Epidermal Growth Factor Receptor (EGFR) Signaling Is a Key Mediator of Hormone-Induced Leukocyte Infiltration in the Pubertal Female Mammary Gland (original) (raw)
Abstract
It is well documented that macrophages and eosinophils play important roles in normal murine pubertal mammary gland development. Although it is accepted that estrogen (E) and progesterone (P) are key players in mammary gland development, the roles these hormones might play in regulating the actions of leukocytes in that process is an understudied area. We show here that P and E, respectively, induce unique, but overlapping, sets of proinflammatory and angiogenic cytokines and chemokines, in the pubertal female BALB/c mammary gland, as well as induce infiltration of macrophages and eosinophils to the mammary periepithelium. This extends earlier studies showing P induction of proinflammatory products in pubertal and adult mammary epithelial organoids and P-induced in vivo infiltration of leukocytes to the adult mammary periepithelium. Importantly, epidermal growth factor receptor-signaling, which is likely mediated by amphiregulin (Areg), a downstream mediator of E and P, is both necessary and sufficient for both E- and P-induced recruitment of macrophages and eosinophils to the pubertal mammary periepithelium. We further show that receptor activator of nuclear factor κB ligand (RANKL), although not sufficient of itself to cause macrophage and eosinophil recruitment, contributes to an optimal response to P. The potency of Areg is highlighted by the fact that it is sufficient to induce macrophage and eosinophil recruitment at levels equivalent to that induced by either E or P. Our finding of a dominant role for Areg in hormonally induced leukocyte recruitment to the pubertal mammary gland parallels its dominance in regulating ductal outgrowth and its role in P-induced proliferation in the pubertal gland.
It is well documented that macrophages and eosinophils play important roles in normal pubertal mammary gland development in the mouse (1). The association of macrophages with the terminal end buds (TEBs) of the developing mammary gland is essential for normal ductal elongation, whereas eosinophil association with the TEBs is required for normal ductal branching. Thus, these 2 cell types perform complimentary roles in pubertal mammary gland development. In experiments using mice carrying a null mutation in the gene encoding macrophage-colony-stimulating factor, macrophages are depleted within the developing mammary gland and morphogenesis is impaired (1). Similarly, mice carrying a null nutation in the gene encoding eotaxin exhibit depletion of eosinophils and impaired morphogenesis within the developing mammary gland (1). Mast cells have also recently been implicated in pubertal mammary gland ductal morphogenesis, with a role independent of that of macrophages (2). They are particularly associated with the TEBs, and mast cell deficiency results in reduced numbers of TEBs.
Although it is accepted that estrogen (E) and progesterone (P) are key players in mammary gland development, the roles that these hormones might play in regulating the actions of leukocytes in that process is an understudied area. We previously identified receptor activator of nuclear factor (NF)-κB ligand (RANKL) and serum amyloid A (SAA)1, SAA2, and SAA3 as proinflammatory products whose mRNA is robustly induced by R5020 (synthetic progestin and progesterone receptor agonist) in BALB/c pubertal and adult mammary epithelial organoids in vitro (3, 4). Additionally, we and others showed progestin induction of RANKL protein expression in adult mammary epithelium in vivo (5–7) and in mammary organoids in vitro (3). Consistent with the potential activity of proinflammatory factors in the recruitment of leukocytes, we demonstrated that P induces in vivo recruitment of cluster of differentiation (CD) 45-positive leukocytes to the mammary periepithelial stroma in adult mice (4). The role of hormones, P in particular, in mediating leukocyte recruitment in the mammary gland had not been previously recognized. Because leukocytes play an essential role in the development of the pubertal mammary gland, extending the study of their hormonal regulation to that life stage is particularly illuminating of the interactions between hormones and mammary gland development.
E has been acknowledged as the primary hormone for pubertal ductal development (8). Although P plays a nonessential role in ductal development (9), it contributes to this process through its activity in regulating amphiregulin (Areg), a role that it shares with E (10). Our previous studies identify Areg as the key mediator of hormonally induced end bud (EB) formation and proliferation at puberty (10). The present studies were directed at examining whether the overlapping activities of P and E extend beyond the regulation of proliferation in the pubertal mammary gland and include the regulation of leukocyte recruitment that is also necessary to development of the pubertal mammary gland. Gene expression, immune, and histochemical analyses revealed that P and E can induce proinflammatory and angiogenic factors in intact mammary glands, as well as induce the recruitment of both macrophages and eosinophils to the periepithelial stroma of the developing mammary gland. The current study expands our understanding of hormonally mediated effects that promote pubertal mammary gland development to previously unrecognized effects of E and P on leukocyte recruitment. Importantly, this study provides mechanistic insight into the manner in which E and P effect leukocyte recruitment, identifying Areg and, to a lesser extent, RANKL as key mediators in this process.
Materials and Methods
Animals
Pubertal female 4-week-old BALB/c mice that had initiated estrous cycling and exhibited TEBs were ovariectomized (OVX). Recovery was allowed for 3 weeks after OVX to permit complete TEB regression before hormone treatments (11). OVX mice were injected daily for 5 days with saline control, 17-β-estradiol (E2) (1 μg/injection (inj)) (Sigma), P (1 mg/inj) (Sigma), or E2+P (1 μg + 1 mg/inj, respectively). These concentrations of E2 and P are physiologically relevant and have been used in previous studies to examine the effects of E2 and P in the pubertal mammary gland (5, 10, 12). To block P receptor (PR)- or E receptor (ER)-mediated effects, antiprogestin mifepristone (RU486) (1.3 mg/inj) (Sigma) or anti-E ICI 182,780 (ICI) (1.1 μg/inj) (Tocris Bioscience) was coinjected with P or E, respectively. To block RANKL-mediated effects, P-treated mice were coinjected with RANK fused to the fragment cystallizable region of immunoglobulin (RANK-Fc) (20 mg/kg dissolved in saline) (R&D Systems) every other day for 5 days. To block epidermal growth factor receptor (EGFR)-mediated effects, E2- and P-treated mice were given Gefitinib (Iressa; ChemieTek) (300 mg/kg dissolved in corn oil) daily for 5 days by oral gavage.
To examine the effects of RANKL or Areg on the mammary gland, OVX mice received 3 daily sc injections of RANKL (3 mg dissolved in saline) (Kingfisher Biotech) or Areg (1 mg dissolved in saline) (Creative Biomart) directly into the fat pad of the inguinal mammary gland on the right side of the mouse. Contralateral inguinal mammary glands were injected with control bovine serum albumin (3 or 1 mg dissolved in saline for the RANKL or Areg experiments, respectively) (Sigma).
All mice were injected with 5-bromo-2′-deoxyuridine (BrdU) (70 μg/g body weight) 2 hours before being killed. Mammary glands were formalin fixed and processed as whole mounts (13), or paraffin embedded for immunohistochemistry (14). All animal experimentation was conducted in accord with accepted standards of humane animal care and approved by the All University Committee on Animal Use and Care at Michigan State University.
Immunofluorescence
Mammary gland sections (5 μm) were prepared and subjected to antigen retrieval and immunofluorescent staining (14). To detect macrophages, sections were incubated with rat monoclonal anti-F4/80 (a mouse macrophage marker) (1:75; AbD Serotec), followed by goat antirat antibody (Ab) conjugated to Alexa Fluor 488 (1:100; Invitrogen). To assess whether macrophages expressed arginase 1 (Arg1), consistent with an M2 (alternatively activated macrophage) phenotype, some sections were additionally incubated with goat anti-Arg1 (1:200; Santa Cruz Biotechnology, Inc), followed by rabbit antigoat Ab conjugated to Alexa Fluor 546 (1:100; Invitrogen). BrdU incorporation was detected by mouse monoclonal anti-BrdU (undiluted; kit from Amersham Biosciences), followed by goat antimouse Ab conjugated to Alexa Fluor 488 (1:200; Invitrogen). To detect IL-17B, sections were incubated with rabbit anti-IL-17B (1:100; Bioss), followed by goat antirabbit Ab conjugated to Alexa Fluor 488 (1:200; Invitrogen). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:10 000; Invitrogen). Sections were visualized and images captured using a Nikon inverted epifluorescence microscope (Mager Scientific) with MetaMorph software (Molecular Devices Corp).
Histochemistry
For identification of eosinophils and mast cells, deparaffinized 5-μm sections were stained with Astra Blue/Vital New Red (15). Sections were visualized using a Nikon Eclipse 400 microscope and a SPOT RT color camera with SPOT software (Diagnostic Instruments). The number of eosinophils (Vital New Red) and mast cells (Astra Blue) is expressed as cells per structure in the mammary gland periepithelial area.
For analysis of blood vessel density, deparaffinized 5-μm sections were incubated in 2% of H2O2 in methanol/PBS (1:1 ratio) for 30 minutes followed by antigen retrieval by boiling at 95°C for 5 minutes. Sections were then treated for 2 hours with rabbit anti-CD31 (1:50 in PBS-0.5% Triton X-100, catalog number AP15436PU-N; Acris Antibodies, Inc) for endothelial cell staining and detection of blood vessels. After a brief wash, sections were incubated with secondary swine antirabbit Ab at room temperature for 30 minutes and then incubated with ABC reagent (PK-7100; Vector Laboratories, Inc) for 30 minutes. The sections were then incubated with metal enhanced 3,3′-diaminobenzidine substrate solution (100 μL of DAB substrate + 900 μL of stable peroxide substrate buffer; Thermo Scientific) for 7 minutes and counterstained with hematoxylin for 2 minutes. The stained sections were visualized with a Nikon Eclipse E400 light microscope. A minimum of 1000 cells was counted for each section, and a minimum of 2–3 tissue sections per animal was analyzed. Digital micrographs were captured, and the images were overlaid with grids containing 240 squares (324 μm2/square). Blood vessel density is expressed as the percentage of CD31-positive squares.
Quantitation and statistical analyses
Macrophages, eosinophils, and mast cells were counted in the periepithelial stroma of small ducts, large ducts, and EBs, from at least 3 mice per treatment. The numbers of cells are presented per mammary gland structure (ie, large duct, small duct, EB). Large ducts were characterized by larger lumen diameter, comprising greater than 50 cells, and by an extensive extracellular matrix and fibroblasts surrounding the epithelium. Small ducts were characterized by smaller lumen diameter, comprising fewer than 50 cells, and by a limited extracellular matrix surrounding the epithelium. EBs were enlarged structures characterized by multiple epithelial cell layers, usually on one side of the structure, leading to a bulb-like appearance. Digital images of inguinal mammary gland whole mounts were analyzed with ImageJ (http://rsb.info.nih.gov/ij/download.html) for epithelial area. To analyze IL-17B fluorescence intensity, the average pixel intensity of all positively stained cells within the ductal epithelium was determined. A threshold was set to exclude background fluorescence, and images were gated to include intensity measurements only from positively staining epithelial cells. For all experiments, a minimum of 3 mice per treatment group was analyzed. Statistical significance was calculated by Student's t test.
RT-PCR analyses
Total RNA was extracted from mouse mammary glands by TRIzol reagent (Invitrogen) and purified using the RT2 qPCR-Grade RNA isolation kit (SABiosciences). Three micrograms of total RNA were used for cDNA synthesis using the RT2 First Strand kit (SABiosciences) following the manufacturer's instructions. cDNAs (20 μL) were diluted to 150 μL with deionized H2O. mRNA levels were quantitated using Inflammatory Cytokines and Receptors and Angiogenesis PCR Arrays (catalog numbers PAMM-011 and PAMM-024, respectively; SABiosciences). In addition to the arrays, primers for the following selected genes were purchased from SABiosciences: Saa1 (PPM05345E), Saa3 (PPM05486E), Tnfs11 (RANKL) (PPM03047E), 18S ribosomal ribonucleic acid (RNA) (rRNA) (PPM57735E), and ribosomal protein L32 (RPL32) (PPM03300B). Primers for Areg were purchased from Applied Biosciences (Mm00437583_m1, catalog number 4331182). For quantitative RT-PCR (qRT-PCR) analysis, each reaction (25 μL) included 12.5 μL of 2× SABiosciences RT2 qPCR Master Mix (SYBR Green), 1 μL of diluted first-stand cDNA synthesis reaction, and 11.5 μL of deionized H2O. qRT-PCR was performed with the ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Inc) using the following program: step 1, 95°C for 10 minutes; step 2, 40 cycles of 95°C for 15 seconds and 60°C for 1 minute; and step 3, dissociation curve 95°C for 1 minute, 65°C for 2 minutes (optics off), 65°C–95°C at 2°C per minute (optics on). The data were analyzed using online software from SABiosciences (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). For Saa1, Saa3, and RANKL, the comparative threshold cycle method was used to calculate the fold change in gene expression after normalization to values for 18S rRNA and RPL32. RNA from the inguinal mammary glands of 3 animals was analyzed from each treatment group.
Tartrate-resistant acid phosphatase 5b (TRAP5b) assay
Blood was collected at the time of harvest, allowed to clot at room temperature for 5 minutes, and then centrifuged at 4000 rpm for 10 minutes. Serum was removed and stored at −80°C. Serum TRAP5b was measured using a MouseTRAP assay kit (SB-TR103; Immunodiagnostic Systems, Inc) according to the manufacturer's protocol.
Results
P and E induce macrophage and eosinophil recruitment to the pubertal mammary periepthelial stroma
We previously found that P induced the recruitment of CD45-positive leukocytes to the mammary periepithelial stroma of adult OVX mice (4). Because E is a key hormone directing pubertal ductal development and the role of P in pubertal leukocyte recruitment is unknown, we examined whether P, and also E, might have similar roles in leukocyte recruitment in the pubertal mammary gland.
Both P and E2 treatment of OVX pubertal animals induced increased numbers of macrophages (Figure 1, A and B) and eosinophils (Figure 1, C and D) in the mammary periepithelial stroma. No significant change was observed for mast cells (Supplemental Figure 1). Combined treatment with both P and E2 produced effects largely similar to that of either hormone alone. Interestingly, P treatment was significantly less effective for eosinophil recruitment to EBs than P and E2 together, suggesting that the effects of the 2 hormones are not entirely identical in this context.
Figure 1.

Both E2 and P regulate macrophage and eosinophil recruitment to the pubertal mammary gland. Pubertal 4-week-old BALB/c mice were OVX, allowed to recover for 3 weeks, and then treated for 5 days with vehicle control (C), E2, P, or E2+P. A, Immunofluorescent detection of F4/80 (green) in C and P-treated mammary gland. Nuclei were counterstained with DAPI (blue). Scale bar, 25 μm. B, F4/80-positive cell recruitment to the periepithelium was analyzed for large ducts, small ducts, and duct ends. The values represent the mean ± SEM (n = 6 animals per treatment). The number of macrophages recruited to periepithelial structures upon treatment with E2, P, and E2+P was above C levels (*, P < .05). C, Histochemical detection of eosinophils (arrows) in C and P-treated mammary gland. Nuclei were counterstained with hematoxylin (blue). Scale bar, 50 μm. D, Eosinophil recruitment to the periepithelium was analyzed for large ducts, small ducts, and duct ends. The number of eosinophils recruited to periepithelial structures upon treatment with E2, P, and E2+P was above C levels (*, P < .05). The number of eosinophils recruited to the periepithelium of P-treated EBs was reduced compared with E2+P-treated EBs (#, P < .05). The values represent the mean ± SEM (n = 6 animals per treatment). E, Macrophage analysis by dual immunofluorescent detection of F4/80 and Arg1. The percentage of F4/80-positive cells coexpressing Arg1 is presented. The values represent the mean ± SEM (n = 3 animals per treatment).
Because macrophages are associated with the tissue-remodeling processes in mammary gland development, we evaluated whether hormone-induced macrophages in the mammary periepithelium might display an M2 or alternative activation phenotype. The phenotype is associated with tissue-remodeling processes (16). To that end, we examined the proportion of F4/80-positive macrophages that were also positive for Arg1, which is expressed at high levels in M2 macrophages (17). Approximately 90% of macrophages across all structures and treatments were Arg1-positive, consistent with M2 polarization (Figure 1E).
P and E induce proinflammatory gene expression in intact mammary glands
Having identified hormone-induced recruitment of macrophages and eosinophils, we assessed the milieu of proinflammatory products that might be directly involved in recruiting these cells to the mammary periepithelial stroma. We previously found that the progestin (R5020) induced proinflammatory gene expression in mammary epithelial cells in vitro (4), in particular, SAA1, SAA2, and SAA3 (Saa1, Saa2, and Saa3) and the NF-κB activating cytokine, RANKL. Analysis of proinflammatory gene expression was carried out in vivo in mammary glands of OVX pubertal BALB/c mice that were treated for 5 days with saline control or hormones. RNA was isolated from their mammary glands and analyzed by qRT-PCR. Similarly to the case with R5020 treatment of cultured mammary epithelial organoids (4), P induced RANKL and Saa1 expression in pubertal animals (Table 1). However, P induced an even broader range of proinflammatory products in intact mammary glands than observed in cell culture. A different, but overlapping, set of cytokine and chemokine mRNAs was observed with E2 treatment.
Table 1.
E2 and P Regulation of Inflammation-Associated Gene Expression
| Symbol | Description | Fold Change | Known P and E Regulation | ||
|---|---|---|---|---|---|
| 5-d E | 5-d P | E | P | ||
| Ccl1 | Chemokine (C-C motif) ligand 1 | No change | 2.7 | ||
| Ccl20 | Chemokine (C-C motif) ligand 20 | No change | 4.5 | ||
| Ccl3 | Chemokine (C-C motif) ligand 3 | 2.1 | No change | ↓ (56) | |
| Ccl8 | Chemokine (C-C motif) ligand 8 | 5.4 | 3.3 | ↑ (70) | |
| Cx3cl1 | Chemokine (C-X3-C motif) ligand 1 | No change | 2.9 | ↑ (71) | |
| Cxcl15 | Chemokine (C-X-C motif) ligand 15 | 12 | 5.7 | ||
| Cxcl5 | Chemokine (C-X-C motif) ligand 5 | 3.1 | No change | ||
| Il17b | IL-17B | 8.6 | 8.9 | ||
| Il8rb | IL-8 receptor, β | No change | −3.0 | ||
| Tnf | TNF | No change | 2.4 | ||
| Areg | Amphiregulin | 24 | 31 | ↑ (10) | ↑ (10) |
| RANKL | Receptor activator of nuclear factor κ B ligand | No change | 630 | ↑ (6, 7) | |
| SAA1 | Serum amyloid A1 | 2.2 | 1.8 | ↑ (4) |
P and E induce angiogenesis in pubertal mammary glands
Leukocyte recruitment has long been associated with angiogenesis (18), and angiogenesis accompanies mammary ductal development with capillaries in close association with epithelial ducts (19). In order to assess whether P and E might also induce factors associated with angiogenesis, we performed a qRT-PCR analysis similar to that for proinflammatory gene products. P and E2 induced differing, but overlapping, sets of factors associated with angiogenesis (Table 2).
Table 2.
E2 and P regulation of Angiogenesis-Associated Gene Expression
| Symbol | Description | Fold Change | Known P and E Regulation | Angiogenic Effect | ||
|---|---|---|---|---|---|---|
| 5-d E | 5-d P | E | P | |||
| Cxcl2 | Chemokine (C-X-C motif) ligand 2 | No change | −2.6 | ↓ (72) | ↓ (73) | |
| Cxcl5 | Chemokine (C-X-C motif) ligand 5 | 2.1 | 2.5 | ↑ (74) | ||
| F2 | Coagulation factor II | No change | 3.2 | ↑ (75) | ||
| Hand2 | Heart and neural crest derivatives expressed transcript 2 | No change | 2.8 | ↑ (76) | ↑ (77) | |
| Itgb3 | Integrin β 3 | No change | 2.1 | ↑ (78) | ||
| Lect1 | Leukocyte cell-derived chemotaxin 1 | No change | 5.0 | ↓ (79) | ||
| Mdk | Midkine | 3.7 | 5.1 | ↑ (80) | ↑ (81) | |
| Sphk1 | Sphingosine kinase 1 | 2.2 | 4.4 | ↑ (82) | ↑ (83) | ↑ (84) |
| Stab1 | Stabilin 1 | 2.1 | No change | ↑ (85) | ||
| Tbx1 | T-box 1 | −2.1 | −2.3 | ↑ (86) | ||
| Thbs1 | Thrombospondin 1 | 2.2 | 5.7 | ↑ (87) | ↑ (88) | ↓ (89) |
| Thbs2 | Thrombospondin 2 | 2.1 | No change | ↓ (89) | ||
| Timp1 | Tissue inhibitor of metalloproteinase 1 | 3.0 | 3.1 | ↑ (90) | ↑ (91) | |
| Tnf | Tumor necrosis factor | No change | 2.5 | ↑ (92) | ↑ (93) | |
| Tnfaip2 | TNF, α-induced protein 2 | −4.8 | No change | ↑ (94) |
We also examined the effects of P and E2 on angiogenesis. Both P and E2 induced increased vascularization of the mammary gland, as evidenced by increased area of CD31 staining (Figure 2). Thus, the observed modulation of genes associated with angiogenesis appears functionally significant.
Figure 2.
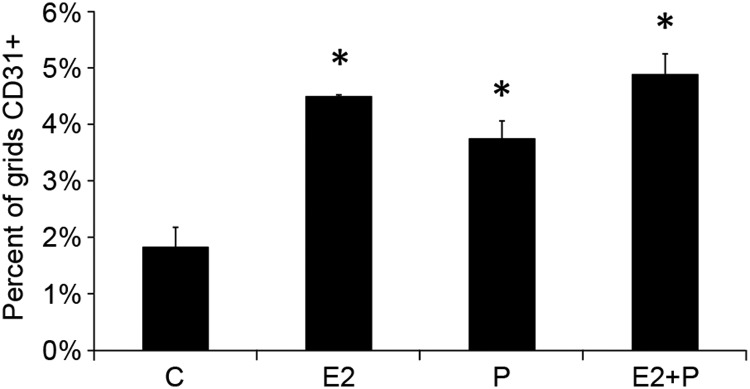
E2 and P increase angiogenesis in the pubertal mammary gland. Pubertal 4-week-old BALB/c mice were OVX, allowed to recover for 3 weeks, and then treated for 5 days with vehicle control (C), E2, P, or E2+P. Blood vessel density was measured, as described in Materials and Methods, by the area occupied by CD31-positive vessels near mammary epithelium. The values represent the mean ± SEM (n = 3 animals per treatment). CD31 staining was greater in E2-, P-, and E2+P-treated mammary glands (*, P < .05) compared with Cs.
Leukocyte recruitment by P and E are mediated through their cognate receptors
The effects of P and E are largely mediated through their cognate receptors, respectively, PR (14) and ERα (8). ERβ appears not to have a role in pubertal mammary gland development, because ERβ-deficient mice have normal mammary gland development (20). In order to confirm that hormone-induced leukocyte recruitment occurred through these classical hormone receptors, we carried out P treatment with and without the PR-binding antagonist RU486, or with E2 with and without the ER-binding antagonist ICI. RU486 effectively blocked P-stimulated macrophage and eosinophil infiltration to the periepithelial stroma surrounding large ducts, small ducts, and EBs (Figure 3, A and B). ICI was similarly effective in blocking E2-stimulated leukocyte infiltration. Therefore, we concluded that the actions of P and E2 in leukocyte recruitment in the mammary gland are mediated through their cognate receptors.
Figure 3.

Areg and RANKL are mediators of hormone-induced macrophage and eosinophil recruitment to mammary epithelium. Pubertal 4-week-old BALB/c mice were OVX, allowed to recover for 3 weeks, and then treated for 5 days with vehicle control (C), E2, or P. A and B, Both macrophage (A) and eosinophil (B) recruitment to mammary gland epithelial structures by E2 and P were inhibited by E2+ICI and P+RU486. The numbers of macrophages and eosinophils recruited to periepithelial structures upon treatment with E2 or P were above that of other treatments (*, P < .05). C and D, Both macrophage (C) and eosinophil (D) recruitment to mammary gland epithelial structures by E2 and P were inhibited by E2+Iressa and P+Iressa. E2- and P-induced macrophage recruitment was completely inhibited in large and small ducts by the addition of Iressa. No EBs were detected in Iressa-treated mammary glands. The numbers of macrophages or eosinophils recruited to periepithelial structures upon treatment with E2 or P were above that of other treatments (*, P < .05). E and F, Both macrophage (E) and eosinophil (F) recruitment to mammary gland epithelial structures by P were inhibited by P+RANK-Fc. The numbers of macrophages or eosinophils recruited to periepithelial structures upon treatment P were above that of C treatments (#, P < .05). P+RANK-Fc decreased macrophage recruitment to large and small ducts compared with P alone (*, P < .05). P+RANK-Fc decreased eosinophil recruitment to large ducts (*, P < .05) and EBs (**, P < .1) compared with P alone. A–F, The values represent the mean ± SEM (n = 3 animals per treatment).
Areg is an essential mediator of both P- and E-induced leukocyte infiltration in the mammary gland
Both P and E contribute to ductal development and proliferation through Areg (10). Although some EGFR ligands, such as TGFα and heparin-binding EGF-like growth factor, have been detected in pubertal end buds (EBs), Areg is the most highly expressed EGFR ligand in ducts and EBs during puberty, and Areg is required for normal ductal morphogenesis (21). Therefore, we hypothesized that Areg might be a mediator of hormonally induced leukocyte recruitment. Stimulation of macrophage (Figure 3C) and eosinophil infiltration (Figure 3D) with either P or E2 in the presence or absence of Iressa, an antagonist of EGFR signaling, fully inhibited E2- and P-stimulated infiltration of macrophages and eosinophils and actually brought their numbers below that of control animals. This demonstrates that EGFR signaling, and likely Areg, is essential to both E- and P-stimulated leukocyte infiltration. Furthermore, Iressa fully inhibited both E2- and P-stimulated EB development and epithelial expansion in the same animals (Figure 4, A–C).
Figure 4.

Iressa inhibits both E2- and P-induced EB formation and epithelial outgrowth. Pubertal 4-week-old BALB/c were OVX, allowed to recover for 3 weeks, and then treated for 5 days with vehicle control (C), E2 ± Iressa (300 mg/kg), or P ± Iressa. A, Whole mounts showing effect of treatment on EB formation after 5 days of C, E2, E2+Iressa, P, or P+Iressa. Note the absence of EBs in Iressa-treated mammary glands. Scale bar, 2 mm. B, EB quantitation from C and treated whole mounts. Values represent the mean ± SEM number of EBs in an inguinal mammary gland (n = 4 mice). E2 and P increased the average EB number (*, P < .05), and this increase was inhibited by the addition of Iressa. C, Epithelial area quantitation from C and treated whole mounts. Values represent the mean ± SEM epithelial area in a pair of inguinal mammary glands (n = 4 mice). E2 and P increased the average epithelial area (*, P < .05), and this increase was inhibited by the addition of Iressa.
To test whether Areg treatment by itself could stimulate leukocyte infiltration to ductal structures, we stimulated macrophage (Figure 5A) and eosinophil infiltration (Figure 5B) in OVX mice with local injection of Areg or a bovine serum albumin (BSA) control into the inguinal mammary gland. Compared with control injections, Areg significantly increased numbers of macrophages and eosinophils to both small and large ducts. The contralateral control-injected gland did not exhibit increased numbers of macrophages or eosinophils, indicating that the effects of Areg are local rather than systemic. As expected, the Areg-injected glands also showed increased numbers of proliferative cells (Figure 5C). Areg was able to induce both the development of proliferating EBs (Supplemental Figure 2A) and macrophage recruitment to EBs (Supplemental Figure 2B).
Figure 5.

Areg increased macrophage and eosinophil recruitment and mammary epithelial cell proliferation. Pubertal 4-week-old BALB/c mice were OVX, allowed to recover for 3 weeks, and then treated for 3 days with bovine serum albumin (BSA) control (C) or Areg. A and B, Both macrophage (A) and eosinophil recruitment (B) were significantly increased by Areg over BSA Cs across all mammary epithelial structures (*, P < .05). C, The percentage of BrdU+ cells was increased by Areg over BSA Cs across all mammary epithelial structures (*, P < .05). A–C, The values represent the mean ± SEM (n = 3 animals per treatment).
RANKL contributes to P-induced leukocyte infiltration in the mammary gland
Most the proinflammatory products identified in hormonally treated mice are at least in part regulated by NF-κB, which is not a primary target of EGFR signaling. We noted that 8 of the 11 hormone-induced proinflammatory cytokine and chemokine mRNAs are known targets of NF-κB transcriptional activation (Table 1). These are chemokine (C-C motif) ligand (Ccl)1 (22), Ccl3 (23), Ccl8 (24), Ccl20 (25), chemokine (C-X3-C motif) ligand 1 (Cx3cl1) (26), chemokine (C-X-C motif) ligand (Cxcl)5 (27), Saa1 (28), and Tnf (29). Conversely, Il17b (30), Tnf (31), and RANKL (32) all mediate their effects, at least in part, through NF-κB activation. Additionally, 5 of the hormone-induced mRNAs associated with angiogenesis are known NF-κB targets (Table 2). These are midkine (33), thrombospondin (Thbs)1 (34), Thbs2 (35), tissue inhibitor of metalloproteinase 1 (36), and TNFα-induced protein (Tnfaip)2 (37). Because RANKL, a potent activator of NF-κB (38), is the most robustly induced of the P-induced cytokines that we examined (Table 1), we investigated the potential role of RANKL in P-induced leukocyte infiltration in the mammary gland.
Macrophage (Figure 3E) and eosinophil (Figure 3F) infiltration was stimulated with P in the presence or absence of RANK-Fc, a decoy receptor and antagonist of RANKL signaling. RANK-Fc significantly reduced macrophage infiltration to large and small ducts (42% and 48%, respectively) but not to EBs. Eosinophil infiltration was also significantly reduced to large ducts and EBs (100% and 43%, respectively) but not to small ducts. We confirmed the activity of RANK-Fc in systemically inhibiting RANKL activity by its suppression of serum TRAP5b levels (Supplemental Figure 3) and its inhibition of ductal and EB proliferation as measured by BrdU incorporation (10). Thus, P-induced RANKL is also a mediator of leukocyte infiltration, in addition to Areg and possibly other P-induced factors that may contribute to this response.
In order to test whether RANKL treatment by itself could stimulate macrophage and eosinophil infiltration to ductal structures, we treated OVX mice with local injection of RANKL vs a BSA control into the inguinal mammary gland and measured macrophage (Supplemental Figure 4A) and eosinophil infiltration (Supplemental Figure 4B). No significant recruitment of macrophages or eosinophils was detected, suggesting that although RANKL is likely necessary for full leukocyte infiltration in response to P, it is not sufficient. There was also no proliferative response to local RANKL injection (Supplemental Figure 4C), consistent with the previously reported dependence of pubertal mammary proliferation on leukocyte infiltration (39).
A possible role for IL-17B in P- and E-induced leukocyte infiltration in the mammary gland
P induction of RANKL provides a potent activator of NF-κB. Because RANK-Fc was not able to fully block macrophage and eosinophil infiltration in response to P, this suggested the role of other factors in this process. Furthermore, because RANKL was not induced by E, this indicated that RANKL was certainly not mediating leukocyte infiltration induced by E. The robust induction of Il17b mRNA by both P and E2 (Table 1) provides another plausible factor for NF-κB activation, because it is capable of inducing Tnf through NF-κB activation (30). Because Areg is induced by both hormones and is likely an essential mediator of hormonally induced leukocyte infiltration in the mammary gland, we tested whether EGFR signaling was responsible for induction of Il17b expression. Mammary gland tissue sections from animals treated with P and E2 in the presence or absence of Iressa (see Figure 2, C and D) were stained for IL-17B (Figure 6A). Either P or E2 treatments were able to increase IL-17B expression, and Iressa was able to fully block this increase. In order to directly test whether Areg treatment by itself might induce IL-17B expression, mammary gland tissue sections from animals subjected to local injection of Areg (see Figure 5) were stained for IL-17B (Figure 6B). Areg was able to increase IL-17B expression in a dosage-dependent fashion to levels equivalent to those seen after 3 days of P treatment. Thus, it is plausible that Il17b is a downstream target of Areg that participates in the induction of NF-κB-dependent cytokine expression in response to both E and P treatment.
Figure 6.
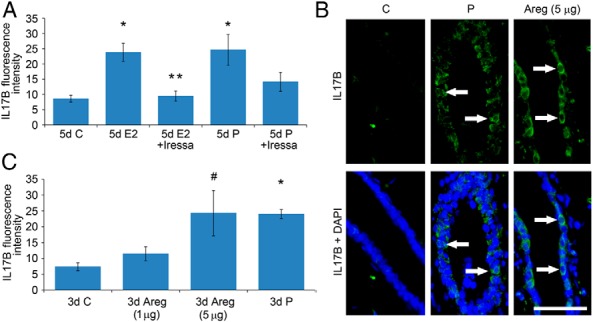
IL-17B is regulated by E2 and P through Areg. A, Pubertal 4-week-old BALB/c mice were OVX, allowed to recover for 3 weeks, and then treated for 5 days with vehicle control (C), E2, E2+Iressa, P, or P+Iressa. A, IL-17B was measured in mammary epithelium by immunofluorescence, as intensity above background. The values represent the mean ± SEM (n = 3 animals per treatment). Five-day E2 and P treatment increased IL-17B compared with C (*, P < .05). E2+Iressa inhibited E2-induced increases in IL-17B (**, P < .05). B and C, Pubertal 4-week-old BALB/c mice were OVX, allowed to recover for 3 weeks, and then treated for 3 days with BSA C, 1 or 3 μg of Areg, or P. B, Immunofluorescent detection of IL-17B (green). White arrows mark examples of cytoplasmic IL-17B expression colocalizing to DAPI staining in mammary epithelial cells. Scale bar, 25 μm. C, IL-17B was measured in mammary epithelium by immunofluorescence, as intensity above background. The values represent the mean ± SEM (n = 3 animals per treatment). Three-day P treatment (*, P < .05) and 3-day 5-μg Areg treatment (#, P < .05) increased IL-17B compared with C treatment.
Discussion
This study examined the roles of P and E in inducing leukocyte infiltration in the pubertal mammary gland that is concomitant with hormonally induced proliferation and ductal outgrowth. It is well established that inflammation-like processes are required for tissue remodeling and angiogenesis, which are essential for normal mammary gland development (39). We now show that P and E, respectively, induce unique, but overlapping, sets of proinflammatory cytokines and chemokines, in the pubertal mammary gland, as well as induce the infiltration of macrophages and eosinophils to the mammary periepithelium. This extends our earlier studies that showed P induction of proinflammatory products in pubertal and adult mammary epithelial organoids and P-induced in vivo infiltration of leukocytes to the adult mammary periepithelium (4). Importantly, we show that EGFR signaling is both necessary and sufficient for both E- and P-induced recruitment of macrophages and eosinophils to the mammary periepithelium in pubertal mice, as well hormonally induced EB development and epithelial expansion. This linkage of leukocyte recruitment and epithelial development to EGFR signaling is consistent with the established role of macrophages and eosinophils in mammary gland morphogenesis (1). Because Areg is the most highly expressed EGFR ligand in ducts and EBs during puberty, and Areg is required for normal ductal morphogenesis (21), it is likely that Areg is responsible for this in vivo requirement for EGFR signaling. Both E (40) and P (41) can up-regulate Areg expression. The potency of Areg is highlighted by the fact that it is sufficient of itself to induce macrophage and eosinophil recruitment at levels equivalent to that induced by either E or P. There is precedent for Areg-induced production of proinflammatory factors in rheumatoid arthritis (42). Our finding of a dominant role for Areg in hormonally induced leukocyte recruitment to the pubertal mammary gland parallels its dominance in regulating ductal outgrowth (21) and parallels its role in P-induced proliferation in the pubertal gland (10).
RANKL, although not sufficient of itself to cause recruitment of macrophages and eosinophils, likely contributes to an optimal response to P, because RANK-Fc significantly blocks P's activity in leukocyte recruitment. This is consistent with data presented here showing that P induces a proinflammatory expression profile with elements unique from those induced by E. The basis for this difference may be the ability of RANKL to activate the noncanonical pathway for NF-κB activation, which targets some genes distinct from those activated by the canonical NF-κB pathway (43). One possible explanation for the fact that the effects of RANK-Fc antagonism are more significant than the inductive effects of RANKL alone is that although leukocyte recruitment may require robust activation of NF-κB, this alone may not be sufficient. Signaling through EGFR is known to activate signal transducer and activator of transcription (Stat) 5 (44) and Stat3 (45), both important regulators of mammary gland development (46). Ductal development is decreased in Stat5 knockout animals (47), and, because leukocyte recruitment plays a critical role in ductal development, we speculate that Stat5 may be involved in the process of leukocyte recruitment. Furthermore, some proinflammatory cytokines similarly require 2 pathways of transcriptional activation for expression. For instance, Ccl20, which is induced by P in our studies, displays synergistic enhancement of expression by Stat3 and NF-κB (48). Ccl3, induced by E in our studies, is regulated by NF-κB but also has a putative Stat5 binding site (49).
RANKL is required for the development of side branches leading to the formation of lobulo-alveolar structures during pregnancy (50), and its targeted expression to mammary epithelial cells in PR-deficient mice is sufficient to induce branching and alveologenesis (51). Our finding that antagonism of the P-specific induction of RANKL has less profound effects than antagonism of Areg signaling is consistent with the reduced ability of the pubertal gland to develop side branches in response to P treatment (10). Indeed, treatment with RANK-Fc only partially suppressed P-induced leukocyte recruitment and proliferation. It may well be that Areg-induced expression of IL-17B, an inducer of proinflammatory cytokines in fibroblasts and macrophages, and presumed NF-κB activator (30), supports Areg induction of proinflammatory cytokines in the absence of RANKL activity. We found that both E and P robustly induce IL-17B expression, probably through Areg. Interestingly, RANK-Fc treatment displayed a more complete suppression of P-induced eosinophil recruitment and proliferation in large ducts. This suggests that RANKL may play a greater role in large ducts than other structures, and this is consistent with its role in side-branching off of large ducts (52). A greater role in large ducts is also consistent with the observation of a higher level of RANKL expression in pubertal ducts (10). It will be interesting to examine whether the dominance of Areg over RANKL in supporting leukocyte infiltration in the pubertal mammary gland is reversed in adult animals, where side-branching and alveologenesis are critical glandular events during pregnancy. In the case of Areg, where inflammation-like effects are closely associated with proliferation, it may also be useful to examine whether inflammation-like effects and proliferation might be dissociated by antagonism of fibroblast growth factor 2 and fibroblast growth factor 7, which have been implicated as downstream proliferative effectors of EGFR signaling in the mammary gland (53).
Treatment with P and E each induce a number of proinflammatory cytokines and chemokines that plausibly support the recruitment of macrophages and eosinophils. Although Areg has been directly implicated in chemotaxis of mesothelioma cells (54) and bone marrow mesenchymal progenitors (55), it is likely that the role of Areg in leukocyte recruitment is largely indirect and through the participation of EGFR signaling and EGFR-dependent induction of IL-17B in the regulation of these proinflammatory cytokines and chemokines. Among the P-induced mRNAs whose expression is associated with inflammation, Ccl1, Ccl20, Cxcl15, Il17b, and Tnf have not previously been reported to be P regulated. None of the genes found to be up-regulated by E here are known to be E regulated. However, opposite to our results, Ccl3 was reported to be down-regulated by E in murine mammary tissue (56). Among the inflammation-associated mRNAs identified as being induced by either P or E, Ccl1 (57), Ccl3 (58), Ccl8 (59), Cx3cl1 (60, 61), RANKL (62), Saa1 (63), and Tnf (64) have all been shown to have chemotactic activity toward monocytes, macrophages, and/or eosinophils. Although the spectrum of cytokines and chemokines induced by P and E, respectively, overlap, some are uniquely induced by P or E. We found that P was significantly less effective than E in inducing eosinophil recruitment to the vicinity of EBs, and this is consistent with the 2 hormones inducing nonidentical sets of proinflammatory factors. For instance, Ccl3 has chemotactic activity toward eosinophils (65) and is induced by E rather than P.
Among the P-induced mRNAs whose expression is associated with angiogenesis, Cxcl5, coagulation factor II, integrin β 3, leukocyte cell-derived chemotaxin 1, stabilin 1, T-box 1, Thbs2, and Tnfaip2 were not previously known to be P or E regulated. Most of these observed changes in expression have been associated in the literature with proangiogenic effects. Consistent with the observation of a largely proangiogenic expression profile, we found that both P and E increased vascularization of the mammary gland.
These studies expand our understanding of hormonally mediated effects that promote pubertal mammary gland development to previously unrecognized effects of E and P on leukocyte recruitment. We provide insight into the mechanisms linking P and E signaling and the inflammation-like and angiogenic processes essential to normal mammary gland development. However, in the context of mammary carcinogenesis, these same processes can also promote tumor development. Animal models of breast cancer have demonstrated that inflammatory processes contribute to tumor proliferation and metastasis (66). It is known that long-term exposure to P alone (67), or in combination with E, promotes the development of mammary cancer in rodents (68). It is noteworthy that most macrophages recruited to the mammary periepithelium in response to P and E express Arg1, consistent with an M2 or alternative activation phenotype. This phenotype is not only associated with tissue-remodeling processes (16) but also tumor progression (69). Thus, the hormonally induced inflammation-like response in the mammary gland presents a plausible mechanism underlying the promotional effects of hormones in mammary tumorigenesis that may be at play in addition to the proliferative effects of E and P.
Acknowledgments
We thank Regina Irwin in the laboratory of Dr Laura McCabe for performing the TRAP5b assays.
This work was supported by the Breast Cancer and the Environment Research Program award 1UO1ESO19434 and the Breast Cancer and the Environment Research Centers award 1U01ES12800 from the National Institute of Environmental Health Sciences and the National Cancer Institute, National Institutes of Health, Department of Health and Human Services. This work was also supported by the Avon Foundation, Ladies Auxiliary to the Veterans of Foreign Wars, and the Helen L. Kay Charitable Trust.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
Abbreviations:
Areg
amphiregulin
Arg1
arginase 1
BrdU
5-bromo-2′-deoxyuridine
BSA
bovine serum albumin
Ccl
chemokine (C-C motif) ligand
CD
cluster of differentiation
Cxcl
chemokine (C-X-C motif) ligand
Cx3cl1
chemokine (C-X3-C motif) ligand 1
DAPI
4′,6-diamidino-2-phenylindole
E
estrogen
E2
17-β-estradiol
EB
end bud
EGFR
epidermal growth factor receptor
ER
E receptor
F4/80
mouse macrophage marker
Fc
fragment cystallizable region of immunoglobulin
ICI
ICI 182,780
M2
alternatively activated macrophage
NF
nuclear factor
OVX
ovariectomized
P
progesterone
PR
P receptor
qRT-PCR
quantitative RT-PCR
R5020
synthetic progestin and progesterone receptor agonist
RANKL
receptor activator of NF-κB ligand
Ru486
mifepristone
SAA
serum amyloid A
Stat
signal transducer and activator of transcription
TEB
terminal end bud
Thbs
thrombospondin
Tnfaip
TNF, α-induced protein
TRAP5b
tartrate-resistant acid phosphatase 5b.
References
- 1.Gouon-Evans V, Rothenberg ME, Pollard JW. Postnatal mammary gland development requires macrophages and eosinophils. Development. 2000;127:2269–2282 [DOI] [PubMed] [Google Scholar]
- 2.Lilla JN, Werb Z. Mast cells contribute to the stromal microenvironment in mammary gland branching morphogenesis. Dev Biol. 2010;337:124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haslam SZ, Drolet A, Smith K, Tan M, Aupperlee M. Progestin-regulated luminal cell and myoepithelial cell-specific responses in mammary organoid culture. Endocrinology. 2008;149:2098–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos SJ, Aupperlee MD, Xie J, et al. Progesterone receptor A-regulated gene expression in mammary organoid cultures. J Steroid Biochem Mol Biol. 2009;115:161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aupperlee MD, Drolet AA, Durairaj S, Wang W, Schwartz RC, Haslam SZ. Strain-specific differences in the mechanisms of progesterone regulation of murine mammary gland development. Endocrinology. 2009;150:1485–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA. 2003;100:9744–9749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srivastava S, Matsuda M, Hou Z, et al. Receptor activator of NF-κB ligand induction via Jak2 and Stat5a in mammary epithelial cells. J Biol Chem. 2003;278:46171–46178 [DOI] [PubMed] [Google Scholar]
- 8.Howlin J, McBryan J, Martin F. Pubertal mammary gland development: insights from mouse models. J Mammary Gland Biol Neoplasia. 2006;11:283–297 [DOI] [PubMed] [Google Scholar]
- 9.Shi HY, Lydon JP, Zhang M. Hormonal defect in maspin heterozygous mice reveals a role of progesterone in pubertal ductal development. Mol Endocrinol. 2004;18:2196–2207 [DOI] [PubMed] [Google Scholar]
- 10.Aupperlee MD, Leipprandt JR, Bennett JM, Schwartz RC, Haslam SZ. Amphiregulin mediates progesterone-induced mammary ductal development during puberty. Breast Cancer Res. 2013;15:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniel CW, Silberstein GB, Strickland P. Direct action of 17 β-estradiol on mouse mammary ducts analyzed by sustained release implants and steroid autoradiography. Cancer Res. 1987;47:6052–6057 [PubMed] [Google Scholar]
- 12.Haslam SZ, Counterman LJ, Nummy KA. Effects of epidermal growth factor, estrogen, and progestin on DNA synthesis in mammary cells in vivo are determined by the developmental state of the gland. J Cell Physiol. 1993;155:72–78 [DOI] [PubMed] [Google Scholar]
- 13.Banerjee MR, Wood BG, Lin FK, Crump LR. Organ culture of whole mammary gland of the mouse. Tissue Culture. 1976;2:457–462 [Google Scholar]
- 14.Aupperlee MD, Smith KT, Kariagina A, Haslam SZ. Progesterone receptor isoforms A and B: temporal and spatial differences in expression during murine mammary gland development. Endocrinology. 2005;146:3577–3588 [DOI] [PubMed] [Google Scholar]
- 15.Duffy JP, Smith PJ, Croker J, Matthews HR. Combined staining method for the demonstration of tissue eosinophils and mast cells. J Histotechnol. 1993;16:143–144 [Google Scholar]
- 16.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346 [DOI] [PubMed] [Google Scholar]
- 17.Rauh MJ, Ho V, Pereira C, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–374 [DOI] [PubMed] [Google Scholar]
- 18.Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997;11:457–465 [PubMed] [Google Scholar]
- 19.Matsumoto M, Nishinakagawa H, Kurohmaru M, Hayashi Y, Otsuka J. Pregnancy and lactation affect the microvasculature of the mammary gland in mice. J Vet Med Sci. 1992;54:937–943 [DOI] [PubMed] [Google Scholar]
- 20.Krege JH, Hodgin JB, Couse JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci USA. 1998;95:15677–15682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luetteke NC, Qiu TH, Fenton SE, et al. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development. 1999;126:2739–2750 [DOI] [PubMed] [Google Scholar]
- 22.Oh CK, Metcalfe DD. Transcriptional regulation of the TCA3 gene in mast cells after Fc epsilon RI cross-linking. J Immunol. 1994;153:325–332 [PubMed] [Google Scholar]
- 23.Grove M, Plumb M. C/EBP, NF-κ B, and c-Ets family members and transcriptional regulation of the cell-specific and inducible macrophage inflammatory protein 1 α immediate-early gene. Mol Cell Biol. 1993;13:5276–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erkel G, Wisser G, Anke T. Influence of the fungal NF-κB inhibitor panepoxydone on inflammatory gene expression in MonoMac6 cells. Int Immunopharmacol. 2007;7:612–624 [DOI] [PubMed] [Google Scholar]
- 25.Kwon JH, Keates S, Bassani L, Mayer LF, Keates AC. Colonic epithelial cells are a major site of macrophage inflammatory protein 3α (MIP-3α) production in normal colon and inflammatory bowel disease. Gut. 2002;51:818–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandrasekar B, Mummidi S, Perla RP, et al. Fractalkine (CX3CL1) stimulated by nuclear factor κB (NF-κB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J. 2003;373:547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith JB, Wadleigh DJ, Xia YR, Mar RA, Herschman HR, Lusis AJ. Cloning and genomic localization of the murine LPS-induced CXC chemokine (LIX) gene, Scyb5. Immunogenetics. 2002;54:599–603 [DOI] [PubMed] [Google Scholar]
- 28.Edbrooke MR, Foldi J, Cheshire JK, Li F, Faulkes DJ, Woo P. Constitutive and NF-κ B-like proteins in the regulation of the serum amyloid A gene by interleukin 1. Cytokine. 1991;3:380–388 [DOI] [PubMed] [Google Scholar]
- 29.Baer M, Dillner A, Schwartz RC, Sedon C, Nedospasov S, Johnson PF. Tumor necrosis factor α transcription in macrophages is attenuated by an autocrine factor that preferentially induces NF-κB p50. Mol Cell Biol. 1998;18:5678–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Chen J, Huang A, et al. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc Natl Acad Sci USA. 2000;97:773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones PL, Ping D, Boss JM. Tumor necrosis factor α and interleukin-1β regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-β and NF-κB. Mol Cell Biol. 1997;17:6970–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darnay BG, Haridas V, Ni J, Moore PA, Aggarwal BB. Characterization of the intracellular domain of receptor activator of NF-κB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-κb and c-Jun N-terminal kinase. J Biol Chem. 1998;273:20551–20555 [DOI] [PubMed] [Google Scholar]
- 33.You Z, Dong Y, Kong X, Beckett LA, Gandour-Edwards R, Melamed J. Midkine is a NF-κB-inducible gene that supports prostate cancer cell survival. BMC Med Genomics. 2008;1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang YL, Chuang LY, Guh JY, et al. Thrombospondin-1 mediates distal tubule hypertrophy induced by glycated albumin. Biochem J. 2004;379:89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adolph KW, Liska DJ, Bornstein P. Analysis of the promoter and transcription start sites of the human thrombospondin 2 gene (THBS2). Gene. 1997;193:5–11 [DOI] [PubMed] [Google Scholar]
- 36.Wilczynska KM, Gopalan SM, Bugno M, et al. A novel mechanism of tissue inhibitor of metalloproteinases-1 activation by interleukin-1 in primary human astrocytes. J Biol Chem. 2006;281:34955–34964 [DOI] [PubMed] [Google Scholar]
- 37.Zhou A, Scoggin S, Gaynor RB, Williams NS. Identification of NF-κ B-regulated genes induced by TNFα utilizing expression profiling and RNA interference. Oncogene. 2003;22:2054–2064 [DOI] [PubMed] [Google Scholar]
- 38.Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179 [DOI] [PubMed] [Google Scholar]
- 39.Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harb Perspect Biol. 2011;3.pii:a003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciarloni L, Mallepell S, Brisken C. Amphiregulin is an essential mediator of estrogen receptor α function in mammary gland development. Proc Natl Acad Sci USA. 2007;104:5455–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das SK, Chakraborty I, Paria BC, Wang XN, Plowman G, Dey SK. Amphiregulin is an implantation-specific and progesterone-regulated gene in the mouse uterus. Mol Endocrinol. 1995;9:691–705 [DOI] [PubMed] [Google Scholar]
- 42.Yamane S, Ishida S, Hanamoto Y, et al. Proinflammatory role of amphiregulin, an epidermal growth factor family member whose expression is augmented in rheumatoid arthritis patients. J Inflamm (Lond). 2008;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Razani B, Reichardt AD, Cheng G. Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol Rev. 2011;244:44–54 [DOI] [PubMed] [Google Scholar]
- 44.Gallego MI, Binart N, Robinson GW, et al. Prolactin, growth hormone, and epidermal growth factor activate Stat5 in different compartments of mammary tissue and exert different and overlapping developmental effects. Dev Biol. 2001;229:163–175 [DOI] [PubMed] [Google Scholar]
- 45.Grandis JR, Drenning SD, Chakraborty A, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J Clin Invest. 1998;102:1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bromberg J. Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res. 2000;2:86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850 [DOI] [PubMed] [Google Scholar]
- 48.Murakami M, Hirano T. A four-step model for the IL-6 amplifier, a regulator of chronic inflammations in tissue-specific MHC class II-associated autoimmune diseases. Front Immunol. 2011;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fung MM, Chu YL, Fink JL, Wallace A, McGuire KL. IL-2- and STAT5-regulated cytokine gene expression in cells expressing the Tax protein of HTLV-1. Oncogene. 2005;24:4624–4633 [DOI] [PubMed] [Google Scholar]
- 50.Fata JE, Kong YY, Li J, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50 [DOI] [PubMed] [Google Scholar]
- 51.Mukherjee A, Soyal SM, Li J, et al. Targeting RANKL to a specific subset of murine mammary epithelial cells induces ordered branching morphogenesis and alveologenesis in the absence of progesterone receptor expression. FASEB J. 2010;24:4408–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez-Valdivia R, Mukherjee A, Ying Y, et al. The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev Biol. 2009;328:127–139 [DOI] [PubMed] [Google Scholar]
- 53.Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Klominek J. Chemotaxis and chemokinesis of malignant mesothelioma cells to multiple growth factors. Anticancer Res. 2004;24:1625–1630 [PubMed] [Google Scholar]
- 55.Zhu J, Siclari VA, Liu F, et al. Amphiregulin-EGFR signaling mediates the migration of bone marrow mesenchymal progenitors toward PTH-stimulated osteoblasts and osteocytes. PLoS One. 2012;7:e50099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fanti P, Nazareth M, Bucelli R, et al. Estrogen decreases chemokine levels in murine mammary tissue: implications for the regulatory role of MIP-1 α and MCP-1/JE in mammary tumor formation. Endocrine. 2003;22:161–168 [DOI] [PubMed] [Google Scholar]
- 57.Reimer MK, Brange C, Rosendahl A. CCR8 signaling influences Toll-like receptor 4 responses in human macrophages in inflammatory diseases. Clin Vaccine Immunol. 2011;18:2050–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DiPietro LA, Burdick M, Low QE, Kunkel SL, Strieter RM. MIP-1α as a critical macrophage chemoattractant in murine wound repair. J Clin Invest. 1998;101:1693–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Proost P, Wuyts A, Van Damme J. Human monocyte chemotactic proteins-2 and -3: structural and functional comparison with MCP-1. J Leukoc Biol. 1996;59:67–74 [DOI] [PubMed] [Google Scholar]
- 60.Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644 [DOI] [PubMed] [Google Scholar]
- 61.Becker Y. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy–a review. Virus Genes. 2006;33:235–252 [DOI] [PubMed] [Google Scholar]
- 62.Mosheimer BA, Kaneider NC, Feistritzer C, Sturn DH, Wiedermann CJ. Expression and function of RANK in human monocyte chemotaxis. Arthritis Rheum. 2004;50:2309–2316 [DOI] [PubMed] [Google Scholar]
- 63.Badolato R, Wang JM, Murphy WJ, et al. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ming WJ, Bersani L, Mantovani A. Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J Immunol. 1987;138:1469–1474 [PubMed] [Google Scholar]
- 65.Post TW, Bozic CR, Rothenberg ME, Luster AD, Gerard N, Gerard C. Molecular characterization of two murine eosinophil β chemokine receptors. J Immunol. 1995;155:5299–5305 [PubMed] [Google Scholar]
- 66.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612 [DOI] [PubMed] [Google Scholar]
- 67.Medina D, Ullrich R, Meyn R, Wiseman R, Donehower L. Environmental carcinogens and p53 tumor-suppressor gene interactions in a transgenic mouse model for mammary carcinogenesis. Environ Mol Mutagen. 2002;39:178–183 [DOI] [PubMed] [Google Scholar]
- 68.Jerry DJ, Kittrell FS, Kuperwasser C, et al. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–1058 [DOI] [PubMed] [Google Scholar]
- 69.Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim Biophys Acta. 2009;1796:11–18 [DOI] [PubMed] [Google Scholar]
- 70.Asselin E, Johnson GA, Spencer TE, Bazer FW. Monocyte chemotactic protein-1 and -2 messenger ribonucleic acids in the ovine uterus: regulation by pregnancy, progesterone, and interferon-tau. Biol Reprod. 2001;64:992–1000 [DOI] [PubMed] [Google Scholar]
- 71.Hannan NJ, Jones RL, Critchley HO, et al. Coexpression of fractalkine and its receptor in normal human endometrium and in endometrium from users of progestin-only contraception supports a role for fractalkine in leukocyte recruitment and endometrial remodeling. J Clin Endocrinol Metab. 2004;89:6119–6129 [DOI] [PubMed] [Google Scholar]
- 72.Kavandi L, Collier MA, Nguyen H, Syed V. Progesterone and calcitriol attenuate inflammatory cytokines CXCL1 and CXCL2 in ovarian and endometrial cancer cells. J Cell Biochem. 2012;113:3143–3152 [DOI] [PubMed] [Google Scholar]
- 73.Keane MP, Belperio JA, Moore TA, et al. Neutralization of the CXC chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J Immunol. 1999;162:5511–5518 [PubMed] [Google Scholar]
- 74.Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2004;172:2853–2860 [DOI] [PubMed] [Google Scholar]
- 75.Yamahata H, Takeshima H, Kuratsu J, et al. The role of thrombin in the neo-vascularization of malignant gliomas: an intrinsic modulator for the up-regulation of vascular endothelial growth factor. Int J Oncol. 2002;20:921–928 [PubMed] [Google Scholar]
- 76.Li Q, Kannan A, DeMayo FJ, et al. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science. 2011;331:912–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamagishi H, Olson EN, Srivastava D. The basic helix-loop-helix transcription factor, dHAND, is required for vascular development. J Clin Invest. 2000;105:261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hayashi H, Sano H, Seo S, Kume T. The Foxc2 transcription factor regulates angiogenesis via induction of integrin β3 expression. J Biol Chem. 2008;283:23791–23800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moses MA, Sudhalter J, Langer R. Identification of an inhibitor of neovascularization from cartilage. Science. 1990;248:1408–1410 [DOI] [PubMed] [Google Scholar]
- 80.Zhang L, Rees MC, Bicknell R. The isolation and long-term culture of normal human endometrial epithelium and stroma. Expression of mRNAs for angiogenic polypeptides basally and on oestrogen and progesterone challenges. J Cell Sci. 1995;108(pt 1):323–331 [DOI] [PubMed] [Google Scholar]
- 81.Choudhuri R, Zhang HT, Donnini S, Ziche M, Bicknell R. An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res. 1997;57:1814–1819 [PubMed] [Google Scholar]
- 82.Sukocheva OA, Wang L, Albanese N, Pitson SM, Vadas MA, Xia P. Sphingosine kinase transmits estrogen signaling in human breast cancer cells. Mol Endocrinol. 2003;17:2002–2012 [DOI] [PubMed] [Google Scholar]
- 83.Jeng YJ, Suarez VR, Izban MG, Wang HQ, Soloff MS. Progesterone-induced sphingosine kinase-1 expression in the rat uterus during pregnancy and signaling consequences. Am J Physiol Endocrinol Metab. 2007;292:E1110–E1121 [DOI] [PubMed] [Google Scholar]
- 84.Nagahashi M, Ramachandran S, Kim EY, et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012;72:726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kzhyshkowska J, Workman G, Cardó-Vila M, et al. Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. J Immunol. 2006;176:5825–5832 [DOI] [PubMed] [Google Scholar]
- 86.Chen L, Mupo A, Huynh T, et al. Tbx1 regulates Vegfr3 and is required for lymphatic vessel development. J Cell Biol. 2010;189:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hyder SM, Liang Y, Wu J. Estrogen regulation of thrombospondin-1 in human breast cancer cells. Int J Cancer. 2009;125:1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iruela-Arispe ML, Porter P, Bornstein P, Sage EH. Thrombospondin-1, an inhibitor of angiogenesis, is regulated by progesterone in the human endometrium. J Clin Invest. 1996;97:403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2:a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li F, Curry TE., Jr Regulation and function of tissue inhibitor of metalloproteinase (TIMP) 1 and TIMP3 in periovulatory rat granulosa cells. Endocrinology. 2009;150:3903–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu H, Chen B, Lilly B. Fibroblasts potentiate blood vessel formation partially through secreted factor TIMP-1. Angiogenesis. 2008;11:223–234 [DOI] [PubMed] [Google Scholar]
- 92.De M, Sanford TR, Wood GW. Interleukin-1, interleukin-6, and tumor necrosis factor α are produced in the mouse uterus during the estrous cycle and are induced by estrogen and progesterone. Dev Biol. 1992;151:297–305 [DOI] [PubMed] [Google Scholar]
- 93.Fràter-Schröder M, Risau W, Hallmann R, Gautschi P, Böhlen P. Tumor necrosis factor type α, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc Natl Acad Sci USA. 1987;84:5277–5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sarma V, Wolf FW, Marks RM, Shows TB, Dixit VM. Cloning of a novel tumor necrosis factor-α-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro. J Immunol. 1992;148:3302–3312 [PubMed] [Google Scholar]