Extraskeletal Myxoid Chondrosarcoma with non-EWSR1-NR4A3 Variant Fusions Correlate with Rhabdoid Phenotype and High Grade Morphology (original) (raw)
. Author manuscript; available in PMC: 2015 May 1.
Abstract
Extraskeletal myxoid chondrosarcomas (EMC) are rare soft tissue sarcomas with distinctive histology and uncertain histogenesis, characterized by EWSR1-NR4A3 fusion in 75% of the cases. A smaller proportion of cases show NR4A3 fused to other gene partners including TAF15, TCF12 and TFG. The impact of various gene fusions on morphology and outcome has not been previously evaluated. We investigated 26 consecutive EMCs and correlated the genetic findings with morphology and clinical outcome. There were 5 females and 21 males (median age of 49.5 years). Mean size of the tumors was 11 cm. FISH analysis showed EWSR1-NR4A3 gene fusion in 16 (62%) cases; TAF15-NR4A3 gene fusion in 7 (27%) cases and TCF12-NR4A3 gene fusion in one (4%) case. Two cases showed only NR4A3 gene rearrangements. Morphologically, most _EWSR1_-rearranged tumors (10 of 16) showed low cellularity, minimal cytologic atypia and low mitotic counts. In contrast, 80% of EMCs with variant (non-EWSR1) NR4A3 gene fusions (TAF15, TCF12) had high grade morphology with increased cellularity, proliferation and cytologic atypia, showing a plasmacytoid / rhabdoid morphology in half the cases. Follow-up showed that only 1 of 16 patients with _EWSR1_-rearranged tumors died of disease, in contrast to 3 of 7 (43%) _TAF15_–rearranged tumors. In conclusion, EMCs with variant NR4A3 gene fusions show a higher incidence of rhabdoid phenotype, high grade morphology and, a more aggressive outcome compared to the EWSR1-NR4A3 positive tumors. Furthermore, FISH assay for NR4A3, along with EWSR1, may be an additional ancillary test to confirm diagnosis of EMCs.
Keywords: NR4A3, CHN, TAF15, EWSR1, TCF12, extraskeletal myxoid chondrosarcoma
INTRODUCTION
Extraskeletal myxoid chondrosarcoma (EMC) is a rare soft tissue sarcoma, occurring with predilection in the deep soft tissue of the limbs in middle-aged adults. [1] Despite having often an indolent course, EMC has a propensity for late recurrence and metastasis, particularly to the lungs.[2] Most tumors show a distinctive morphology, composed of monotonous oval to short spindled cells arranged in cords or reticular pattern, in a diffusely myxoid extracellular matrix.[1] However, in a subset of cases the tumors are more cellular, with a predominant epithelioid or rhabdoid phenotype and higher degree of cytologic atypia. This latter appearance in particular is under-recognized and often misdiagnosed with a variety of other soft tissue tumors, including myoepithelial tumors (parachordoma), extra-axial chordoma, and myxofibrosarcoma, among others.
The genetic hallmark of EMC is a pathognomonic NR4A3 gene rearrangement, which in most cases is being fused to EWSR1 as a result of a t(9;22)(q22;q12). [3-6] However, in a smaller percentage of cases NR4A3 is fused instead to alternative gene partners, including TAF15, TCF12 and TFG. [7-10] Despite this rather comprehensive catalog of genetic abnormalities, most clinical labs have only limited molecular tests available to be applied in daily practice, such as EWSR1 break-apart assay by FISH or the EWSR1-NR4A3 fusion by RT-PCR. [11, 12] These tests typically are not informative for the 25-30% _EWSR1_-negative subset of EMC, which often remain mis-diagnosed. Thus, the purpose of our study is to investigate the prevalence of these genetic abnormalities in a series of consecutive EMCs with typical morphology, using a comprehensive panel of FISH probes covering the entire spectrum of reported fusions in EMC and to correlate the transcript variant to morphology and clinical outcome.
MATERIALS AND METHODS
Twenty-six cases of EMC were identified between 2000 and 2013 from the Department of Pathology files at Memorial Sloan-Kettering Cancer Center. The criteria for the selection included a typical morphology and available tissue for molecular studies. Hematoxylin and eosin sections and immunohistochemical stains performed at the time of diagnosis were reviewed. The gross and microscopic findings, including tumor size, anatomic location, tumor borders, surgical margins, cellularity, percentage of myxoid component, nuclear pleomorphism, mitoses (per 10 high power fields), and presence of necrosis were recorded. Clinical and follow-up data were obtained from the clinical database. The study was approved by the Institutional Review Board (IRB# 02-060 MSKCC).
Fluorescence in situ hybridization (FISH)
FISH on interphase nuclei from paraffin embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking NR4A3 (CHN), EWSR1, TAF15, TCF12, TFG and FUS genes. (Supplementary Table 1) BAC clones were chosen according to USCS genome browser (http://genome.uscs.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI; Oakland, CA; http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). A positive score was interpreted when at least 20% of the nuclei showed a breakapart signal. Nuclei with incomplete set of signals were omitted from the score.
Reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted in 3 cases with available frozen tumor tissue, using the Trizol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). The quality of RNA was assessed by RT-PCR for PGK housekeeping gene (as evidenced by the presence of a 247 bp amplified product). RT–PCR was performed by SuperScriptVR First–Strand Synthesis System (Invitrogen). Primers used for RT-PCR validation included: TAF15 exon 6 forward primer (5’-GAGCACCTTCCTATGACCAGCCAG-3’) and CHN exon 3 reverse primer (5’-CGAACTCAAGCCTTCCTGCGTGTA-3’). Primers used to detect the EWSR1-NR4A3 fusions are as follows: EWSR1 exon 12 forward (5’-AAGGCGATGCCACAGTGTC-3’) and NR4A3 reverse A exon 3 (5’-CCTGGAGGGGAAGGGCTAT-3’) for the type 1 EWSR1- NR4A3 fusion (109 base pair product), and EWSR1 exon 7 forward (5’-CGTAAGCTTTCCTACAGCCAAGCTCCAAGTC-3’) and NR4A3 reverse B exon 2 (5’-TCTCTCCAGCGGAGGCTGAGA-3’) for the type 2 fusion (201 base pair product). The PCR products were analyzed by electrophoresis. The amplified PCR products were sequenced using the Sanger’s method.
RESULTS
Clinical Features
Clinical features are summarized in Table 1. The study was comprised of twenty-six patients including 5 females and 21 men, with a median age of 49.5 yrs (range, 33-81 yrs). The anatomic location of the tumors included: thigh (14), elbow (1), buttock (2), back (1), arm (2), groin (1), perineum (1), pelvis (1), calf (1), foot (1) and inguinal (1). The samples available for molecular analyses originated from the primary lesions in 23 cases, from the local recurrences in 2 cases and from the lung metastasis in one case. The primary tumor of the case of the lung metastasis sample was located in the inguinal region. All patients were treated by surgical excision of the mass.
Table 1.
Clinicopathologic and cytogenetic findings and follow-up
| Case# | Age/Sex | Site | Size (cm) | Cellularity | Atypia⊕ | Mitoses (/ 10 HPF) | Fluorescence In-situ hybridization (FISH) | LR/DR | Follow-up Status | Duration (months) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NR4A3 (CHN) | EWSR1 | TAF15 | TCF12 | TFG | FUS | ||||||||||
| 1 | 56 / M | elbow | 2 | low | minimal | 0 | + | + | NED | 88 | |||||
| 2 | 51 / F | thigh | 18.5 | low | minimal | 0 | + | + | LR | NED | 56 | ||||
| 3 | 81 / M | thigh | 14 | low | mild | 0 | + | + | DUK | 96 | |||||
| 4* | 64 / M | thigh | 16.5 | moderate | moderate | 4 | + | + | NED | 11 | |||||
| 5 | 41 / F | thigh | 14.8 | low | minimal | 0 | + | + | DR | AWD | 46 | ||||
| 6 | 58 / F | thigh | 24 | low | mild | 0 | + | + | DR | AWD | 41 | ||||
| 7 | 38 / M | thigh | 7 | low | mild | 1 | + | + | NED | 25 | |||||
| 8*# | 77 / M | back | 6.5 | high | moderate | 10 | + | + | NED | 5 | |||||
| 9 | 46 / M | lung | 1 | low | mild | 1 | + | + | DR | AWD | 95 | ||||
| 10 | 78 / M | thigh | 11 | low | mild | 3 | + | + | DR | DOD | 4 | ||||
| 11*# | 68 / F | thigh | 10.5 | high | severe⊕ | 6 | + | + | NED | 2 | |||||
| 12* | 49 / M | thigh | 12.5 | low | mild | 1 | + | + | AWD | 55 | |||||
| 13 | 33 / M | thigh | 15.2 | moderate | moderate | 1 | + | + | DR | AWD | 66 | ||||
| 14 | 42 / M | arm | 6 | low | mild | 0 | + | + | DR | AWD | 62 | ||||
| 15 | 50 / M | thigh | 11 | low | mild | 1 | + | + | NED | 7 | |||||
| 16* | 74 / M | calf | 11 | low | mild | 0 | + | + | NED | 34 | |||||
| 17# | 63 / M | perineum | 19 | moderate | moderate⊕ | 5 | + | - | + | NED | 3 | ||||
| 18 | 39 / M | thigh | 4.5 | low | mild | 2 | + | - | + | NED | 18 | ||||
| 19* | 46 / M | buttock | 16.5 | high | moderate | 9 | + | - | + | LR/DR | DOD | 12 | |||
| 20 | 40 / M | foot | 7 | low | mild | 6 | + | - | + | DR | DOD | 99 | |||
| 21# | 63 / M | groin | 12 | high | moderate⊕ | 12 | + | - | + | NED | 11 | ||||
| 22* | 46 / M | pelvis | 12.2 | high | severe⊕ | 5 | + | - | + | LR | DOD | 7 | |||
| 23*# | 70 / M | buttock | 12 | high | moderate | 10 | + | - | + | NED | 2 | ||||
| 24 | 35 / M | thigh | 2.7 | high | severe⊕ | 10 | + | - | - | + | - | NED | 30 | ||
| 25* | 41 / M | thigh | 8.7 | high | severe⊕ | 3 | + | - | - | - | - | - | DR | AWD | 40 |
| 26 | 39 / M | arm | 3.5 | moderate | moderate | 4 | + | - | - | - | - | - | LR | NED | 36 |
Pathologic Findings
The primary tumors ranged in size from 2-24 cm (mean, 11 cm). All tumors had well-circumscribed borders and involved the deep soft tissues. (Fig 2A) Microscopically, all tumors showed a multi-nodular and lobular growth pattern. The tumors were diffusely (100%) myxoid in 19 cases, predominantly myxoid (>80%) in 6 cases. Only one case showed scant myxoid matrix (10%). The tumor cells ranged from small to medium in size and from ovoid to epithelioid in shape. The tumor cells were arranged in cords and reticular patterns in the predominantly myxoid areas (Fig 1A), while in tumors with increased cellularity throughout, the tumor cells were distributed in trabeculae and closely packed cords (Figs. 2B-E). Six cases showed an epithelioid / rhabdoid morphology (Figs 2F, 3B). Mitoses ranged from 0-1 to 10 per 10 high power fields. Necrosis, ranging from focal to 90% of the tumor, was seen in 9 cases.
Figure 2. Pathologic spectrum of TAF15-NR4A3 fusion positive tumors.

(A) Gross appearance of a well-circumscribed tumor, involving soft tissue, with a myxoid to hemorrhagic cut surface (EMC#22); (B) Low magnification commonly show a distinctive lobular and multinodular growth pattern (EMC#23, 40x); Higher magnification showing a more solid and less myxoid appearance, with vague nest formation (C; EMC#22; H&E, 200x), undifferentiated morphology with monotonous small round cells in a vague myxoid stroma (D, EMC#21,H&E, 200x); solid and more anaplastic / high grade appearance (E, EMC#18, H&E, 200x); distinctive rhabdoid appearance, with epithelioid cells showing densely eosinophilic cytoplasm and eccentric nuclei, reminiscent of a plasmacytoid phenotype (F, EMC#17, H&E, 200x). (G) Strong S100 protein expression in the latter case with rhabdoid morphology, occurring in the perineum of a 63 year-old man (EMC#17, 200x); (H) RT-PCR identified the presence of a TAF15-NR4A3 (CHN) fusion transcript. The encoded protein sequence is shown below; (I) FISH showing rearrangement (arrows) of TAF15 gene (red, centromeric, green, telomeric).
Figure 1. EWSR1-NR4A3 positive EMC (EMC#2).

(A) Morphologic appearance showing a low grade myxoid lesion composed of bland ovoid cells arranged in a reticular pattern (H&E, 200x); FISH analysis shows break-apart signals (arrows) for NR4A3 (CHN) gene (B) and EWSR1 (C)(red, centromeric; green telomeric)
Figure 3. TCF12-NR4A3 rearranged high grade EMC (EMC#24).
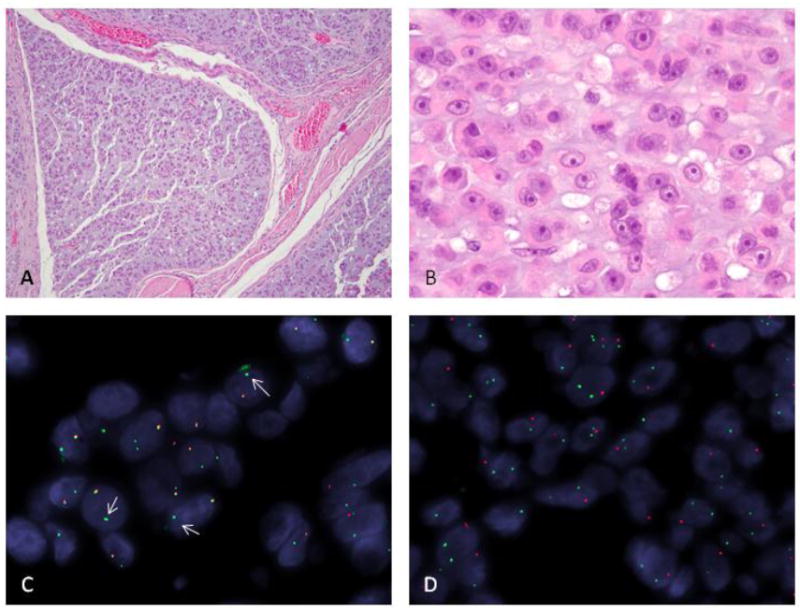
(A) Low power showing nodular growth pattern (H&E, 40x); (B) higher power showing a solid growth of epithelioid cells with densely eosinophilic cytoplasm and vague rhabdoid morphology; prominent nucleoli are noted within a vesicular chromatin (H&E, 200x); FISH showing an unbalanced rearrangement (arrows) for NR4A3 gene with loss of the centromeric part (red) (C) and a more complex pattern of rearrangement of the TCF12 locus (D) with deletion of the centromeric part (red) as well as an additional break/intra-chromosomal inversion on the other chromosome 15 (green, telomeric, and red, centromeric, are apart rather than fused signals)
Immunohistochemical stains were performed at diagnosis on 14 cases. Review of the IHC stains revealed no specific immunoprofile with most tumors (8 of 14) being negative for S100, EMA, pan-keratin, SMA, Desmin and CD34. Five cases showed focal scattered positivity for S100 protein. Only one case (EMC#17) was positive diffusely positive for S100 protein (Fig. 2 G) but was negative for other markers. This latter case occurred in the perineum and had a distinctive rhabdoid morphology (Fig. 2 F), reminiscent of a proximal type epithelioid sarcoma. However, INI1 (BAF47) expression was retained in this case as well as four additional cases tested (2 with EWSR1-NR4A3 and 3 with TAF15-NR4A3 fusions), including 4 cases with rhabdoid features.
FISH and Molecular findings
FISH for NR4A3 and EWSR1 was performed on all 26 tumors. All cases showed rearrangement of the NR4A3 gene confirming the diagnosis of EMC (Fig. 1B, 3C). Rearrangements of the EWSR1 and NR4A3 genes were seen in 16 of 26 cases (62%) indicating a t(9:22) translocation (Fig. 1 B,C). FISH for TAF15, TFG, TCF12 and FUS was performed on the tumors that were NR4A3 positive and EWSR1 negative. Rearrangements of both NR4A3 and TAF15 genes were identified in 7 cases (27%) indicating a t(9:17) translocation (Fig. 2I). Only one case was positive for TCF12 gene rearrangement in keeping with a t(9:15) translocation (Fig. 3C). Two cases showed rearrangement of the NR4A3 gene only. FISH for TFG and FUS gene rearrangements were negative in the three cases tested.
In EMC#22 with available frozen tissue, RT-PCR showed the fusion of exon 3 of NR4A3 with exon 6 of the TAF15 gene (Fig. 2H). In addition, RT-PCR for EWSR1-NR4A3 gene fusion performed in two cases (EMC# 1 &10) detected EWSR1-NR4A3 type 1 gene fusion (exon 12 of EWSR1 fused to exon 3 of NR4A3) in both the cases.
Correlation of the clinical features and morphology with the genetic findings
EMCs with EWSR1-NR4A3 gene fusion
There were 16 tumors in this group, occurring in 11 males and 5 females. Eleven of the 16 tumors (69%) originated in the muscles of the thigh. These cases predominantly showed low cellularity (Fig. 1A) with only 4 of 16 cases (25%) showing moderate to high cellularity. Only 1 of the 4 cases with increased cellularity showed a rhabdoid / epithelioid phenotype. Mitotic counts for most cases were 0-1/ 10 HPFs with only 3 of 16 cases (19%) showing increased mitoses of more than or equal to 4 per 10 high power fields. Tumor necrosis ranging from focal to 40% was seen in 4 of 16 cases.
EMCs with variant non-EWSR1 NR4A3 fusions
There were 10 tumors with NR4A3 variant gene fusions all occurring in males and only 3 (30%) occurred in the thigh. Morphologically, 8 of 10 cases (80%) showed moderate to increased cellularity with moderate to severe cytologic atypia (Figs. 2,3). The tumor cells showed a plasmacytoid / rhabdoid morphology in half (50%) of the cases (Figs. 2F and 3B). Eight of 10 cases showed increased mitotic activity of >4 per 10 high power fields with an average of 7 MF/ 10 HPFs. Tumor necrosis of 15% and 90% was seen in 2 of 10 cases, respectively.
Clinical Follow-up
Follow up information was available in all patients, ranging from 2-99 months (mean follow-up for survivors – 35 months). Four patients (15%) developed local recurrence (LR) and 9 cases (35%) developed distant recurrence / metastasis (DR). Local recurrences occurred from 3-36 months after the primary surgical resection, while metastases occurred at 2-70 months following original tumor resection. At the time of last follow-up, 14 patients had no evidence of disease (NED), 7 were alive with disease (AWD), 1 patient died of unknown cause (DUK) and 4 patients died of disease (DOD).
Of the 16 patients with tumors exhibiting EWSR1-NR4A3 gene fusion, follow-up ranged from 2 to 96 months (mean follow-up – 43.3 months). LR and DR were seen in 1 (6%) and 6 (37%) patients, respectively. Only one of the 16 patients (6%) with this fusion variant DOD. Six patients are AWD with a mean follow-up of 60.8 months. Of the 4 cases with increased cellularity and mitotic activity, 3 were NED at last follow-up ranging from 2 – 11 months and 1 was AWD at a follow-up of 95 months.
Of the 7 patients with TAF15-NR4A3 gene fusion-positive tumors, follow-up ranged from 2 to 99 months (mean follow-up – 21.7 months). Two (28%) patients each developed LR and DR. Three (43%) of the 7 patients with this fusion DOD at 7, 12 and 99 months. Among the 5 tumors with variant non-EWSR1-NR4A3 fusions having an epithelioid/rhabdoid phenotype there was no distinct correlation with outcome, with 3 patients being NED at 3-30 months follow-up, one patient AWD at 40 months and one patient DOD at 7 months.
DISCUSSION
The terminology and histogenesis of extraskeletal myxoid chondrosarcoma (EMC) has been long controversial. Originally described as ‘chordoid tumor’ by Dr Stewart in 1948,[13] to reflect its resemblance to a chordoma and lack of definitive chondroid lineage, was later defined as EMC by Enzinger and Shiraki into a distinct clinicopathologic entity.[1] Despite its name, there is no convincing evidence of cartilaginous differentiation,[14] and previous studies have shown distinct genetic abnormalities between EMC and most skeletal chondrosarcomas with extensive myxoid changes.[12]
Patients with EMC follow a protracted clinical course with a relatively high incidence of distant spread, typically to the lung,[15] and poor response rates to most conventional chemotherapeutic agents.[16] In one of the larger clinicopathologic studies to date including 42 patients with EMC, the overall survival was 100% at 5 years and 69% at 10 years.[15] The correlation between histologic grade in EMC and behavior has been controversial. Most cases of EMC are predominantly myxoid, with low cellularity and classified as low grade tumors. Cellular forms of EMC have been reported, although the detailed criteria for truly high-grade tumors are limited. Antonescu et al. and Lucas et al. reported that histologically high-grade EMCs showed adverse clinical outcomes, including higher metastatic rates.[12, 17] The study by Lucas et al pointed out specifically to a distinctive and striking histological feature in their study group of numerous large epithelioid cells, which they define as ‘high grade EMC’, with all four patients succumbing of disease in a relatively short interval.[17] In contrast, studies by Saleh et al and Jambhekar et al found no relation between cellularity and prognosis.[2, 18] Furthermore, Meis-Kindblom et al. showed that clinical parameters, but not histologic features, such as old age, large tumor size, and proximal tumor locations, were associated with decreased survival.[19] Oliveira et al. defined large tumor size, high cellularity, presence of anaplasia or rhabdoid features, high mitotic activity, and high Ki-67 expression as adverse prognostic factors.[20] In the study by Kawaguchi et al., neither the histologic grade nor clinical parameters including age, gender, tumor size, location, and depth of the tumor showed significant impact on disease-free, metastasis-free, or recurrence-free survival.[15] A significant caveat is that most of these prior studies analyzing clinical adverse factors in EMC are based on morphology alone. As the histologic overlap between EMC and other epithelioid and myxoid soft tissue neoplasms is noteworthy, especially at the high grade end of the spectrum, it is difficult to exclude that some of the tumors included in these series may represent alternative diagnoses. As our study includes only genetically confirmed EMC, it provides a unique platform to investigate the impact of the variability of fusion structure on morphologic traits and behavior, not previously analyzed in these clinicopathologic studies.
About two-thirds of EMC harbor the chromosomal reciprocal translocation t(9;22)(q22;q12),[3] resulting in the fusion of EWSR1 gene to the nuclear receptor subfamily 4, group A, member 3 (NR4A3, a.k.a. CHN, TEC and NOR1).[4-6] An additional subset is characterized by a t(9;17)(q22;q11), resulting in the fusion of TATA binding protein-associated factor 15 (TAF15) to NR4A3.[7, 8] Rare cases with either (9;15)(q22;q21)[9] or t(3;9)(q12;q22)[10] have also been reported, in which NR4A3 is fused to transcription factor 12 (TCF12) or TRK-fused gene (TFG), respectively. TAF15 (a.k.a. TAF2N, RBP56 and YAF(II)68), together with EWSR1 and FUS belong to the TET multifunctional proteins that bind both RNA and DNA.[21] The N-terminal region of EWSR1, FUS and TAF15 contains degenerate repeats of the SYGQ motif and mediates powerful transcriptional activation when fused to heterologous DNA-binding domains of a variety of transcription factors.[22] Although FUS may substitute EWSR1 gene in a variety of translocation-associated sarcomas, EMC is the only tumor type to date in which TAF15 substitutes the EWSR1 for NR4A3 fusions. Furthermore, the NR4A3 gene fusions have not been described in other sarcoma subtypes and are considered pathognomonic for this disease. Both EWSR1-NR4A3 and TAF15-NR4A3 fusion proteins have been shown to be strong transcriptional activators.[23, 24]
Our study confirms that EWSR1-NR4A3 fusion is the most common genetic event in EMC, occurring in 62% of cases with classic morphology. The morphologic appearance of this molecular subset showed predominantly low cellularity, minimal cytologic atypia and low mitotic activity, with 12 cases (75%) being classified as low grade and only one patient (6%) succumbing to the disease. In contrast, 27% of cases had a TAF15-NR4A3 fusion with this subset predominantly (71%) showing features of ‘high grade morphology’ as described in prior studies. Also, three of the 7 patients with _TAF15-NR4A3_-positive tumors died of disease. Interestingly, EMCs with non-_EWSR1_variant translocations seem to occur more frequently outside the common thigh anatomic location and are more often associated with a rhabdoid phenotype microscopically. Although epithelioid/rhabdoid morphology did not seem to correlate with clinical outcome, a confounding factor could be the short follow-up duration in the patients with variant gene fusions. Furthermore, the rates of distant recurrences appear to be quite similar regardless of the fusion type.
As the morphologic spectrum of EMC varies from a low grade myxoid tumor to a more solid high grade epithelioid neoplasm, so is the histologic overlap with other myxoid and epithelioid soft tissue tumors. Particularly difficult is the distinction with the recently described soft tissue myoepithelial tumors.[25] Although myoepithelial neoplasms are defined by their positivity for EMA/Keratin and S100, the lack of a consistent immunohistochemical profile in EMCs makes the distinction rather difficult based on morphology and immunohistochemistry alone. Furthermore, both these entities can show EWSR1 gene rearrangements[25], adding to the challenge in diagnosis. In these difficult cases, FISH for the presence of a NR4A3 gene rearrangement can confidently distinguish between these two tumor types.[11, 26]
Extra-axial or soft tissue chordomas (a.k.a. chordoma periphericum) also show similar morphologic features with EMC. Immunohistochemical reactivity for the notochordal specific protein T-Brachyury is extremely helpful in confirming a diagnosis of extra-axial chordomas over an EMC. Although predominantly an extra-skeletal disease, EMCs can involve the bone occasionally. Rare reports of such tumors with the EWSR1-NR4A3 fusions have been reported (14). In osseous locations, differential diagnosis includes primary skeletal chondrosarcoma with secondary myxoid changes, which lack EWSR1-NR4A3 fusions.[12]
The presence of EMC with predominantly rhabdoid phenotype has raised the possibility of a genetic relationship with the prototypical rhabdoid soft tissue tumor, the proximal epithelioid sarcoma. Latest reports of EMC showing loss of INI1 expression immunohistochemically further questioned this hypothesis.[14, 27] As such, we tested 5 _NR4A3_-fusion positive (2 with EWSR1-NR4A3 and 3 with TAF15-NR4A3 fusions) tumors with predominantly rhabdoid phenotype (4 of the 5 cases tested) for INI1 expression by immunohistochemistry and 2 cases (cases’ 21 and 25) for SMARCB1 deletions by FISH, however, none showed loss of protein expression or gene deletion (data not shown).
In summary, our study highlights the association between variant translocations and morphology in EMCs. EMCs with TAF15-NR4A3 fusions appear to have ‘high grade’ histology. Although the findings suggest that EMCs with variant translocations may have a more aggressive behavior compared to EMCs with the more common _EWSR1_-related fusions, additional studies with longer follow-up data are required to confirm this finding. We have also emphasized the importance of applying genetic ancillary techniques for a more accurate classification of EMC from its morphologic mimics and to confirm a NR4A3 gene rearrangement in challenging diagnoses. The presence of an EWSR1 gene rearrangement in this context of tumors with overlapping morphologies is not specific. In our opinion, FISH assay for NR4A3 break-apart, alone or supplementing EWSR1, is more useful to differentiate EMC from its morphologic look-a-likes, especially in tumors with high grade, epithelioid morphology.
Supplementary Material
01
Acknowledgments
The authors would like to thank Alyne Manzo for preparation of composite figures and Milagros Soto for editorial assistance.
Supported in part by: P01CA47179 (CRA, SS), P50 CA 140146-01 (CRA, SS).
Footnotes
Disclosure / Conflict of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Enzinger FM, Shiraki M. Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol. 1972;3:421–435. doi: 10.1016/s0046-8177(72)80042-x. [DOI] [PubMed] [Google Scholar]
- 2.Saleh G, Evans HL, Ro JY, Ayala AG. Extraskeletal myxoid chondrosarcoma. A clinicopathologic study of ten patients with long-term follow-up. Cancer. 1992;70:2827–2830. doi: 10.1002/1097-0142(19921215)70:12<2827::aid-cncr2820701217>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 3.Hinrichs SH, Jaramillo MA, Gumerlock PH, Gardner MB, Lewis JP, Freeman AE. Myxoid chondrosarcoma with a translocation involving chromosomes 9 and 22. Cancer Genet Cytogenet. 1985;14:219–226. doi: 10.1016/0165-4608(85)90187-6. [DOI] [PubMed] [Google Scholar]
- 4.Gill S, McManus AP, Crew AJ, Benjamin H, Sheer D, Gusterson BA, Pinkerton CR, Patel K, Cooper CS, Shipley JM. Fusion of the EWS gene to a DNA segment from 9q22-31 in a human myxoid chondrosarcoma. Genes Chromosomes Cancer. 1995;12:307–310. doi: 10.1002/gcc.2870120412. [DOI] [PubMed] [Google Scholar]
- 5.Labelle Y, Zucman J, Stenman G, Kindblom LG, Knight J, Turc-Carel C, Dockhorn-Dworniczak B, Mandahl N, Desmaze C, Peter M, et al. Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum Mol Genet. 1995;4:2219–2226. doi: 10.1093/hmg/4.12.2219. [DOI] [PubMed] [Google Scholar]
- 6.Clark J, Benjamin H, Gill S, Sidhar S, Goodwin G, Crew J, Gusterson BA, Shipley J, Cooper CS. Fusion of the EWS gene to CHN, a member of the steroid/thyroid receptor gene superfamily, in a human myxoid chondrosarcoma. Oncogene. 1996;12:229–235. [PubMed] [Google Scholar]
- 7.Panagopoulos I, Mencinger M, Dietrich CU, Bjerkehagen B, Saeter G, Mertens F, Mandahl N, Heim S. Fusion of the RBP56 and CHN genes in extraskeletal myxoid chondrosarcomas with translocation t(9;17)(q22;q11) Oncogene. 1999;18:7594–7598. doi: 10.1038/sj.onc.1203155. [DOI] [PubMed] [Google Scholar]
- 8.Sjogren H, Meis-Kindblom J, Kindblom LG, Aman P, Stenman G. Fusion of the EWS-related gene TAF2N to TEC in extraskeletal myxoid chondrosarcoma. Cancer Res. 1999;59:5064–5067. [PubMed] [Google Scholar]
- 9.Sjogren H, Wedell B, Meis-Kindblom JM, Kindblom LG, Stenman G. Fusion of the NH2-terminal domain of the basic helix-loop-helix protein TCF12 to TEC in extraskeletal myxoid chondrosarcoma with translocation t(9;15)(q22;q21) Cancer Res. 2000;60:6832–6835. [PubMed] [Google Scholar]
- 10.Hisaoka M, Ishida T, Imamura T, Hashimoto H. TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2004;40:325–328. doi: 10.1002/gcc.20044. [DOI] [PubMed] [Google Scholar]
- 11.Noguchi H, Mitsuhashi T, Seki K, Tochigi N, Tsuji M, Shimoda T, Hasegawa T. Fluorescence in situ hybridization analysis of extraskeletal myxoid chondrosarcomas using EWSR1 and NR4A3 probes. Hum Pathol. 2010;41:336–342. doi: 10.1016/j.humpath.2009.04.028. [DOI] [PubMed] [Google Scholar]
- 12.Antonescu CR, Argani P, Erlandson RA, Healey JH, Ladanyi M, Huvos AG. Skeletal and extraskeletal myxoid chondrosarcoma: a comparative clinicopathologic, ultrastructural, and molecular study. Cancer. 1998;83:1504–1521. doi: 10.1002/(sici)1097-0142(19981015)83:8<1504::aid-cncr5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 13.Stewart FW. Case 211. Division of Laboratories and Research; Albany, New York: 1948. [Google Scholar]
- 14.Fletcher CD, Bridge JA, Hogendoorn PCW, Mertens F. WHO Classification of Tumours of Soft tissue and Bone. WHO Press. 2013 [Google Scholar]
- 15.Kawaguchi S, Wada T, Nagoya S, Ikeda T, Isu K, Yamashiro K, Kawai A, Ishii T, Araki N, Myoui A, Matsumoto S, Umeda T, Yoshikawa H, Hasegawa T. Multi-Institutional Study of 42 Cases in J. Extraskeletal myxoid chondrosarcoma: a Multi-Institutional Study of 42 Cases in Japan. Cancer. 2003;97:1285–1292. doi: 10.1002/cncr.11162. [DOI] [PubMed] [Google Scholar]
- 16.Drilon AD, Popat S, Bhuchar G, D’Adamo DR, Keohan ML, Fisher C, Antonescu CR, Singer S, Brennan MF, Judson I, Maki RG. Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer. 2008;113:3364–3371. doi: 10.1002/cncr.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucas DR, Fletcher CD, Adsay NV, Zalupski MM. High-grade extraskeletal myxoid chondrosarcoma: a high-grade epithelioid malignancy. Histopathology. 1999;35:201–208. doi: 10.1046/j.1365-2559.1999.00735.x. [DOI] [PubMed] [Google Scholar]
- 18.Jambhekar NA, Baraniya J, B R, Joshi U, Badhwar R. Extraskeletal Myxoid Chondrosarcoma: Clinicopathologic, Histochemical, and Immunohistochemical Study of 10 Cases. INT J SURG PATHOL. 1997;5:77–81. [Google Scholar]
- 19.Meis-Kindblom JM, Bergh P, Gunterberg B, Kindblom LG. Extraskeletal myxoid chondrosarcoma: a reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am J Surg Pathol. 1999;23:636–650. doi: 10.1097/00000478-199906000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Oliveira AM, Sebo TJ, McGrory JE, Gaffey TA, Rock MG, Nascimento AG. Extraskeletal myxoid chondrosarcoma: a clinicopathologic, immunohistochemical, and ploidy analysis of 23 cases. Mod Pathol. 2000;13:900–908. doi: 10.1038/modpathol.3880161. [DOI] [PubMed] [Google Scholar]
- 21.Riggi N, Cironi L, Suva ML, Stamenkovic I. Sarcomas: genetics, signalling, and cellular origins. Part 1: The fellowship of TET. The Journal of pathology. 2007;213:4–20. doi: 10.1002/path.2209. [DOI] [PubMed] [Google Scholar]
- 22.Arvand A, Denny CT. Biology of EWS/ETS fusions in Ewing’ family tumors. Oncogene. 2001;20:5747–5754. doi: 10.1038/sj.onc.1204598. [DOI] [PubMed] [Google Scholar]
- 23.Kim S, Lee HJ, Jun HJ, Kim J. The hTAF II 68-TEC fusion protein functions as a strong transcriptional activator. Int J Cancer. 2008;122:2446–2453. doi: 10.1002/ijc.23379. [DOI] [PubMed] [Google Scholar]
- 24.Labelle Y, Bussieres J, Courjal F, Goldring MB. The EWS/TEC fusion protein encoded by the t(9;22) chromosomal translocation in human chondrosarcomas is a highly potent transcriptional activator. Oncogene. 1999;18:3303–3308. doi: 10.1038/sj.onc.1202675. [DOI] [PubMed] [Google Scholar]
- 25.Antonescu CR, Zhang L, Chang NE, Pawel BR, Travis W, Katabi N, Edelman M, Rosenberg AE, Nielsen GP, Dal Cin P, Fletcher CD. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–1124. doi: 10.1002/gcc.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang WL, M E, Czerniak BA, et al. Fluorescence in situ hybridization is a useful ancillary diagnostic tool for extraskeletal myxoid chondrosarcoma. Modern Pathology. 2008;21(11):1303–1310. doi: 10.1038/modpathol.2008.114. [DOI] [PubMed] [Google Scholar]
- 27.Kohashi K, Oda Y, Yamamoto H, Tamiya S, Oshiro Y, Izumi T, Taguchi T, Tsuneyoshi M. SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol. 2008;32:1168–1174. doi: 10.1097/PAS.0b013e318161781a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01