the Cys residue.
AraF Forms a Disulfide-linked Homodimer
While performing experiments on the purified AraF protein, we observed that AraF forms a high molecular mass complex migrating in SDS-polyacrylamide gels at the size expected for a dimer (Fig. 4A, lane 1), which was confirmed by gel filtration and mass spectrometry. As shown in Fig. 4A (lane 2), the AraF dimer is sensitive to DTT, which indicates that it results from an intermolecular disulfide bond between two AraF molecules, most likely following the oxidation of the cysteine residue of one of the two subunits to a sulfenic acid (see “Discussion”). To establish the physiological relevance of the dimer formation, it was important to determine whether the AraF homodimer also forms in vivo. The Δ_araBAD_ mutant was grown in LB at 37 °C, l-arabinose was added to induce AraF expression, and samples were taken at different time points. Free thiols were blocked with IAM to prevent nonphysiological oxidation of cysteine thiols, and proteins were precipitated with TCA. Then, the samples were resuspended in a denaturing buffer and analyzed by Western blotting using a specific anti-AraF antibody. As shown in Fig. 4B, a DTT-sensitive band migrating at the size expected for an AraF homodimer (∼70 kDa) appeared in stationary phase, indicating that AraF forms a disulfide-linked homodimer in vivo. As dimer formation was observed in stationary phase, i.e. after 5 h of induction (Fig. 4C, lane 1), a growth condition known to accumulate ROS, we investigated whether exposing AraF-expressing cells to H2O2 or HOCl, two oxidizing agents commonly encountered by bacteria, leads to increased dimer formation. Whereas addition of H2O2 does not cause dimer formation, addition of HOCl leads to a significant accumulation of the AraF dimer (Fig. 4D, lanes 1 and 2).
FIGURE 4.

AraF forms a disulfide-linked homodimer reduced by DsbC. A, DTT and DsbC can fully reduce the disulfide-linked dimer of AraF in vitro, whereas DsbG catalyzes the reduction only to a certain extent. For this experiment, AraF was incubated for 1 h at 37 °C either alone (1), with DTT (2), DsbC (3), or DsbG (4). The samples were analyzed by SDS-PAGE. B, AraF forms a DTT-sensitive dimer in E. coli cells grown to stationary phase. C, dimer formation in wild-type and Δ_dsbC_ strains is shown. Aliquots were taken 5 h after l-arabinose addition. Deletion of dsbC leads to an increased accumulation of the AraF dimer. D, dimer formation in wild-type and Δ_dsbC_ strains is shown. Expression of AraF was induced for 90 min in wild-type and Δ_dsbC_ strains. Then, cells were exposed to 3 mm HOCl for 20 min, showing that HOCl treatment leads to dimer formation in both strains. We observed that dimer formation is significantly increased in cells lacking dsbC. The band migrating faster than monomeric AraF, which is observed in both the wild-type and the Δ_dsbC_ strain following HOCl addition, probably results from a proteolytic cleavage of AraF. For images in B–D, samples were normalized by _A_600 and were analyzed by immunoblotting using an anti-AraF antibody. The molecular mass markers (in kDa) are indicated on the left.
Formation of the Dimer Prevents l-Arabinose Binding by AraF
The fact that the cysteine residue of AraF is located in the vicinity of the sugar binding site raises the question of whether its involvement in an intermolecular disulfide bond influences the binding properties of AraF toward l-arabinose. To address this question, we purified the AraF homodimer by gel filtration, and we compared its affinity constant (Kd) for l-arabinose with that of the monomeric protein. Remarkably, whereas we found a Kd of 0.41 μm for the wild-type protein, in agreement with previous data (16, 17), the disulfide-linked dimer was unable to bind l-arabinose, indicating that formation of the intermolecular disulfide disturbs substrate binding in AraF (Fig. 5).
FIGURE 5.
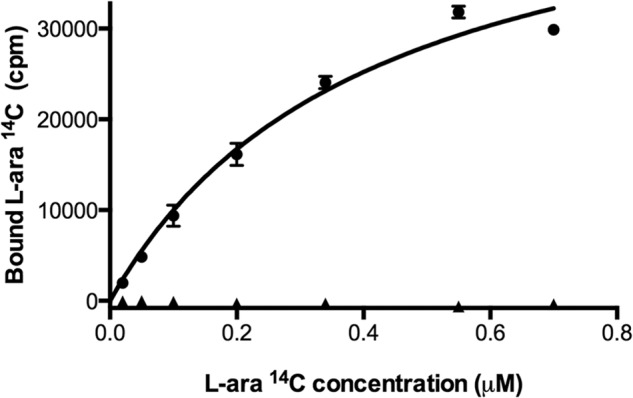
The disulfide-linked AraF homodimer cannot bind l-arabinose. The monomeric (●) and dimeric (▴) forms of AraF were incubated overnight with increasing concentrations of l-[1-14C]arabinose. l-Arabinose binding was then measured as explained under “Experimental Procedures.” Whereas we found a Kd of 0.41 μm for the monomeric protein, the disulfide-linked dimer was found unable to bind l-arabinose.
The AraF Homodimer Is Specifically Reduced by DsbC
The identification of an AraF-DsbC complex suggests that DsbC is able to reduce the intermolecular disulfide of the AraF homodimer. This is indeed what we observed when the AraF homodimer was incubated in vitro with DsbC in excess (Fig. 4A, lane 3). Furthermore, we found that DsbC regulates dimer formation in living cells by comparing the amount of the AraF dimer detected in a Δ_dsbC_ mutant and in a wild-type strain. As shown in Fig. 4, C and D (lanes 3 and 4), we observed that deletion of dsbC leads to an increased accumulation of the AraF dimer, especially during HOCl-induced oxidative stress. Thus, these results indicate that DsbC is involved in the reduction of the AraF intermolecular disulfide bond in vivo.
As explained above, DsbG, a homodimeric protein sharing a similar three-dimensional structure and a similar C_XX_C catalytic motif with DsbC, controls the sulfenylation level of certain periplasmic proteins. To test whether DsbG is also able to interact with AraF, we expressed a DsbGC_XX_A mutant in a Δ_dsbG_ strain and purified the protein by affinity chromatography. However, no band corresponding to AraF was released from DsbG upon DTT treatment (Fig. 1C), indicating that DsbG and AraF do not interact in vivo. In agreement with this result, we found that, although DsbG appears to be able to reduce the disulfide-linked AraF dimer in vitro (Fig. 4A, lane 4), it does it less efficiently than DsbC, further indicating that AraF is a specific DsbC substrate. Interestingly, DsbC was previously shown to inefficiently catalyze the in vitro reduction of the transpeptidase YbiS, a specific DsbG substrate (6). Thus, it seems that although the homologous proteins DsbC and DsbG have favorite substrates in vivo, they are both able to catalyze to a certain extent the reduction of one another's substrates in vitro.
DISCUSSION
The function of DsbC as a periplasmic protein-disulfide isomerase is well documented. The protein has indeed been shown to assist the folding of several envelope proteins containing disulfides formed between cysteine residues that are not consecutive in the sequence. The list of DsbC substrates includes RNase I, MepA (22), AppA (23), and RcsF (24), as well as eukaryotic recombinant proteins expressed in E. coli such as RNase A, bovine pancreatic trypsin inhibitor, and urokinase (12, 25, 26). In this study, we show that, in addition to its role in helping proteins with multiple cysteine residues to fold, DsbC is also involved in the defense mechanisms against oxidative stress by reducing a disulfide-linked dimer formed by the l-arabinose-binding protein AraF under oxidative stress conditions. This finding further highlights the importance of protecting periplasmic proteins from oxidative damage, a concept recently put forward by the identification of several reducing pathways involved in the protection of bacterial envelope proteins (27). Indeed, in E. coli, DsbG has been shown to control the level of sulfenylation in a family of l,d-transpeptidases (6), whereas in Neisseria meningitidis, PilB repairs oxidized methionine residues (28). Moreover, in Caulobacter crescentus, PrxP has been identified as the first periplasmic peroxiredoxin, directly reducing ROS in the cell envelope (29). Noteworthy, the activity of DsbC, DsbG, PilB, and PrxP depends on electrons provided by DsbD, an inner membrane protein that transfers reducing equivalents from the cytoplasmic thioredoxin system to the periplasm (12, 27–30).
We also report that the cysteine residue of AraF can be oxidized to a sulfenic acid, both in vitro and in vivo, which indicates that it is sensitive to ROS oxidation. It is likely that the formation of the sulfenic acid is the first step toward the formation of the AraF disulfide-linked homodimer. Indeed, although we do not have a direct evidence to support this hypothesis, the dimer most probably results from the reaction between the sulfenic acid of one AraF molecule and the thiol group of a second one.
In the structure shown in Fig. 2A, the single cysteine residue of AraF is buried in the cleft of the protein, near the l-arabinose binding site, and is therefore poorly accessible (21). This raises the intriguing question of how this buried cysteine residue can mediate the formation of a disulfide-linked dimer. However, previous work (32, 33) showed that the hinge that links the two globular domains of AraF is remarkably flexible (19, 20, 32), allowing the l-arabinose-free protein to adopt an open conformation (Fig. 2B) (34). Thus, it is likely that the conformational flexibility of AraF facilitates the formation of a disulfide bond between two cysteine residues of two neighboring AraF monomers.
Another intriguing question is why the single cysteine residue of AraF has been conserved during evolution. A plausible hypothesis is that this cysteine residue is important for the regulation of l-arabinose transport in the periplasm. Indeed, the import of l-arabinose fuels the tricarboxylic acid cycle and therefore the electron transport chain, from which electrons can escape and promote the formation of ROS. Thus, when ROS accumulate, oxidizing AraF to a dimer unable to bind l-arabinose might be a way to shut off l-arabinose import. The function of DsbC would be to reduce the dimer and to restore the l-arabinose uptake when ROS concentrations decrease.
Altogether, our results further highlight the sensitivity of single cysteine residues in oxidizing environments such as the bacterial periplasm, where they can function as switch to tune protein activity in response to changes in the oxidative state of the cell. Moreover, our work shows that even cysteine residues that are buried in the protein structure can become susceptible to oxidation by ROS, probably following conformational changes. Protecting these single cysteine residues requires reducing systems. We postulate that, in the oxidizing environment of the endoplasmic reticulum, proteins with single cysteine residues are also sensitive to oxidation and require the presence of reducing pathways for correct functioning.
Acknowledgments
We thank Veronica Tamu Dufe for help with crystallization studies, Asma Boujtat and Gaëtan Herinckx for technical assistance, and Géraldine Laloux and Alexandra Gennaris for critical reading of the manuscript.
*
This work was supported by the European Research Council (FP7/2007–2013) ERC Independent Researcher Starting Grant 282335–Sulfenic (to J.-F. C.).
2
The abbreviations used are:
ROS
reactive oxygen species
IAM
iodoacetamide
MS/MS
tandem mass spectrometry
Ni-NTA
nickel-nitrilotriacetic acid
SRM
single reaction monitoring.
REFERENCES
- 1.Goemans C., Denoncin K., Collet J. F. (2013) Folding mechanisms of periplasmic proteins. Biochim. Biophys. Acta, in press, 10.1016/j.bbamcr.2013.10.014 [DOI] [PubMed] [Google Scholar]
- 2.Messens J., Collet J. F. (2006) Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 38, 1050–1062 [DOI] [PubMed] [Google Scholar]
- 3.Dutton R. J., Boyd D., Berkmen M., Beckwith J. (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddie K. G., Carroll K. S. (2008) Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 12, 746–754 [DOI] [PubMed] [Google Scholar]
- 5.Roos G., Messens J. (2011) Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic. Biol. Med. 51, 314–326 [DOI] [PubMed] [Google Scholar]
- 6.Depuydt M., Leonard S. E., Vertommen D., Denoncin K., Morsomme P., Wahni K., Messens J., Carroll K. S., Collet J. F. (2009) A periplasmic reducing system protects single cysteine residues from oxidation. Science 326, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 7.Rietsch A., Belin D., Martin N., Beckwith J. (1996) An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 93, 13048–13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shevchik V. E., Condemine G., Robert-Baudouy J. (1994) Characterization of DsbC, a periplasmic protein of Erwinia chrysanthemi and Escherichia coli with disulfide isomerase activity. EMBO J. 13, 2007–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collet J. F., Messens J. (2010) Structure, function, and mechanism of thioredoxin proteins. Antiox. Redox Signal. 13, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 10.Casadaban M. J. (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage Lambda and Mu. J. Mol. Biol. 104, 541–555 [DOI] [PubMed] [Google Scholar]
- 11.Miller J. H. (1992) A Short Course in Bacterial Genetics: Laboratory Manual (Cold Spring Harbor Laboratory, ed), Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 12.Rietsch A., Bessette P., Georgiou G., Beckwith J. (1997) Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol. 179, 6602–6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawazu S., Takemae H., Komaki-Yasuda K., Kano S. (2010) Target proteins of the cytosolic thioredoxin in Plasmodium falciparum. Parasitol. Int. 59, 298–302 [DOI] [PubMed] [Google Scholar]
- 14.Sturm N., Jortzik E., Mailu B. M., Koncarevic S., Deponte M., Forchhammer K., Rahlfs S., Becker K. (2009) Identification of proteins targeted by the thioredoxin superfamily in Plasmodium falciparum. PLoS Pathog. 5, e1000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denoncin K., Vertommen D., Paek E., Collet J. F. (2010) The protein-disulfide isomerase DsbC cooperates with SurA and DsbA in the assembly of the essential β-barrel protein LptD. J. Biol. Chem. 285, 29425–29433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newcomer M. E., Miller D. M., 3rd, Quiocho F. A. (1979) Location of the sugar-binding site of l-arabinose-binding protein: sugar derivative syntheses, sugar binding specificity, and difference Fourier analyses. J. Biol. Chem. 254, 7529–7533 [PubMed] [Google Scholar]
- 17.Declerck N., Abelson J. (1994) Novel substrate specificity engineered in the arabinose-binding protein. Protein Eng. 7, 997–1004 [DOI] [PubMed] [Google Scholar]
- 18.Horazdovsky B. F., Hogg R. W. (1989) Genetic reconstitution of the high-affinity l-arabinose transport system. J. Bacteriol. 171, 3053–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newcomer M. E., Gilliland G. L., Quiocho F. A. (1981) l-Arabinose-binding protein-sugar complex at 2.4 Å resolution: stereochemistry and evidence for a structural change. J. Biol. Chem. 256, 13213–13217 [PubMed] [Google Scholar]
- 20.Quiocho F. A., Vyas N. K. (1984) Novel stereospecificity of the l-arabinose-binding protein. Nature 310, 381–386 [DOI] [PubMed] [Google Scholar]
- 21.Miller D. M., 3rd, Newcomer M. E., Quiocho F. A. (1979) The thiol group of the l-arabinose-binding protein: chromophoric labeling and chemical identification of the sugar-binding site. J. Biol. Chem. 254, 7521–7528 [PubMed] [Google Scholar]
- 22.Hiniker A., Bardwell J. C. (2004) In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279, 12967–12973 [DOI] [PubMed] [Google Scholar]
- 23.Berkmen M., Boyd D., Beckwith J. (2005) The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein-disulfide isomerase, DsbC. J. Biol. Chem. 280, 11387–11394 [DOI] [PubMed] [Google Scholar]
- 24.Leverrier P., Declercq J. P., Denoncin K., Vertommen D., Hiniker A., Cho S. H., Collet J. F. (2011) Crystal structure of the outer membrane protein RcsF, a new substrate for the periplasmic protein-disulfide isomerase DsbC. J. Biol. Chem. 286, 16734–16742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun X. X., Wang C. C. (2000) The N-terminal sequence (residues 1–65) is essential for dimerization, activities, and peptide binding of Escherichia coli DsbC. J. Biol. Chem. 275, 22743–22749 [DOI] [PubMed] [Google Scholar]
- 26.Joly J. C., Swartz J. R. (1997) In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry 36, 10067–10072 [DOI] [PubMed] [Google Scholar]
- 27.Cho S. H., Collet J. F. (2013) Many roles of the bacterial envelope reducing pathways. Antiox. Redox Signal. 18, 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brot N., Collet J. F., Johnson L. C., Jönsson T. J., Weissbach H., Lowther W. T. (2006) The thioredoxin domain of Neisseria gonorrhoeae PilB can use electrons from DsbD to reduce downstream methionine sulfoxide reductases. J. Biol. Chem. 281, 32668–32675 [DOI] [PubMed] [Google Scholar]
- 29.Cho S. H., Parsonage D., Thurston C., Dutton R. J., Poole L. B., Collet J. F., Beckwith J. (2012) A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. mBio. 3, e00291–002911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bessette P. H., Cotto J., Gilbert H. F., Georgiou G. (1999) In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem. 274, 7784–7792 [DOI] [PubMed] [Google Scholar]
- 31.Vertommen D., Depuydt M., Pan J., Leverrier P., Knoops L., Szikora J. P., Messens J., Bardwell J. C., Collet J. F. (2008) The disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner. Mol. Microbiol. 67, 336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermersch P. S., Tesmer J. J., Lemon D. D., Quiocho F. A. (1990) A Pro to Gly mutation in the hinge of the arabinose-binding protein enhances binding and alters specificity: sugar-binding and crystallographic studies. J. Biol. Chem. 265, 16592–16603 [PubMed] [Google Scholar]
- 33.Mao B., McCammon J. A. (1983) Theoretical study of hinge bending in l-arabinose-binding protein: internal energy and free energy changes. J. Biol. Chem. 258, 12543–12547 [PubMed] [Google Scholar]
- 34.Schreier B., Stumpp C., Wiesner S., Höcker B. (2009) Computational design of ligand binding is not a solved problem. Proc. Natl. Acad. Sci. U.S.A. 106, 18491–18496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]