Chlorpheniramine Analogues Reverse Chloroquine Resistance in Plasmodium falciparum by Inhibiting PfCRT (original) (raw)
Abstract
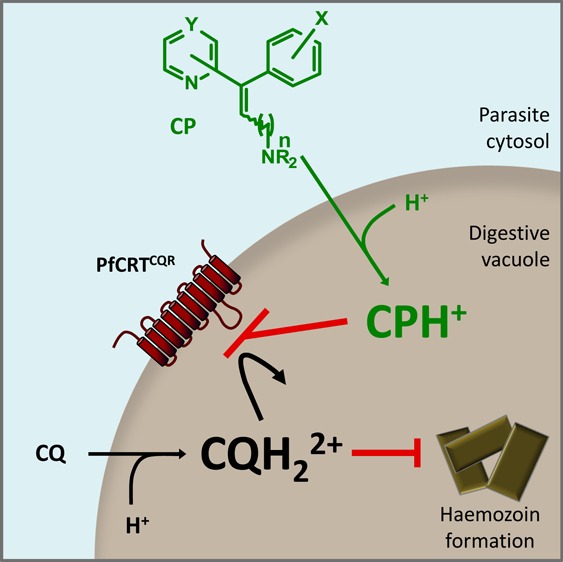
The emergence and spread of malaria parasites that are resistant to chloroquine (CQ) has been a disaster for world health. The antihistamine chlorpheniramine (CP) partially resensitizes CQ-resistant (CQR) parasites to CQ but possesses little intrinsic antiplasmodial activity. Mutations in the parasite’s CQ resistance transporter (PfCRT) confer resistance to CQ by enabling the protein to transport the drug away from its site of action, and it is thought that resistance-reversers such as CP exert their effect by blocking this CQ transport activity. Here, a series of new structural analogues and homologues of CP have been synthesized. We show that these compounds (along with other in vitro CQ resistance-reversers) inhibit the transport of CQ via a resistance-conferring form of PfCRT expressed in Xenopus laevis oocytes. Furthermore, the level of PfCRT-inhibition was found to correlate well with both the restoration of CQ accumulation and the level of CQ resensitization in CQR parasites.
Keywords: Malaria parasite, chloroquine resistance, chemosensitization, chlorpheniramine, PfCRT, Xenopus oocytes
The malaria parasite Plasmodium falciparum has proven largely refractory to the vaccine approaches trialled to date and reliance on chemotherapy is under serious threat with the recent emergence of parasites that are resistant to the current mainstay of malaria treatment (the artemisinin-based combination therapies).1 As a result, malaria remains a global health problem; there are around 225 million clinical cases and over 1.2 million deaths each year,2 and the disease also imposes considerable socio-economic burdens upon afflicted countries. Chloroquine (CQ, Figure 1A) was a cheap, safe, and efficacious treatment for the disease until the eventual emergence and spread of resistant parasites. Resistance to CQ is caused primarily by mutations in the P. falciparum CQ resistance transporter, PfCRT.3 Within the _P. falciparum_-infected erythrocyte, PfCRT is found in the membrane of the parasite’s digestive vacuole4 (DV; pH 5–5.5); the organelle in which CQ accumulates and exerts its antimalarial effect. Mutations in PfCRT that confer CQ resistance result in a marked reduction in the accumulation of CQ within the DV. Using the Xenopus laevis oocyte expression system, a CQ resistance-conferring form of PfCRT (PfCRTCQR) was shown to transport CQ, whereas the wild-type form found in CQ-sensitive (CQS) parasites (PfCRTCQS) lacked this ability.5
Figure 1.

Chemical structures and synthesis. (A) The structures of chloroquine (CQ), verapamil (VP), chlorpheniramine (CP), promethazine (PZ), and primaquine (PQ). (B) Retrosynthesis of CP analogues. (C) Synthesis of ketones 1–6. Reagents and conditions: (a) _n_-BuLi, THF, ether, −85 °C warming to −10 °C, 15 min, then −110 °C warming to r.t., 24 h (50–77%); (b) _n_-BuLi, ether, −78 °C, 1 h warming to r.t., 1.5 h (91%); (c) MnO2, 11 days (78%); (d) ether, toluene, 80 °C, 1.75 h then 2 M H2SO4, 0.5 h (64–66%). (D) Synthesis of CP analogues 12–21c. Reagents and conditions: (e) R2NH(aq), MeOH, 80 °C, 18 h (96–99%); (f) LiN(SiMe3)2, THF, 0 °C, 15 min, then 1–6, r.t., 24 h (33–83%); (g) Pd/C, H2, EtOH, 6.5–13 h (46–50%); (h) PtO2, H2, EtOH, 3.5–6.5 h (77–99%).
Mutations in PfCRT have been associated with decreases or increases in the parasite’s susceptibility to other antimalarials,6 and the transporter has been shown to be one of the key determinants of the parasite’s response to a diverse set of novel antiplasmodial compounds.7 Whether these effects are due to PfCRT-mediated drug efflux and/or the inhibition of the transporter’s normal function (which is essential for the survival of the parasite, but currently unknown) remains unclear.8 A better understanding of the interactions between PfCRTCQR and its substrates and inhibitors could inform the development of new antimalarial chemotherapies and strategies.
CQ-resistant (CQR) parasites can be partially resensitized to CQ in vitro by a range of weak bases including the calcium channel blocker verapamil (VP), the antihistamines promethazine and chlorpheniramine (PZ and CP, respectively), and the liver-stage antimalarial primaquine (PQ) (Figure 1A).9 The phenomenon of CQ resistance-reversal is characterized by both an increase in CQ accumulation and a decrease in the CQ IC50 in CQR parasites (the CQ IC50 is the concentration of CQ at which the inhibition of parasite growth is half-maximal).10 Reversers of CQ resistance generally have no effect on either CQ accumulation or the CQ IC50 in CQS parasites10 and possess little or no intrinsic antimalarial activity against the intraerythrocytic stage of the parasite. It is thought that resistance-reversers accumulate in the acidic environment of the DV via weak-base trapping and that they may exert their CQ resistance-reversing activity by binding to PfCRTCQR and thereby inhibiting the efflux of CQ from the DV.9 Consistent with this mechanism of action, VP and a series of dibemethin derivatives have recently been shown to interact directly with PfCRTCQR to inhibit CQ transport in the oocyte system.5,11
Resistance-reversers generally feature a secondary or tertiary amine tethered to two aromatic hydrophobic residues by a flexible aliphatic side chain.9 The failure of early in vivo animal trials with VP12 and the apparent lack of activity of the in vitro resistance-reversers cyproheptadine13 and desipramine14 in human trials have been attributed to a combination of (1) the low availability of the resistance-reversers due to binding to serum protein, (2) the inability to achieve the concentrations required for efficacy without risking toxic side-effects, and (3) low potency of the resistance-reversing effect.15 Some of these properties may be inherent to weakly basic hydrophobic compounds. However, the successful treatment of CQR parasite infections with the combination of CQ and CP in Nigeria16−18 suggests that these obstacles may be overcome. Given the promising clinical use of CP as a CQ resistance-reverser, we used the molecular framework of this compound as a basis for a series of derivatives, with the aim of developing structure–activity relationships, investigating their mode of action, and potentially increasing the resistance-reversing activity of CP. The structural changes included increases to the side-chain length, alterations to the heteroaromatic ring, and changes to the position of attachment of the chlorine atom on the phenyl ring.
The desired CP analogues were prepared using a Wittig olefination reaction to install the amine side-chain from pyridyl- or pyrazinyl- chlorophenyl ketones (Figure 1B). This enabled the synthesis of a range of analogues with the desired changes. The 2-pyridyl ketones 1–3 were prepared using the method described by Popova et al.19 from 2-bromopyridine and methyl 2-, 3-, or 4-chlorobenzoate (Figure 1C). The 3-pyridyl ketone 4 was prepared in a two-step procedure from the reaction of lithiated 3-bromopyridine with 3-chlorobenzaldehyde, followed by manganese dioxide oxidation,20 which was slow but high-yielding and clean. Because of the problems with the stability of 4-bromopyridine, 5 was prepared using an alternate strategy from the Grignard reaction of 4-cyanopyridine with 3-chlorophenylmagnesium bromide and acidic aqueous workup.21 Pyrazine ketone 6 was prepared in a similar fashion from 2-cyanopyrazine.
The Wittig salts 7–8 were prepared using the method of Rao and Reddy,22 via (2-methoxyethyl)triphenylphosphonium bromide (Figure 1B). Compounds 9–11 were prepared from the corresponding (ω-bromoalkyl)triphenyl phosphonium bromides using the procedure of Sano et al.23
Wittig olefination utilizing LiHMDS proceeded smoothly to produce olefins 12–21a,b, although yields decreased with increasing chain length (Figure 1D). Hydrogenation was performed in excellent conversion, utilizing palladium on carbon or platinum dioxide catalysts, as illustrated by the disappearance of the distinctive olefinic triplet between 6.23 and 6.87 ppm and the appearance of a benzylic triplet between 4.07 and 4.72 ppm. The use of Pd/C promoted cleavage of the chloro group, whereas platinum dioxide could be used to catalyze hydrogenation of alkenes, suppressing cleavage of the chloro groups. Compounds 12a–21a, 12b–21b, 12c–21c, and 22 were prepared in this manner, and the compounds tested are presented in Table 1. Compounds 23a and 23b were prepared from the Horner–Wadsworth–Emmons (HWE) olefination between diethylcyanomethyl phosphonate and 2-benzoyl-pyridine.24 Diastereomers were separated by either flash column chromatography or HPLC, and all CP analogues were purified by HPLC prior to testing (see Supporting Information for details).
Table 1. Analogues Prepared and Tested in This Study.
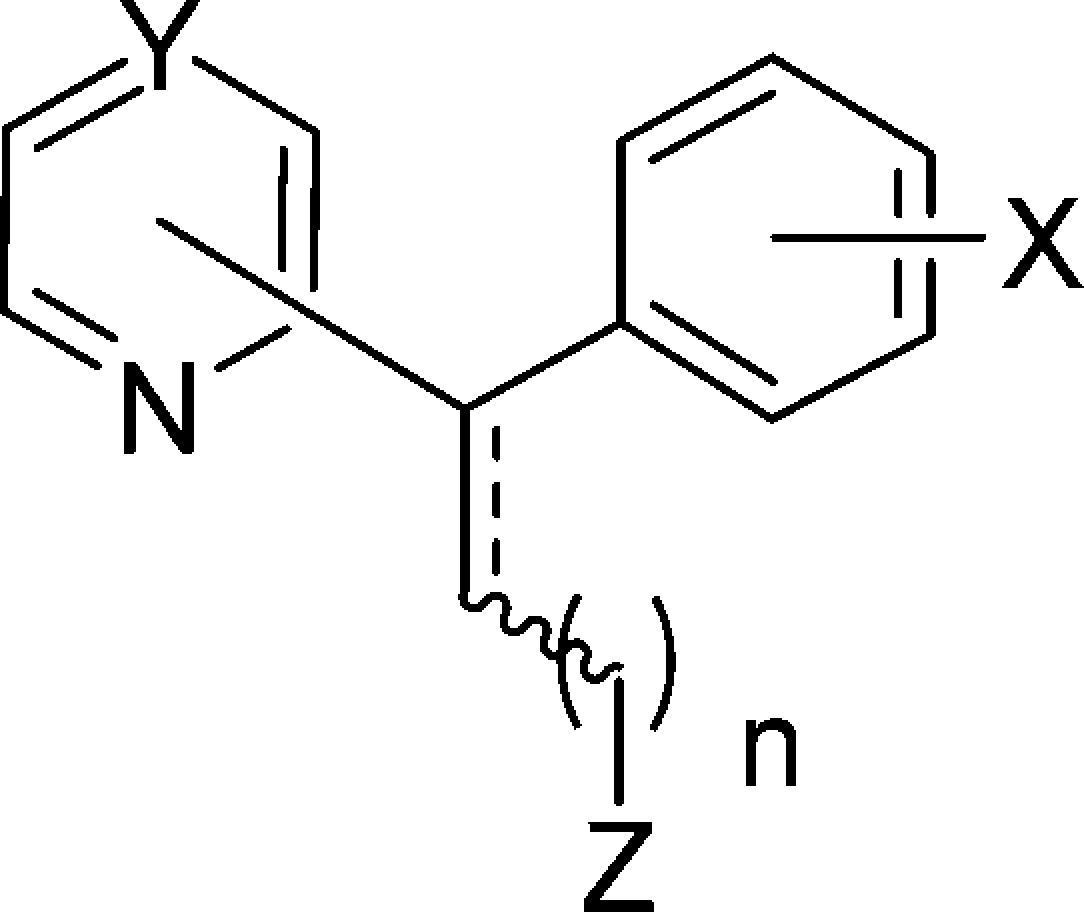
| | config | subst | Y | X | n | Z | | | --------- | ----- | --- | --- | ---- | --- | ---- | | 12a | E | 2 | CH | 4-Cl | 1 | NEt2 | | 12b | Z | 2 | CH | 4-Cl | 1 | NEt2 | | 12c | rac | 2 | CH | 4-Cl | 1 | NEt2 | | 13a | E | 2 | CH | 3-Cl | 1 | NMe2 | | 13b | Z | 2 | CH | 3-Cl | 1 | NMe2 | | 13c | rac | 2 | CH | 3-Cl | 1 | NMe2 | | 14a | E | 2 | CH | 4-Cl | 2 | NMe2 | | 14b | Z | 2 | CH | 4-Cl | 2 | NMe2 | | 14c | rac | 2 | CH | 4-Cl | 2 | NMe2 | | 15a | E | 2 | CH | 3-Cl | 2 | NMe2 | | 15b | Z | 2 | CH | 3-Cl | 2 | NMe2 | | 15c | rac | 2 | CH | 3-Cl | 2 | NMe2 | | 16a | E | 2 | CH | 2-Cl | 2 | NMe2 | | 16c | rac | 2 | CH | 2-Cl | 2 | NMe2 | | 17a | E | 2 | CH | 4-Cl | 3 | NMe2 | | 17b | Z | 2 | CH | 4-Cl | 3 | NMe2 | | 17c | rac | 2 | CH | 4-Cl | 3 | NMe2 | | 18c | rac | 2 | CH | 4-Cl | 4 | NMe2 | | 19c | rac | 3 | CH | 3-Cl | 2 | NMe2 | | 20c | rac | 4 | C | 3-Cl | 2 | NMe2 | | 21c | rac | 2 | N | 3-Cl | 2 | NMe2 | | 22 | rac | 2 | CH | H | 2 | NMe2 | | 23a | E | 2 | CH | H | 0 | CN | | 23b | Z | 2 | CH | H | 0 | CN | | CP | rac | 2 | CH | 4-Cl | 1 | NMe2 | | CP | (+) | 2 | CH | 4-Cl | 1 | NMe2 |
The ability of the CP analogues to inhibit the PfCRTCQR-mediated transport of CQ was assessed using the Xenopus oocyte system. When present at an extracellular concentration of 100 μM, CP, PQ, PZ, and VP all inhibited the transport of [3H]CQ via PfCRTCQR (Figure 2A). Each of the 24 CP analogues also inhibited PfCRTCQR-mediated [3H]CQ transport when tested at 100 μM, albeit to different degrees (Figure 2B; P < 0.01) and with less potency than the benchmark resistance-reverser VP (_P_ < 0.05). None of the compounds affected the diffusion of CQ into oocytes expressing PfCRTCQS (_P_ > 0.05). The structural changes investigated here mostly resulted in reduced PfCRTCQR inhibition relative to CP, with the exception of 13c (the 3-chlorophenyl derivative of CP) and 19c (containing 3-chlorophenyl and 3-pyridyl groups). Both of these compounds appeared to be slightly more potent than CP, but the differences were not statistically significant (P > 0.05). The general trends derived from this data were (1) the reduced compounds (present as racemates) were more active than their olefinic counterparts (e.g., 15a–c), (2) lengthening the side-chain was not beneficial (e.g., 14c, 17c, and 18c), and (3) having a 3-chlorophenyl group was preferable to 2-chloro, 4-chloro, or unsubstituted phenyl groups (e.g., 15c, 14c, 16c, and 22).
Figure 2.
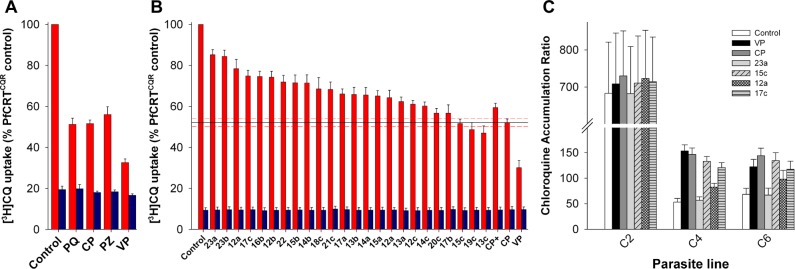
Inhibition of PfCRTCQR-mediated CQ transport by the resistance-reversers PZ, CP, PQ, VP, and by analogues of CP. (A) [3H]CQ uptake into oocytes expressing PfCRTCQR (red bars) or PfCRTCQS (blue bars) in the presence of 100 μM PQ, CP, PZ, or VP. (B) [3H]CQ uptake into oocytes expressing PfCRTCQR (red bars) or PfCRTCQS (blue bars) in the presence of CP analogues (100 μM). The level of inhibition by CP is shown as a solid line with SEM (n = 5) represented by red dashed lines. (C) Effects of VP, CP, and the CP analogues 23a, 15c, 12a, and 17c on [3H]CQ accumulation by erythrocytes infected with C2GCO3, C4Dd2, or C67G8 parasites. [See Supporting Information for further details.]
An analysis of the concentration-dependent inhibition of PfCRTCQR by a selection of representative derivatives (12a, 13c, 15c, and 17c; Figure S1 d) and resistance reversers (CP, PZ, and PQ; Figure S1 a–c) yielded the IC50 values reported in Table 2. The IC50 value of 12a was significantly greater than those of the other compounds tested (P < 0.05). Compound 17c was less potent than PZ or PQ, but more active than 12a. The activities of 13c and 15c were comparable to that of CP but were significantly lower than that of VP (30 ± 3 μM;5P < 0.01).
Table 2. Inhibition of PfCRTCQR-Mediated CQ Transport in Oocytes, Intrinsic Antiplasmodial Activity, and in Vitro Resistance-Reversal Activity of Four CP Analogues.
| | | parasite growth relative to control (%)b | CQ IC50 (nM) (RMI)c | | | | | | | ------- | ---------------------------------------------------------- | ----------------------------- | ----------------- | ----------------- | --------------------- | --------------------- | --------------------- | | compd | IC50 values (μM) for the inhibition of PfCRTCQRa | C2GC03 | C4Dd2 | C67G8 | C2GC03 | C4Dd2 | C67G8 | | CQ | | | | | 14.5 ± 1.7 (1.00) | 154 ± 8 (1.00) | 110 ± 15 (1.00) | | VP | 33 ± 3 | 43 ± 6 | 46 ± 3 | 55 ± 5d | 13.1 ± 1.6 (0.91) | 36.2 ± 2.8 (0.23) | 71 ± 8 (0.65) | | CP | 54 ± 5 | 97 ± 2 | 78 ± 5 | 76 ± 1d | 15.6 ± 1.7 (1.09) | 36.6 ± 0.7 (0.24) | 39.8 ± 3.1 (0.37) | | 15c | 66 ± 8 | 99 ± 2 | 67 ± 3d | 72 ± 4d | 13.8 ± 1.8 (0.95) | 41 ± 2 (0.27) | 36.8 ± 1.9 (0.35) | | 12a | 321 ± 24 | 100 ± 4 | 84 ± 5d | 90 ± 1 | 14.8 ± 2.4 (1.01) | 75 ± 2 (0.49) | 75 ± 5 (0.69) | | 17c | 180 ± 29 | 100 ± 4 | 74 ± 5d | 79 ± 4d | 13.4 ± 1.9 (0.92) | 44 ± 2 (0.28) | 42 ± 3 (0.39) | | 13c | 63 ± 7 | 99 ± 4 | 75 ± 2d | 74 ± 3d | 14.3 ± 1.6 (0.99) | 42 ± 4 (0.27) | 38.3 ± 4.1 (0.35) | | PZ | 85 ± 7 | | | | | | | | PQ | 68 ± 8 | | | | | | |
CP derivatives 12a, 15c, 17c, and 23a were tested for the ability to behave as CQ resistance-reversers in CQR parasites. In the first set of experiments, CQ accumulation was measured in the C2GCO3, C4Dd2, and C67G8 parasite lines in the presence or absence of VP, CP, and the CP analogues. These isogenic parasite lines express either the wild-type pfcrt allele (C2GCO3) or the CQ-resistance-conferring pfcrt alleles from the CQR strains Dd2 or 7G8 (C4Dd2 and C67G8, respectively). Under control conditions, the CQ accumulation ratios for the CQR C4Dd2 and C67G8 lines were 13.3 ± 2.8 and 11.6 ± 1.6 times lower, respectively, than that measured in the CQS C2GCO3 line (Figure 2C). VP (1 μM) had no effect on CQ accumulation in the C2GCO3 parasites, but caused a 3.1 ± 0.5 fold increase in the CQ accumulation ratio in the C4Dd2 line and a 1.9 ± 0.2 fold increase in the C67G8 line (P < 0.05; Figure 2C). These findings are consistent with previous reports.25
Both CP and 15c caused marked increases in the accumulation of CQ in the CQR lines; CQ levels were increased 2.7–2.9-fold in the C4Dd2 parasites and 2.0–2.2-fold in the C67G8 parasites (P > 0.05). Compounds 17c and 12a had less dramatic effects on CQ accumulation, causing increases of 2.4- and 1.6-fold, respectively, in the C4Dd2 line, and fold-increases of 1.8 and 1.4 in the C67G8 line.
Compound 23a had no effect on CQ accumulation in either the CQS or CQR lines (P > 0.05), which is consistent with our finding that it is a poor inhibitor of CQ transport via PfCRTCQR (Figure 2B). None of the compounds tested altered CQ accumulation in the C2GC03 parasite line (P > 0.05).
In the second set of experiments, the CP analogues 12a, 13c, 15c, and 17c were tested for the ability to modulate CQ susceptibility in the pfcrt transfectant lines using parasite proliferation assays. In the absence of CQ, 800 nM VP was found to have a negative effect on proliferation in all three lines (Table 2; P < 0.05). By contrast, at the same concentration, CP and its derivatives had no effect on the proliferation of CQS C2GC03 parasites, yet all displayed significant inhibitory activity against the CQR C4Dd2 and C67G8 lines (Table 2; P < 0.05). This finding is consistent with previous observations of resistance-reversers displaying modest but significant levels of intrinsic antiplasmodial activity against CQR strains26 (while exerting little or no effect in CQS strains) and provides further support for the idea that PfCRTCQR could be viewed as a drug target.8
None of the test compounds affected the IC50 of CQ against CQS C2GC03 parasites (Table 2; P > 0.2). However, VP, CP, and the four CP analogues each caused a significant decrease in the IC50 of CQ against the CQR lines (Table 2; P < 0.01). In all cases, the resistance modification index (RMI) calculated for the C67G8 line was greater than that measured for the C4Dd2 line, indicating a weaker resistance-reversing effect against the C67G8 line; this was particularly noticeable for VP (Table 2).
The haplotype of PfCRTCQR expressed in the oocyte system is the same as that carried by the C4Dd2 parasite line. It was therefore relevant to compare the IC50 values obtained for each compound in the oocyte system with the corresponding (1) fold-increases in CQ accumulation and (2) RMI values for CQ susceptibility determined for the C4Dd2 parasites. As shown in Figure 3A, there was a strong positive correlation between the ability to inhibit PfCRTCQR-mediated CQ transport in oocytes and the restoration of CQ accumulation in erythrocytes infected with C4Dd2 parasites (_R_2 = 0.94, P = 0.0064). Likewise, the IC50 values for PfCRTCQR inhibition were strongly correlated with the CQ RMI values derived for C4Dd2 parasites (Figure 3B; _R_2 = 0.869, P = 0.0067). These analyses indicate that the degree of anti-PfCRTCQR activity exhibited by a CQ resistance-reverser is an important determinant of its in vitro chemosensitization activity. While the number of compounds compared here was small, these findings suggest that the extent to which a compound inhibits PfCRT-mediated CQ transport in the oocyte system is a relatively reliable indicator of its ability to inhibit PfCRTCQR in situ.
Figure 3.

Correlation between the anti-PfCRTCQR activities of resistance-reversers in oocytes and their respective CQ-chemosensitizing activities in C4Dd2 parasites. The IC50 values for the inhibition of PfCRTCQR-mediated CQ transport by CP, VP, 12a, 13c, 15c and 17c were plotted against (A) the fold increase in the CQ accumulation ratio (_R_2 = 0.94, P = 0.0064) or (B) the CQ resistance modification index (RMI) (_R_2 = 0.869, P = 0.0067).
In summary, a library of CP analogues was produced, and all of these molecules were shown to be inhibitors of PfCRTCQR in the oocyte system. The most active of these compounds carried a 3-chlorophenyl group in place of the 4-chlorophenyl group of CP and had in vitro resistance-reversing activity similar to that of CP. Within this series of analogues, the extent of PfCRTCQR inhibition observed in the oocyte system was found to correlate well with the compound’s ability to modulate the CQ-susceptibility of CQR parasites. This finding indicates that the degree of anti-PfCRT activity exhibited by a resistance-reverser is an important determinant of its in vitro chemosensitization activity in CQR parasites. Moreover, the oocyte system was able to detect relatively small differences in the inhibitory activities of the 24 CP analogues, indicating that this assay is well suited to assessing potential blockers of PfCRTCQR. The fact that previous work27 has shown that CP, PZ, and PQ appear to be substrates of PfCRTCQR in situ, together with the finding here that they inhibit the transporter in a concentration-dependent manner, indicates that these compounds exert their CQ resistance-reversing activity by competing with CQ for transport via PfCRTCQR. CQ remains a first-line treatment and prophylactic in 21 countries and a CQ-azithromycin combination is in phase IIb/III development for preventative treatment in pregnant women. It is therefore possible that inhibitors of mutant PfCRT could play a role in CQ-based combination therapies, or in therapies based on related quinolines such as amodiaquine, to boost the activity of the quinoline partner drug against CQR parasites. Alternatively, inhibitors of mutant PfCRT could be combined with the antimalarial pharmacophore of CQ28,29 or with other antimalarial moieties to create dual-function antimalarials that are active against drug-resistant parasites.30
Acknowledgments
We are grateful to the Canberra Branch of the Australian Red Cross Blood Service for the provision of blood.
Glossary
ABBREVIATIONS
CQ
chloroquine
CP
chlorpheniramine
CQR
chloroquine-resistant
CQS
chloroquine-sensitive
PfCRT
Plasmodium falciparum chloroquine resistance transporter
PQ
primaquine
PZ
promethazine
VP
verapamil
Supporting Information Available
Experimental details for the synthesis and purification of the compounds, the full spectral data, the in vitro assay conditions, and Figure S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ K.J.D. and R.L.S. contributed equally to this work. R.A.B. and R.E.M. are cosenior authors. All authors contributed to the writing of the manuscript.
This work was supported by the Australian National Health and Medical Research Council (NHMRC; grant 1007035 to R.E.M.) and the Research School of Chemistry, ANU. R.E.M. was supported by NHMRC fellowships 520320 and 1053082 and the L’Oréal Australia For Women in Science program. A.M.L. was supported by an NHMRC Overseas Biomedical Fellowship [585519]. K.J.D. was supported by a Rod Rickards scholarship.
The authors declare no competing financial interest.
Supplementary Material
References
- Phyo A. P.; Nkhoma S.; Stepniewska K.; Ashley E. A.; Nair S.; McGready R.; ler Moo C.; Al-Saai S.; Dondorp A. M.; Lwin K. M.; Singhasivanon P.; Day N. P.; White N. J.; Anderson T. J.; Nosten F. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 2012, 37998301960–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. J.; Rosenfeld L. C.; Lim S. S.; Andrews K. G.; Foreman K. J.; Haring D.; Fullman N.; Naghavi M.; Lozano R.; Lopez A. D. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 2012, 3799814413–431. [DOI] [PubMed] [Google Scholar]
- Fidock D. A.; Nomura T.; Talley A. K.; Cooper R. A.; Dzekunov S. M.; Ferdig M. T.; Ursos L. M. B.; bir Singh Sidhu A.; Naudé B.; Deitsch K. W.; Su X.-Z.; Wootton J. C.; Roepe P. D.; Wellems T. E. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 2000, 64861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R. A.; Ferdig M. T.; Su X.-Z.; Ursos L. M. B.; Mu J.; Nomura T.; Fujioka H.; Fidock D. A.; Roepe P. D.; Wellems T. E. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol. Pharmacol. 2002, 61135–42. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Marchetti R. V.; Cowan A. I.; Howitt S. M.; Bröer S.; Kirk K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science 2009, 32559481680–1682. [DOI] [PubMed] [Google Scholar]
- Petersen I.; Eastman R.; Lanzer M. Drug-resistant malaria: Molecular mechanisms and implications for public health. FEBS Lett. 2011, 585111551–1562. [DOI] [PubMed] [Google Scholar]
- Yuan J.; Cheng K. C.-C.; Johnson R. L.; Huang R.; Pattaradilokrat S.; Liu A.; Guha R.; Fidock D. A.; Inglese J.; Wellems T. E.; Austin C. P.; Su X.-Z. Chemical genomic profiling for antimalarial therapies, response signatures, and molecular targets. Science 2011, 3336043724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers R.; Nash M.; Martin R. Know your enemy: understanding the role of PfCRT in drug resistance could lead to new antimalarial tactics. Cell. Mol. Life Sci. 2012, 69121967–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schalkwyk D. A.; Egan T. J. Quinoline-resistance reversing agents for the malaria parasite Plasmodium falciparum. Drug Resist. Update 2006, 94211–226. [DOI] [PubMed] [Google Scholar]
- Bray P. G.; Ward S. A. A comparison of the phenomenology and genetics of multidrug resistance in cancer cells and quinoline resistance in Plasmodium falciparum. Pharmacol. Ther. 1998, 7711–28. [DOI] [PubMed] [Google Scholar]
- Zishiri V. K.; Hunter R.; Smith P. J.; Taylor D.; Summers R.; Kirk K.; Martin R. E.; Egan T. J. A series of structurally simple chloroquine chemosensitizing dibemethin derivatives that inhibit chloroquine transport by PfCRT. Eur. J. Med. Chem. 2011, 4651729–1742. [DOI] [PubMed] [Google Scholar]
- Kyle D. E.; Milhous W. K.; Rossan R. N. Reversal of Plasmodium falciparum resistance to chloroquine in Panamanian Aotus monkeys. Am. J. Trop. Med. Hyg. 1993, 481126–133. [DOI] [PubMed] [Google Scholar]
- Björkman A.; Willcox M.; Kihamia C. M.; Mahikwano L. F.; Phillips Howard P. A.; Hakansson A.; Warhurst D. Field study of cyproheptadine/chloroquine synergism in falciparum malaria. Lancet 1990, 336870659–60. [DOI] [PubMed] [Google Scholar]
- Warsame M.; Wernsdorfer W. H.; Björkman A. Lack of effect of desipramine on the response to chloroquine of patients with chloroquine-resistant falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 1992, 863235–236. [DOI] [PubMed] [Google Scholar]
- Ward S. A.; Bray P. G. Is reversal of chloroquine resistance ready for the clinic?. Lancet 2001, 3579260904. [DOI] [PubMed] [Google Scholar]
- Sowunmi A.; Oduola A. M. J.; Ogundahunsi O. A. T.; Falade C. O.; Gbotosho G. O.; Salako L. A. Enhanced efficacy of chloroquine–chlorpheniramine combination in acute uncomplicated falciparum malaria in children. Trans. R. Soc. Trop. Med. Hyg. 1997, 91163–67. [DOI] [PubMed] [Google Scholar]
- Ogungbamigbe T.; Ojurongbe O.; Ogunro P.; Okanlawon B.; Kolawole S. Chloroquine resistant Plasmodium falciparum malaria in Osogbo Nigeria: efficacy of amodiaquine + sulfadoxine-pyrimethamine and chloroquine + chlorpheniramine for treatment. Mem. Inst. Oswaldo Cruz 2008, 103, 79–84. [DOI] [PubMed] [Google Scholar]
- Nakornchai S.; Konthiang P. Potentiation of antimalarial drug action by chlorpheniramine against multidrug-resistant Plasmodium falciparum in vitro. Parasitol. Int. 2006, 553195–199. [DOI] [PubMed] [Google Scholar]
- Popova I. S.; Formanovsky A. A.; Mikhura I. V. Synthesis of 2- and 3-pyridinyl(aryl)methanones. Russ. Chem. Bull. 2002, 513540–543. [Google Scholar]
- Harrowven D. C.; Sutton B. J.; Coulton S. Intramolecular radical additions to pyridines. Org. Biomol. Chem. 2003, 1224047–4057. [DOI] [PubMed] [Google Scholar]
- Hoegberg T.; Ulff B.; Renyi A. L.; Ross S. B. Synthesis of pyridylallylamines related to zimelidine and their inhibition of neuronal monoamine uptake. J. Med. Chem. 1981, 24121499–1507. [DOI] [PubMed] [Google Scholar]
- Rao G. V.; Reddy G. C. Synthesis of 2-(_N_-disubstituted amino)ethyltriphenylphosphonium bromides. Tetrahedron Lett. 2008, 495824–826. [Google Scholar]
- Sano T.; Sugaya T.; Kasai M. Process improvement in the production of a pharmaceutical intermediate using a reaction calorimeter for studies on the reaction kinetics of amination of a bromopropyl compound. Org. Process Res. Dev. 1998, 23169–174. [Google Scholar]
- Lee D.; Yang Y.; Yun J. Copper-catalyzed asymmetric reduction of 3,3-diarylacrylonitriles. Org. Lett. 2007, 9142749–2751. [DOI] [PubMed] [Google Scholar]
- Martin R. E.; Butterworth A. S.; Gardiner D. L.; Kirk K.; McCarthy J. S.; Skinner-Adams T. S. Saquinavir inhibits the malaria parasite’s chloroquine resistance transporter. Antimicrob. Agents Chemother. 2012, 5652283–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. J.; Thacker D.; Tan J. C.; Pleeter P.; Checkley L.; Gonzales J. M.; Deng B.; Roepe P. D.; Cooper R. A.; Ferdig M. T. Chloroquine susceptibility and reversibility in a Plasmodium falciparum genetic cross. Mol. Microbiol. 2010, 783770–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehane A. M.; Kirk K. Efflux of a range of antimalarial drugs and ‘chloroquine resistance reversers’ from the digestive vacuole in malaria parasites with mutant PfCRT. Mol. Microbiol. 2010, 77, 1039–1051. [DOI] [PubMed] [Google Scholar]
- Zishiri V. K.; Joshi M. C.; Hunter R.; Chibale K.; Smith P. J.; Summers R. L.; Martin R. E.; Egan T. J. Quinoline antimalarials containing a dibemethin group are active against chloroquinone-resistant Plasmodium falciparum and inhibit chloroquine transport via the P. falciparum chloroquine-resistance transporter (PfCRT). J. Med. Chem. 2011, 54196956–6968. [DOI] [PubMed] [Google Scholar]
- Burgess S. J.; Selzer A.; Kelly J. X.; Smilkstein M. J.; Riscoe M. K.; Peyton D. H. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 2006, 49185623–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. X.; Smilkstein M. J.; Brun R.; Wittlin S.; Cooper R. A.; Lane K. D.; Janowsky A.; Johnson R. A.; Dodean R. A.; Winter R.; Hinrichs D. J.; Riscoe M. K. Discovery of dual function acridones as a new antimalarial chemotype. Nature 2009, 4597244270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.