Platinum compounds for high-resolution in vivo cancer imaging (original) (raw)
. Author manuscript; available in PMC: 2015 Jun 1.
Published in final edited form as: ChemMedChem. 2014 Feb 6;9(6):1131–1135. doi: 10.1002/cmdc.201300502
Abstract
Platinum(II) compounds, principally cisplatin and carboplatin, are commonly used front-line cancer therapeutics. Despite widespread use and the interest of developing new derivatives, including nanoformulations with improved properties, it has been difficult to visualize Pt compounds in live subjects, in real-time, and with subcellular resolution. Here we present four novel cisplatin- and carboplatin-derived fluorescent imaging compounds for quantitative intravital cancer imaging. We conjugate boron dipyromethene FL (BODIPY-FL) to PtII complexes for robust in vivo fluorescence, and show retained DNA-damaging and cytotoxic properties. We image pharmacokinetics and tumor uptake in a xenograft cancer mouse model. Finally, we present a genetic reporter of single-cell DNA damage for in vivo imaging, and simultaneously monitor Pt drug accumulation and resultant DNA damage in individual tumor cells, at subcellular resolution, and in real-time in a live animal model of cancer.
Keywords: intravital imagingagents, pharmacology, fluorescent probes, DNA damage, pharmacokinetics
Platinum(II) compounds, most prominently cisplatin and carboplatin, have been used for decades as effective antineoplastic agents for a variety of solid malignancies. Nonetheless, side effects combined with limited pharmacokinetic (PK) properties defined by a short initial plasma half-life remain significant drawbacks. PtII therapies chiefly work through binding DNA and forming adducts such as intrastrand crosslinks[1]. Drug resistance often emerges as a result of both an altered pharmacodynamic (PD) response to this DNA damage[2-3], along with modified pathways of intracellular Pt transport[4] and metabolism[5]. New strategies are under development for overcoming toxicity and efficacy issues, including nano-encapsulation for controlled and targeted drug release[6], generation of novel Pt and other transition-metal compounds displaying alternative toxicity[7], and design of combination therapies[8]. PK plays an essential role in the success of all three of these strategies. For example with combination therapy, adjusting dosing schedule within hours has been shown capable of impacting efficacy[9-10]. Nevertheless, it has been challenging to directly image Pt compounds in live subjects, in real-time, and with subcellular resolution, which would be advantageous for understanding and efficiently guiding PK/PD drug properties.
Deep-tissue imaging of living subjects using intravital fluorescence microscopy provides a platform for observing combined PK/PD drug properties at high spatiotemporal resolution within a relevant tumor micro-environmental niche[11]. Even heavily examined chemotherapies such as paclitaxel can act unexpectedly in vivo compared to in vitro, and intravital imaging has elucidated many of these differences[12]. Furthermore, intratumoral heterogeneity affects drug response, particularly through emergent drug resistance and metastasis[13]. To this end, intravital imaging enables measurements of drug distribution at subcellular resolution while simultaneously tracking mitotic arrest[12], apoptosis[14], and DNA damage (reported herein) at the single-cell level. Although previous fluorescent-Pt compounds have been described elsewhere[15-17], they use fluorescein derivatives, which are generally not ideal for intravital imaging due to photobleaching and poor fluorescence in intracellular environments[18]. In contrast, BODIPY-FL (BFL) has been shown to exhibit excellent properties for in vivo imaging[19], including environmentally robust fluorescence; structural stability in vivo; high photostability; high brightness; and favorable cell permeability[20-22]. Therefore, here we developed four new BFL-conjugated Pt compounds and demonstrated their applicability to high-resolution intravital imaging of PK/PD in a xenograft model of cancer.
The PtII compounds 1 and 2 were synthesized as previously reported[23], but using BFL as the imaging reporter. The fluorescent analogue 3 was obtained by reaction of the free amine with BFL succinimidyl ester (Scheme 1). The carboplatin analogue (6) was synthesized by introducing the carbo ligand via reaction with the corresponding silver salt Ag2R(COO)2, successive deprotection, and amine NHS coupling. The monoamine trichloride salt PPh4[PtCl3(NH3)] reacts readily with free amines[24] and allowed the selective introduction of N-BOC-ethylenediamine. After deprotection, the small linker was used to attach the fluorescent tag (complex 10). Complex 11 was prepared by direct reaction with amino-BODIPY. All complexes were characterized by fluorimetry (Figure 1A) and by ESI-MS, 1H and 195Pt NMR (Supplementary Figures S1-S25). Chromatographically pure aliquots (no further Pt species or fluorescent impurities detectable by 195Pt NMR or fluorescence channel of the HPLC) were purified by preparative HPLC-MS and stored at -80°C before using in biological experiments.
Scheme 1.
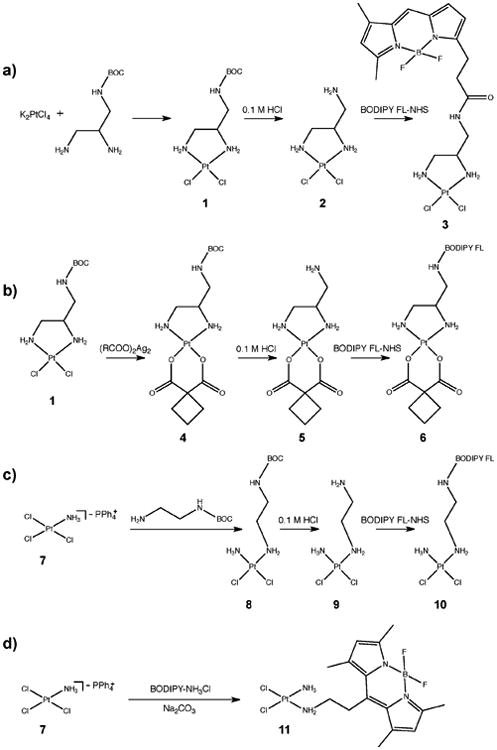
Synthesis routes for compounds 3, 6, 10, and 11.
Figure 1.
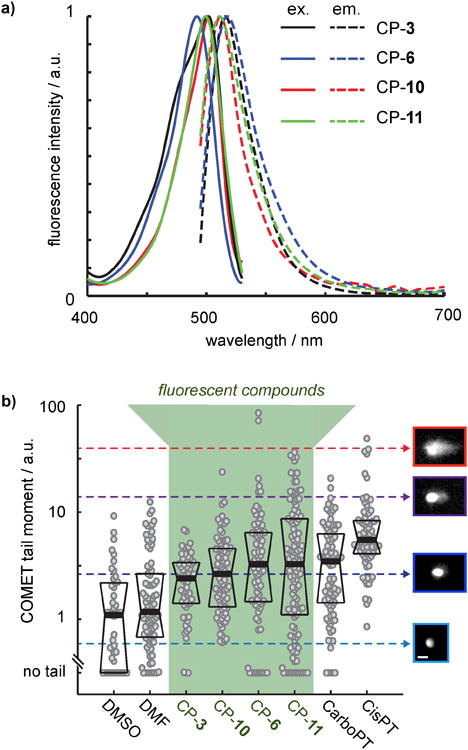
Characterization of in vitro fluorescence and DNA damaging properties for BFL-Pt compounds. a) Ex/em spectra for compounds 3, 6, 10, and 11 (λex = 488nm) at 30μM in PBS. b) Alkaline comet assay results, measured 24 hr after drug treatment. Each dot represents a single cell, with data combined from n=2 separate experiments, and with thick and thin lines representing the median +/- one quartile. Cell images of propidium iodide staining at right exemplify tail-moment measurements, indicating DNA damage. Scale bar = 40μm in all images.
We tested the in vitro cellular response to BFL-Pt compounds by assessing DNA damage and cytotoxicity. We examined whether the BFL-Pt compounds were capable of damaging DNA in human ovarian carcinoma cells using the OVCA429 cell line and the alkaline single-cell gel electrophoresis (comet) assay (Figure 1B). Under alkaline conditions, the comet assay detects a spectrum of DNA damage events at 24 hr post-treatment, including single- and double-strand breaks. Although DNA damage was heterogeneous from cell-to-cell, all BFL-Pt compounds elicited a significant increase in DNA damage when averaged across the cell populations (p<0.05; Mann-Whitney U test), with greatest effects for platinum-compound 11 (CP-11). To measure BFL-Pt cytotoxicity, we treated two ovarian cancer cell lines (OVCA429 and SKOV3) for 48 hr and measured viability using a resazurin-based assay. Similar to comet results, CP-11 was most potent among BFL-Pt compounds (Table 1). Although the carboplatin-related compound CP-6 was less potent than CP-11, it only exhibited a 1.3-2.7 fold loss in potency compared to its parent drug carboplatin, depending on the cell line (Table 1). Of note, carboplatin sensitivity observed here is highly consistent with previously published results[25].
Table 1.
Biological and chemical properties of BFL-Pt and parent compounds. Measurement uncertainty corresponds to ± S.E.M., with the exception of the initial plasma half-life (t1/2 initial) measurements, which report standard deviation over n>4 blood vessels across n≥2 mice.
| Compound] | CP-3 | CP-6 | CP-10] | CP-11 | Cisplatin | Carboplatin |
|---|---|---|---|---|---|---|
| MW (g/mol) | 629 | 700 | 617 | 573 | 301 | 371 |
| logP | 0.58 ± 0.02 | 0.64 ± 0.06 | 0.33 ± 0.07 | 0.53 ± 0.05 | -2.5[30] | -2.3[30] |
| % Albumin Binding | 53 ± 1 | >95 | >95 | >95 | 95[29] | 40[29] |
| Ex/Em (nm) | 501/515 | 492/518 | 501/512 | 500/510 | n/a | n/a |
| Φf (quantum yield) | 0.79 ± 0.01 | 0.61 ± 0.08 | 0.76 ± 0.01 | 0.63 ± 0.05 | n/a | n/a |
| Imaging Dose (mg/kg) | 3.1 | 3.5 | 3.1 | 4.3 | n/a | n/a |
| OVCA429 IC50 (μm) | >700 | 280 ± 50 | 210 ± 70 | 260 ± 10 | 15 ± 1 | 210 ± 6 |
| SKOV3 IC50 (μm) | >700 | 270 ± 10 | 280 ± 5 | 230 ± 20 | 3 ± 0.3 | 100 ± 6 |
| t1/2 initial (min); nu/nu mice | 8 ± 7 | 2 ± 1.5 | 4 ± 3 | 13 ± 6 | 15[28] | 5[32-33] |
We next characterized the in vitro cellular uptake and distribution of the most potent BFL-Pt compound, CP-11. On OVCA429 cancer cells, CP-11 localized to the cytoplasm near the nucleus, consistent with previous reports using fluoresceinderived Pt compounds[4,15-17] (Figure 2A). We also imaged CP-11 in live tumor cells expressing a fluorescently-tagged DNA-repair protein 53BP1, which localizes to the nucleus and forms punctate fluorescence at sites of DNA double-strand-breaks (DSBs)[26-27]. 53BP1 was truncated and fused with mApple fluorescent protein within a lentiviral vector to yield a new genetic construct with efficient protein expression and high fluorescence ideally suited to intravital imaging. Although some CP-11 nuclear staining was detectable in vitro, we found no apparent co-localization with 53BP1 puncta, underscoring drug promiscuity (Figure 2B).
Figure 2.
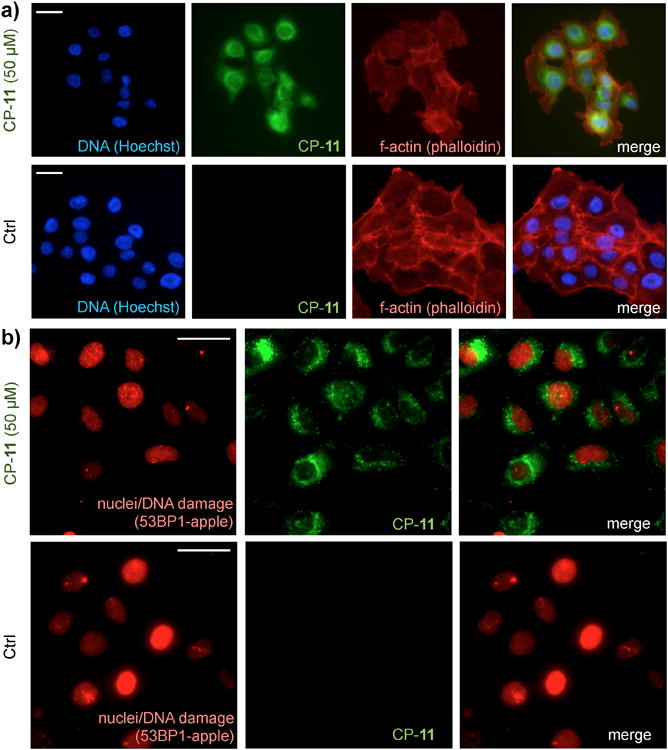
In vitro BFL-Pt imaging of ovarian cancer cells. a) Fixed-cell immunofluorescence of OVCA429 treated with 50μM CP-11 for 24 hr. b) Live-cell immunofluorescence of OVCA429-53BP1-apple treated with 50μM CP-11 for 1 hr. Scale bar = 25μm in all images.
We next assessed the in vivo PK properties of all four BFL-Pt compounds in nu/nu mice. For CP-11, we measured PK using a window-chamber tumor model that allows for the simultaneous time-course fluorescence microscopy of fluorescent drug in both the mouse vasculature and tumor tissue11. With this approach, we found CP-11 displays comparable plasma kinetics as reported for cisplatin, which was previously determined by atomic absorption spectroscopy[28]. CP-11 showed an initial plasma half-life of 13 min (Figure 3A; Table 1) and rapid distribution into tumor cells and stroma (Figure 3B). Following the initial decay, plasma drug levels stabilized with no detectable signal decrease over 30 min of imaging for CP-11, again consistent with the late plasma half-life of >12 hr for cisplatin[28]. We also measured plasma PK for CP-3, CP-6, and CP-10 using time-lapse fluorescence microscopy of mouse ear vasculature. Similar to CP-11, initial plasma half-lives ranged from 2-8 min for the other compounds (Table 1). BFL-Pt compounds exhibited a maximum 8-15 fold signal increase over background fluorescence during real-time in vivo imaging experiments, depending on the compound. Figure 3B depicts a peak 12-fold increase in fluorescence signal for CP-11. With one exception, BFL-Pt compounds all displayed albumin binding in excess of 95%, bearing similarity to the parent compounds (Table 1)[29]. CP-3 protein binding was lower, perhaps suggesting a lower reactivity reflected by low potency in the DNA damage (Figure 1B) and cytotoxicity (Table 1) assays. Although the BFL-Pt compounds displayed significantly lower logP values compared to the parent drugs, their PK properties nonetheless resembled those of the parent drugs, particularly between CP-11 and cisplatin. Furthermore, previous reports have shown that fluorescent labeled Pt compounds generally retain key intracellular trafficking properties despite altered lipophilicity[15-17]. Nevertheless, increased lipophilicity and decreased cytotoxicity of the BFL-Pt compounds should be taken into account when designing experiments and interpreting results.
Figure 3.
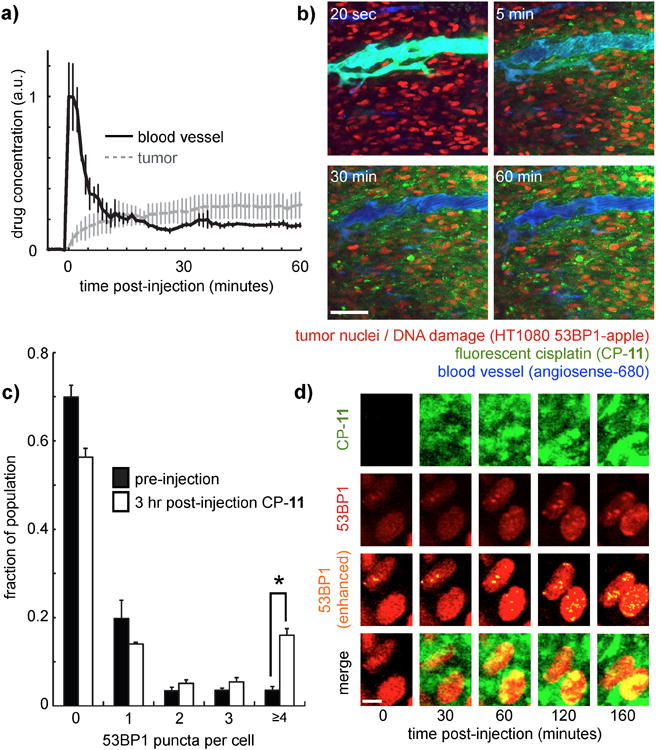
Real-time in vivo imaging of BFL-Pt pharmacokinetics and resulting DNA damage in tumor cells within a xenograft animal model. a-b) Concentration of in vivo CP-11 over time (a), corresponding to time-series window-chamber images shown at right (b). Scale bar = 50μm. c) CP-11 treatment causes a significant increase in the fraction of tumor cells showing ≥4 53BP1 puncta, 3 hr following drug injection. Values were measured from >10 images and >500 individual tumor cells across two mice (*p=0.002, student's t-test). d) Zoomed-in image of two tumor cells showing accumulation of 53BP1 puncta over time. Scale bar = 5μm.
To match in vivo PK with PD response, we used intravital imaging of fluorescent 53BP1 puncta to track DNA damage in tumor cells following CP-11 treatment. As with the comet assay (Figure 1B), we found heterogeneity in DNA damage across the population of tumor cells. Nonetheless, when examined across the tumor-cell population, CP-11 treatment caused a significant increase in the fraction of cells with greater than four double-strand breaks, as visualized by 53BP1 puncta (Figure 3C). Figure 3D presents two example cells tracked over time that exhibit an accumulation of CP-11 and that subsequently show an increase in 53BP1 puncta. In this model we found little apparent correlation between 53BP1 puncta and local CP-11 accumulation, suggesting that drug response heterogeneity may arise from factors besides differential drug transport, such as cell-cycle[30] and cellular redox state[34].
In sum, here we report four novel Pt compounds for high resolution intravital imaging. We demonstrate these compounds i) can cause DNA damage and cell death in cancer cells; ii) show comparable PK with parent compounds; and iii) can be used for high-resolution imaging of PK/PD in a live animal model of cancer. This work presents a proof-of-principle exhibition of the ability to visualize drug accumulation and subsequent DNA damage in single tumor cells monitored in real time following drug administration. We anticipate that the imaging tools presented herein will be useful in guiding the effort to understand the role of Pt distribution and transport in various models of disease; to study the role of intratumoral heterogeneity in influencing drug response and emergent drug resistance; to guide development of targeted/time-released Pt-drug delivery via nanoformulations; and perhaps to image real-time tumor-stroma interactions as Pt-compounds distribute to various cell types within the tumor microenvironment.
Experimental Section
See Supplementary Information for extended experimental methods. Complex 1 and 2[23], [PtCl3NH3]PPh4[24], Ag2R(COO)2(R = cyclobutane-1,1-dicarboxylic acid)[35] and Amino-BODIPY[36] were synthesized as previously described. For 53BP1 transgene expression, a fragment corresponding to amino acids 1220-1711 was subcloned into the pLVX lentiviral vector (Clontech)[26]. A DeltaVision (Applied Precision) modified Olympus BX63 microscopy system with an environmental chamber was used for live-cell microscopy. Intravital microscopy was performed on an Olympus FV1000 multiphoton imaging system and using animals in accordance with guidelines from the Institutional Subcommittee on Research Animal Care. Following previously described procedures[19], tumors were injected in nu/nu mice (Cox7, MGH) 30 min following dorsal window chamber implantation and imaged 2 weeks later. Mice were injected via tail vein catheter with Angiosense-680 (Perkin-Elmer) to identify vasculature, 150 nmol CP-11, and imaged.
Supplementary Material
Supporting Information
Acknowledgments
Part of this work was supported by NIH U54-CA151884 and a fellowship to B.A. from Deutsche Akademie der Naturforscher Leopoldina. We thank Alex Zaltsman, Matthew Sebas, and Olivier Kister for technical assistance. We thank Drs. Lippard, Farokhzad, and Langer for helpful discussions.
Footnotes
Contributor Information
Dr. Miles A. Miller, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114 (USA).
Dr. Bjorn Askevold, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114 (USA).
Dr. Katherine S. Yang, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114 (USA)
Prof. Ralph Weissleder, Email: rweissleder@mgh.harvard.edu, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114 (USA); Harvard Medical School, 200 Longwood Avenue, Boston, MA 02115 (USA).
References
- 1.Zhu G, Song L, Lippard SJ. Cancer Res. 2013;73:4451. doi: 10.1158/0008-5472.CAN-13-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouwman P, Jonkers J. Nat Rev Cancer. 2012;12:587. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 3.Oliver TG, Mercer KL, Sayles LC, Burke JR, Mendus D, Lovejoy KS, Cheng MH, Subramanian A, Mu D, Powers S, Crowley D, Bronson RT, Whittaker CA, Bhutkar A, Lippard SJ, Golub T, Thomale J, Jacks T, Sweet-Cordero EA. Genes Dev. 2010;24:837. doi: 10.1101/gad.1897010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blair BG, Larson CA, Adams PL, Abada PB, Pesce CE, Safaei R, Howell SB. Mol Pharmacol. 2011;79:157. doi: 10.1124/mol.110.068411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang XJ, Shen DW, Chen KG, Wincovitch SM, Garfield SH, Gottesman MM. J Cell Physiol. 2005;202:635. doi: 10.1002/jcp.20253. [DOI] [PubMed] [Google Scholar]
- 6.Johnstone TC, Kulak N, Pridgen EM, Farokhzad OC, Langer R, Lippard SJ. ACS Nano. 2013;7:5675. doi: 10.1021/nn401905g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suntharalingam K, Johnstone TC, Bruno PM, Lin W, Hemann MT, Lippard SJ. J Am Chem Soc. 2013;135:14060. doi: 10.1021/ja4075375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelland L. Nat Rev Cancer. 2007;7:573. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 9.Xu JM, Azzariti A, Colucci G, Paradiso A. Cancer Chemother Pharmacol. 2003;52:442. doi: 10.1007/s00280-003-0687-8. [DOI] [PubMed] [Google Scholar]
- 10.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Cell. 2012;149:780. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pittet MJ, Weissleder R. Cell. 2011;147:983. doi: 10.1016/j.cell.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Cancer Res. 2011;71:4608. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holzel M, Bovier A, Tuting T. Nat Rev Cancer. 2013;13:365. doi: 10.1038/nrc3498. [DOI] [PubMed] [Google Scholar]
- 14.Earley S, Vinegoni C, Dunham J, Gorbatov R, Feruglio PF, Weissleder R. Cancer Res. 2012;72:2949. doi: 10.1158/0008-5472.CAN-11-4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katano K, Safaei R, Samimi G, Holzer A, Tomioka M, Goodman M, Howell SB. Clin Cancer Res. 2004;10:4578. doi: 10.1158/1078-0432.CCR-03-0689. [DOI] [PubMed] [Google Scholar]
- 16.Liang XJ, Finkel T, Shen DW, Yin JJ, Aszalos A, Gottesman MM. Mol Cancer Res. 2008;6:1499. doi: 10.1158/1541-7786.MCR-07-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalayda GV, Zhang G, Abraham T, Tanke HJ, Reedijk J. J Med Chem. 2005;48:5191. doi: 10.1021/jm050216h. [DOI] [PubMed] [Google Scholar]
- 18.Hama Y, Urano Y, Koyama Y, Bernardo M, Choyke PL, Kobayashi H. Bioconjug Chem. 2006;17:1426. doi: 10.1021/bc0601626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, Weissleder R. Nat Commun. 2013;4:1504. doi: 10.1038/ncomms2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulrich G, Ziessel R, Harriman A. Angew Chem Int Ed Engl. 2008;47:1184. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- 21.Karolin J, Johansson LBA, Strandberg L, Ny T. Journal of the American Chemical Society. 1994;116:7801. [Google Scholar]
- 22.Hinkeldey B, Schmitt A, Jung G. Chemphyschem. 2008;9:2019. doi: 10.1002/cphc.200800299. [DOI] [PubMed] [Google Scholar]
- 23.Molenaar C, Teuben JM, Heetebrij RJ, Tanke HJ, Reedijk J. J Biol Inorg Chem. 2000;5:655. doi: 10.1007/s007750000153. [DOI] [PubMed] [Google Scholar]
- 24.Abrams MJ, Giandomenico CM, Vollano JF, DA S. Inorg Chim Acta. 1987;131:3. [Google Scholar]
- 25.Bicaku E, Xiong Y, Marchion DC, Chon HS, Stickles XB, Chen N, Judson PL, Hakam A, Gonzalez-Bosquet J, Wenham RM. British journal of cancer. 2012;106:1967. doi: 10.1038/bjc.2012.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimitrova N, Chen YC, Spector DL, de Lange T. Nature. 2008;456:524. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karanam K, Kafri R, Loewer A, Lahav G. Mol Cell. 2012;47:320. doi: 10.1016/j.molcel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnsson A, Olsson C, Nygren O, Nilsson M, Seiving B, Cavallin-Stahl E. Cancer Chemother Pharmacol. 1995;37:23. doi: 10.1007/BF00685625. [DOI] [PubMed] [Google Scholar]
- 29.Sooriyaarachchi M, Narendran A, Gailer J. Metallomics. 2011;3:49. doi: 10.1039/c0mt00058b. [DOI] [PubMed] [Google Scholar]
- 30.Olive PL, Banath JP. Cytometry B Clin Cytom. 2009;76:79. doi: 10.1002/cyto.b.20450. [DOI] [PubMed] [Google Scholar]
- 31.Screnci D, McKeage MJ, Galettis P, Hambley TW, Palmer BD, Baguley BC. Br J Cancer. 2000;82:966. doi: 10.1054/bjoc.1999.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Li M, Rinehart JJ, Zhang R. Clin Cancer Res. 2004;10:1633. doi: 10.1158/1078-0432.ccr-0829-3. [DOI] [PubMed] [Google Scholar]
- 33.Elferink F, van der Vijgh WJ, Klein I, Vermorken JB, Gall HE, Pinedo HM. Cancer Treat Rep. 1987;71:1231. [PubMed] [Google Scholar]
- 34.Simons AL, Ahmad IM, Mattson DM, Dornfeld KJ, Spitz DR. Cancer Res. 2007;67:3364. doi: 10.1158/0008-5472.CAN-06-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rochon FD, Massarweh G. Inorganica chimica acta. 2006;359:4095. [Google Scholar]
- 36.Donnelly P, Paterson B. WO Patent 2010066010. 2010
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information