Statistical Design for Biospecimen Cohort Size in Proteomics-based Biomarker Discovery and Verification Studies (original) (raw)
. Author manuscript; available in PMC: 2014 May 30.
Published in final edited form as: J Proteome Res. 2013 Oct 28;12(12):5383–5394. doi: 10.1021/pr400132j
Abstract
Protein biomarkers are needed to deepen our understanding of cancer biology and to improve our ability to diagnose, monitor and treat cancers. Important analytical and clinical hurdles must be overcome to allow the most promising protein biomarker candidates to advance into clinical validation studies. Although contemporary proteomics technologies support the measurement of large numbers of proteins in individual clinical specimens, sample throughput remains comparatively low. This problem is amplified in typical clinical proteomics research studies, which routinely suffer from a lack of proper experimental design, resulting in analysis of too few biospecimens to achieve adequate statistical power at each stage of a biomarker pipeline. To address this critical shortcoming, a joint workshop was held by the National Cancer Institute (NCI), National Heart, Lung and Blood Institute (NHLBI), and American Association for Clinical Chemistry (AACC), with participation from the U.S. Food and Drug Administration (FDA). An important output from the workshop was a statistical framework for the design of biomarker discovery and verification studies. Herein, we describe the use of quantitative clinical judgments to set statistical criteria for clinical relevance, and the development of an approach to calculate biospecimen sample size for proteomic studies in discovery and verification stages prior to clinical validation stage. This represents a first step towards building a consensus on quantitative criteria for statistical design of proteomics biomarker discovery and verification research.
Keywords: Statistical Experiment Design, Biomarker, Proteomics, Unbiasedness, Power Calculation
INTRODUCTION
Current Challenges in Clinical Proteomics
Through the identification of novel plasma and tissue protein biomarkers, clinical proteomics has the potential to enable advances in multiple clinical challenges in cancer, including the improved detection of cancers and the prediction of efficacy for therapeutic treatments. Clinical proteomic studies for diagnostic biomarker discovery in a multistage biomarker pipeline typically begin with the identification and measurement of a large number of proteins in a set of source biospecimens. Differentially expressed proteins identified in case and control source samples form the initial protein candidate list. By confirming differential expression in clinically useful samples such as blood, or serially measuring fewer proteins with increasing precision on greater numbers of samples, candidates can be progressively credentialed to yield a select few proteins that may warrant assessment in an often time-consuming and costly large-scale clinical validation trial [1–2].
All stages of the proteomics biomarker pipeline present challenges to clinical proteomics. These challenges include (1) measurement standardization and optimization for discovery and verification stages; (2) development of high throughput, high precision, low cost per sample assays for clinical validation stage; (3) understanding the analytical and clinical validation designs acceptable to the FDA; and (4) statistical experimental design at discovery and verification stages of the pipeline that are poorly understood in comparison to clinical validation. Insufficient attention to statistical design has often resulted in the analysis of too few samples to achieve any reasonable statistical power to detect biomarkers. Here we address the last challenge with regard to sample sizes for protein discovery and verification, and derive statistical and clinical criteria for advancing a biomarker candidate within the pipeline.
In collaboration with other efforts, the NCI’s Clinical Proteomic Technologies for Cancer initiative (NCI-CPTC), launched in 2006, helped address some of these barriers, including standardizing proteomic platform measurements, clarifying FDA requirements for analytical validation [3] through a joint NCI-FDA workshop [4]. The introduction of “verification” between discovery and clinical validation stages efficiently triages identified candidates for further assessment in the clinical validation stage where a handful of verified candidates are quantitatively measured in thousands of clinical samples reflecting the full spectrum of biomarker heterogeneity in the target population. Verification bridges discovery and validation using an intermediate number of candidates, precision of measurement, length of time for assay development, and sample throughput. To achieve this, verification relies on analytically robust, targeted proteomic platforms and well-characterized affinity reagents (if needed) to measure relative concentrations of a moderate number of biomarker candidates in a moderate number of patient samples.
To illustrate an example of such a proteomics biomarker pipeline, we present a proposed scheme for discovery and verification of plasma biomarkers for differential diagnosis of ovarian cancers. In the discovery stage, untargeted mass spectrometry analysis (“shotgun proteomics”) of benign and malignant ovarian cyst fluids will yield a large number of differentially expressed candidate proteins from a small number of case and control patient samples. These proteins are ranked for differential expression by a _t_-test on the log-intensity scale. The top 50 candidates are passed onto the verification stage using quantitative targeted assays in plasma based on multiple reaction monitoring mass spectrometry (MRM-MS). This technique, commonly used for small molecule and metabolite analysis in clinical labs, has recently been optimized using proteotypic peptides (unique to each target protein) as surrogates for protein concentration measurement. For some of these candidates with low plasma concentrations (nM or less), MRM-MS assays may require the development of anti-peptide antibodies against selected proteotypic peptides prior to MS analysis on a triple quadrupole mass spectrometer (i.e., immuno-MRM) [5]. Using MRM-MS, plasma from a moderate number of cases with malignant ovarian tumors and controls with benign ovarian disease is measured for the top 50 candidates. Subsequently, a _t_-test of the log-concentration ranks each candidate and 5 top-ranked, “verified” candidates are further passed to the clinical validation stage using high throughput, reliable assays, such as ELISAs. A caveat for developing ELISAs for “verification” is their higher cost and lead time (> 12 months) compared to immuno-MRMs (~ 3 months). Thousands of validation samples are measured for these top 5 protein targets in plasma from cases and controls obtained in an unbiased way by drawing blood prior to diagnosis. This sample set may be split randomly into a training set and a validation set. A classifier using the 5 best candidates is trained on the training set and unbiased operating characteristics are estimated on the validation set. If the classifier’s characteristics reach clinical utility, the biomarker pipeline has identified a biomarker classifier. A forebear of this example pipeline produced OVA1 [6], while a more distant forebear produced ROMA [7]; both are FDA authorized tests for differential diagnosis of pelvic masses.
Statistical Design for a Multistage Proteomics Biomarker Pipeline
Many publications have addressed experimental design of clinical validation studies, sample size calculations in multistage genome-wide association studies [8–9], diagnostic studies in radiology, or comparison of medical tests [10–13]. However, sample sizes for a multistage proteomics biomarker pipeline, such as the example described above, have not been fully addressed. The workshop entitled “Experimental Design Considerations in Research Studies Using Proteomic Technologies” addressed this challenge. Statistical design at each stage of a proteomics biomarker pipeline with an intended clinical use of an assay (or medical device) as required by the FDA [14] is vital to inform whether there is sufficient power to justify moving candidates to the next stage. A case study for the detection of ovarian cancers using a multistage pipeline was used to illustrate statistical design considerations involving MS-based technologies [15–16] (Methods and Results). In addition to sample size, designs for unbiased selection of patient cohorts [17] were proposed at this workshop that will not be described in this manuscript. Compared to previous publications, this report is novel in three aspects: (1) it addresses sample sizes for the discovery and verification stages of a proteomics biomarker pipeline (not clinical validation); (2) the modeling accounts for the marker being shed only by a subset of tumors as would be predicted by tumor heterogeneity and as occurs with known cancer biomarkers; and (3) the selection of candidates for successive stages of development includes clinical criteria to assess whether a candidate has a chance to have clinical utility.
Defining Statistical Design Goals
Guided by the context of an intended clinical use, the aim is to identify experimental designs that will achieve acceptable statistical power for a true biomarker with specified characteristics to reach clinical validation stage from discovery and verification stages. Verification in the ovarian case study uses MRM-MS, which has previously demonstrated intra- and inter-laboratory precision of %CV < 20% [18]. The statistical design and sample sizes presented here, however, apply broadly to biomarkers other than proteins and technologies other than MS. For instance, autoantibodies are promising alternative biomarkers, and protein arrays that detect autoantibodies in blood can be used in discovery and verification stages to triage candidates using the approaches described herein [19].
Each stage in the pipeline winnows the candidate list. Given current MS instrumentation, the discovery stage may detect ~1,000–10,000 proteins from clinical samples. If each stage achieves a 10-fold winnowing, a three-stage pipeline [20–22] achieves a combined 1,000-fold reduction in the number of candidates to approximately 1–10 validated biomarkers. Given the current lack of standardization for statistical design, this report focuses on designing discovery and verification stages to achieve a given power for a true biomarker to reach clinical validation stage.
The decision on whether a candidate proceeds from discovery to verification stage depends on multiple factors often specific to the discovery effort, including throughput of sample processing, and both the biological relevance and the statistical significance of the candidate. One additional criterion advocated here but not often addressed by the community is the potential contribution to a clinical question. If the potential contribution is very small, one might consider not advancing such a candidate to the next stage, even if it were statistically significant. In a clinical example of early detection of ovarian cancer where acceptable performance is most readily defined by the positive predictive value (PPV), we derive the specificity corresponding to the lower limit acceptable (actionable) for the PPV, and incidence of the disease, assuming a sensitivity of 100%. Candidate sensitivities less than 5% will not likely be significant contributors to the ultimate goal of 100% sensitivity. By applying such clinical quantitative criteria in concert with conventional statistical criteria at each stage of biomarker development, we ensure that biomarker candidates emerging at the end of the pipeline will have a reasonable expectation of contributing significantly to the sensitivity of a biomarker panel while retaining clinically actionable specificity and PPV. This approach, first introduced at this workshop and subsequently presented at a National Institute of Diabetes and Digestive and Kidney Diseases workshop [23], is greatly expanded in this report. Though introduced in the context of ovarian cancer detection, it is readily generalized to other diagnostic situations.
METHODS
The Statistical Model
The statistical model aims to simulate the discovery and verification stages to estimate the probability of detecting one biomarker whose distribution separates cases and controls among the thousands of matrix proteins detectable in the source biospecimens whose distribution does not separate cases from controls. The protein molecules in the sample source (e.g., plasma or cystic fluid) except for the biomarker are referred to as the background matrix. In addition to the biological variation between patients, the measurement processes in discovery and verification stages add analytical variation. For the discovery phase, the measurement CV increases as the concentration decreases from 20% for the most abundant proteins to 60% for the least abundant, detectable proteins. Thus, a model for the number of proteins at each decade of concentration is required. Hortin and Anderson previously surveyed the first four decades of plasma proteins [24]. As the number of proteins approximately doubles with each decade, the extrapolation of this trend to eight decades provides the model for the distribution of protein concentrations for a total of ~8,000 matrix proteins. We modeled the measurement variation by the CV = 15% + 5*decade, where decade = 1,…, 8, and the proteins are distributed across the concentration decades as described in Figure 1. The biological CV between patients is more complicated. High abundance proteins have low biological CVs (e.g., 9% for albumin), whereas low abundant proteins have a range of biological CVs. There are plasma proteins with low concentration and a tight range of concentrations between people (e.g., many hormones), while other plasma proteins with large variation at low concentrations. To derive a statistical model for the biological CV as a function of concentration, we estimated biological CVs from a database on normal biological variation for laboratory tests and their normal ranges [25]. The details of the statistical modeling are provided in the supplement but in essence consist of increasing expected CV and increasing variation in CV with increasing decade of concentration.
Figure 1. Distribution of Proteins in Blood (Plasma/Serum) by Concentration Decade.

This is a discrete version of a triangular distribution of the number of plasma proteins with increasing concentration decade (adapted from Horton and Anderson et al. [24]). Until a human protein quantitation project is completed, the distribution of plasma proteins as a function of concentration below 4 logs of concentration is based on an extrapolation.
The total CV in the simulations due to measurement (e.g., 50%) and biological variation (e.g., 60%) was then calculated as 78% = √(50%2 + 60%2). To first order the CV is equivalent to the standard deviation (SD) on the log-concentration scale. Each decade of proteins was simulated with the number of proteins given in Figure 1 (except for the last decade in the figure) for a total of 7,899 plasma proteins. Let Y denote the log-concentration of a (non-biomarker) protein in a discovery biospecimen if it could be measured without error, X denote variation in log-concentration due to the measurement process, and Z denote the log-concentration of a biomarker which has a different distribution in cases compared to controls. The biological distribution across subjects of log-concentration for each matrix protein i = 1, …, P, in cases (j=2) and controls (j=1), and in subjects n = 1, …, N was modeled by:
Yijn~N(μi,σi),μi=decade=1,…,8withprobabilityofselectingtheithdecadeproportionaltothenumberofproteinsinthedecade(Figure1),andlog(σi)~N(3.60-0.0757∗decade,0.784-0.596∗decade).
Measurement variation for discovery stage was modeled by:
Xijn~N(0,τi),τi=15%+5∗decade,decade=1,…,8
The biological variation of the biomarker’s concentration between subjects was modeled by:
Zjkn~pkN(μ+Δ,σ)+(1-pk)N(μ,σ)(j=2=case)biomarkerhassignal=Δ=sσfors=2,3,4,or5inpk=10%,20%,30%,50%and80%ofcases,k=1,2,3,4,5.Zjkn~N(μ,σ)(j=1incontrolsubjects).
The over-expression of a protein in a proportion pk of subjects is Δ = s*σ, where s = 2, 3, 4, or 5 is the number of SDs separating the mean of the log-concentration of the biomarker in the over-expressed cases from mean for the biomarker in the control subjects. For proximal fluids and tissue, the signal/noise ratio of the biomarker is likely to be greater than in a fluid more remote from the disease such as plasma. Therefore, the separation was modeled as s = 2, 3, 4, or 5 for proximal fluids and tissue in the discovery stage, and s = 1, 2, 3, or 4 in plasma for the verification stage. Since it is likely that undiscovered biomarkers have low concentration, the average concentration μ for the biomarker was modeled at the lowest decade of concentration. Based on variations in known plasma biomarkers, the biological CV σ for the as-yet-undiscovered cancer biomarker was assumed to be above average compared to other plasma proteins at the same decade. Thus, log(σ) was set at 1 SD above its expected value. The measurements on P proteins (P ~ 7,899 discovery) were simulated in N cases and N control subjects, and the _t_-statistic comparing cases to controls was calculated for each of the P matrix proteins with log-concentration (Yijn + Xijn) where there is no difference in the distribution between the cases and controls. However, there will be a few proteins amongst 7,899 proteins where the N simulated results in the cases appear to be significantly elevated compared to those in the controls due to chance. The one (biomarker) protein Z with signal Δ in a proportion p of the N cases and no signal in a proportion (1-p) of N cases will have results also simulated in N cases and N controls where there is likely to be a significant difference. If the _t_-statistic for Z is within the top C1 results for discovery stage, or C2 results for verification stage, then the biomarker is passed onto the next stage. C1 is the number of targeted assays (e.g., immuno-SRMs) that the investigator is planning to develop for the verification stage (C1 = 20, 50, and 100), and C2 is the number of high throughput reliable assays that the investigator plans to develop or obtain for the clinical validation stage. C2 is expected to be proportional to the number of targeted assays (C1) and set at 10% of C1 for this simulation. The process was repeated 1,000 times and the proportion of times that Z was successfully passed to the next stage was calculated. This estimate was calculated for each combination of the proportion of cases over-expressing the biomarker p (10%, 20%, 30%, 50%, 80%) with over-expression s = 2, 3, 4, or 5 SDs (or 1, 2, 3, or, 4 SDs for plasma) in the cases above the controls, and sample sizes of n = 10, 25, 50, and 100 in the discovery stage, while n = 25, 50, 100, and 250 in the verification stage. The results are the power given in Tables 1 and 2 with a minimum Monte-Carlo accuracy of 1.5% around 50%, and 1% or less when the power is 80% or greater.
Table 1.
Probability of Biomarkers Successfully Passing Discovery Stage
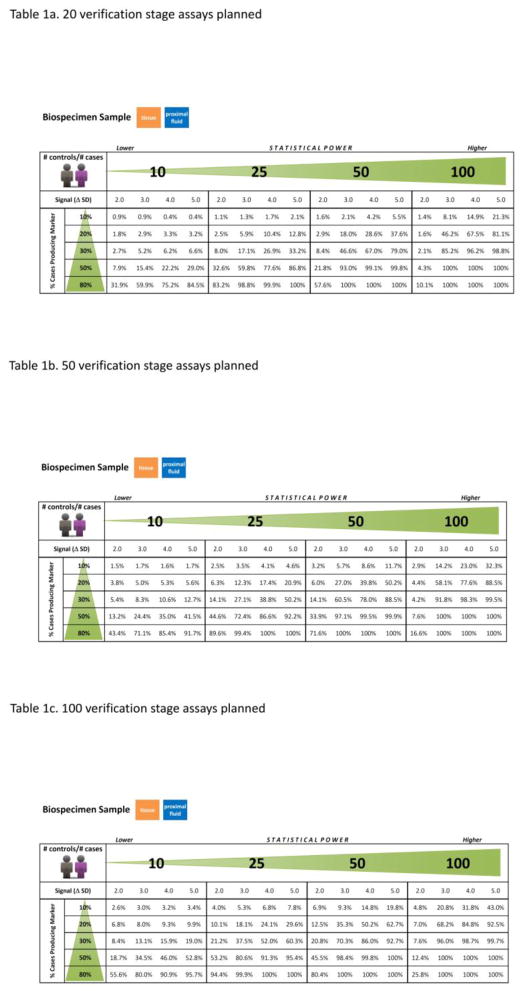
Table 2.
Probability of Biomarkers Successfully Passing Verification Stage

Factors affecting required sample size to achieve a specified power to identify the biomarker include the biomarker’s amount of separation between cases and controls (Figure 3), and the bandwidth of the pipeline. The bandwidth is measured by the number of targeted assays planned to be developed in the verification stage, and the number of high throughput low measurement CV assays to be developed in the clinical validation stage. The number of false positive candidate proteins will affect power and will increase as the analytical depth of the discovery instrument increases. Finally, between subject variation in protein log-concentration, and variation due to the measurement process (analytical variation), increase the required sample sizes. These factors are discussed more fully in the supplement.
Figure 3. Separation of Biomarker Distribution between Cases Shedding the Biomarker and Controls, Crossed with Fraction of Cases Shedding Biomarker.

This figure is a simulation example provided to biomarker researchers in choosing the expected separation between cases and controls (rows) provided by the target biomarker, and the fraction of cases shedding the biomarker (column). These two parameters are instrumental in determining the required number of samples. The biomarker distribution in controls is given by the blue histogram with density represented by the dashed line. Cases are a mixture of tumors that shed the biomarker and have a distribution (light red) shifted to the right from the biomarker distribution in controls by 5, 4, 3, and 2 SDs for the 1st, 2nd, 3rd and 4th rows, respectively. The proportion of cases shedding the biomarker changes by column from 80% to 50% to 20% in the 1st, 2nd and 3rd column, respectively. Cases that do not shed the biomarker have the same biomarker distribution as controls. The red histogram represents the mixture of the cases shedding the biomarker (solid line on right, light red) and the cases not shedding the biomarker (solid line on left under the dashed line, dark red). The top left corner (5 SDs of separation with 80% cases shedding biomarker) illustrates the most extreme and easy-to-discover tumor biomarker (CA125). Hence, this situation forms the extreme of the spectrum of separation and fraction of cases shedding the biomarker with subsequent examples of decreasing the separation, or the fraction shedding the biomarker, or both. Biomarker discoverers need to judge where the “to-be-discovered” biomarker lies within this spectrum and obtain an estimate as to sample size in the discovery and verification stages of a multistage proteomic pipeline.
Defining Quantitative Criteria Based on Clinical Judgments
In addition to requiring candidates to meet statistical criteria, a biomarker should also have clinical utility. One reasonable approach to define quantitative criteria based on clinical requirements for candidate biomarkers is as follows:
- Define the intended clinical use, including a full description of the clinical management pathway within which the diagnostic test will be applied;
- Calculate an acceptable benefits-to-harms (B/H) ratio, that is, benefit of true positive and true negative compared to the harm resulting from false positive and false negative tests [38];
- Derive the required specificity assuming 100% sensitivity to achieve an acceptable B/H ratio within the clinical pathway;
- Evaluate the sensitivity of biomarker at the required specificity. This sensitivity is likely an upper limit on the contribution to a panel that aims at 100% sensitivity at the required specificity.
The first two steps apply generally, while the third and fourth steps focus on specificity because in the context of early detection for ovarian cancer, harm occurs mostly when false positive results lead to unnecessary surgery. For intended clinical uses where the greatest harm occurs due to false negative results, step three should focus on sensitivity and step four on evaluating specificity at the required sensitivity. This general approach is approximate and appropriate when disease incidence is low, such as for the early detection of cancer. The Supplemental Methods provide the exact formulae when disease incidence is not low such as differential diagnosis of pelvic masses where incidence of malignancy is 20%. Harm due to false negatives will be negligible if the ovarian cancers missed by screening are still detected clinically due to symptoms at the same time as if screening did not occur. By benefits and harms, we implicitly mean the net benefits and net harms, which assess the difference between outcomes using the diagnostic test to outcomes under usual care without the diagnostic test.
Defining the minimally acceptable B/H ratio draws a somewhat arbitrary boundary, namely the maximum number of major harms that are clinically acceptable for each major benefit provided by the test. Although specifics can be challenging to rigorously defend, such a “line in the sand” has previously been drawn for the early detection of ovarian cancers, where the minimally acceptable B/H ratio was determined to be no more than 10 surgeries to find 1 screen-detected ovarian cancer [39]. Setting the discussion within the context of clinical B/H ratio as a formative criterion has the distinct advantage of using a scale on which physicians, patients and payers can collaborate to form judgments informed by their respective tolerance of risk and benefit. Scales such as fold increase in average biomarker level from controls to cases (as commonly used in proteomics), or specificity, or positive likelihood ratio, can subsequently be derived from this B/H ratio. In contrast, setting seemingly high but arbitrary fold increases or specificities create difficulties for most physicians, patients and payers in forming judgments as to what is clinically relevant and acceptable. For example, while 85% specificity may be adequate for one intended use, the specificity may need to be 95%, or 98%, or 99.8% to achieve clinical utility in the context of other intended uses, even with a clinically acceptable level of sensitivity.
For the initial discovery stage, it is reasonable for the B/H judgments to consider only major effects and for the clinically motivated criteria to be estimated to first order with refinement occurring in parallel with each stage of the pipeline. If these criteria are developed at the outset and systematically applied throughout the biomarker pipeline, resulting candidates will have a better chance of meeting the clinical criteria for their intended use.
For ovarian cancers, combined surgery and chemotherapy can be curative if disease is detected early. However, disease usually presents when a patient becomes symptomatic, most often at late stage. Currently, although multiple early detection ovarian cancer trials are underway, no method has yet been shown to significantly reduce either the number of cancers diagnosed at late rather than early stage, or mortality. Based on the aforementioned approach (steps 1–4), quantitative criteria for an early detection ovarian cancer biomarker or a panel are set as follows:
- The intended clinical use is the early detection of ovarian cancers; where a clinical management pathway [35, 40] is:
- annual blood test on all postmenopausal women;
- ultrasound for women with positive blood test;
- referral for surgery for women with positive ultrasound.
- The benefit of a true positive is the earlier detection of an ovarian cancer, while the benefit of a true negative is reassurance (small). The harm of false positive results is surgery on women with no ovarian cancers, whereas the harm of a false negative is to miss ovarian cancers. In this case, we do not consider a volunteer’s experience of an ultrasound when the woman does not have the disease a major harm compared with unnecessary surgery. With a false negative blood test, an ovarian cancer will most likely be detected with usual clinical care at the same point in time as when no screening is performed; therefore, the net harm of a false negative is likely to be small compared to no screening. Taking account of only the main contributions, the B/H ratio is the early detection of ovarian cancers compared to the surgeries performed as a result of false positive tests. Hence, the acceptable B/H ratio is established by determining the number of surgeries required to detect one case of ovarian cancer, that is, a PPV of 20% [35, 40];
- The required specificity, derived by assuming 100% sensitivity, is determined through the following considerations: Since the annual incidence of ovarian cancers in postmenopausal population in the U.S. is 1 in 2,500, a screening process needs to increase the incidence in the test-positive population by 500-fold to achieve the goal of 5 surgeries for 1 ovarian cancer. In screening trials, ultrasound as a second line test reduces false positives by 10-fold, thus a plasma biomarker test needs to reduce false positives by 50-fold to achieve an overall reduction of 500-fold. This makes the reasonable assumption that a false positive ultrasound is statistically independent of a false positive blood test. Hence, the required specificity (ReqSP) for the blood test is 98%, which achieves the target false positive rate of 1 in 50 (2%). A candidate for an ovarian cancer blood test with less than 5% sensitivity at 98% specificity at either the transition from discovery stage or verification stage would likely not be continued within the pipeline.
RESULTS
Discovery Stage Power
The two probabilities for discovery and verification are independent of each other due to separate sets of samples used in these two stages. In order to achieve an overall probability of detection of 0.8, the probability of success in each stage needs to be 0.9 (0.9 × 0.9 = 0.81). For signals where the number of SDs separating cases producing the biomarker from controls ranges across Δ = 2, 3, 4 and 5, the proportion p of cases producing the marker of ranges across 10%, 20%, 30%, 50%, and 80%, and the number of candidates being passed from discovery to verification stage ranges across C1 = 20, 50, and 100, the probability of success in discovery stage has been estimated through simulation and summarized in Table 1 (n1 = number of cases = number of controls = 10, 25 and 50). For example, with only 10 cases and 10 controls and 20 candidates planned for verification, a biomarker must be expressed in at least 80% of the cases, and cases and controls must be separated by at least 5 SDs to achieve greater than 80% probability of reaching verification. If 50 targeted assays are planned for verification, then the power exceeds 90% with the same parameters. As the number of biospecimens increases, the requirement for the proportion of cases shedding the biomarker and/or the separation in SDs can relax (decrease) while retaining comparable power. When 25 cases and 25 controls are analyzed, a marker can be expressed in only 50% of cases at 2 SDs to achieve an 80% probability of passing through discovery to verification (Table 1). An increase of discovery sample sets to 50 cases and 50 controls substantially increases the probability of passing the marker to verification stage, even if the marker is present in as few as 30% of cases.
Verification Stage Power
Due to generally lower measurement CV of targeted proteomics approaches used at the verification stage, with all other parameters being equal, verification will yield a greater chance than discovery of identifying the true biomarker. The more candidates assessed at the verification stage, the greater the chance of passing the biomarker onto the clinical validation stage. The simulations quantify the chance of a biomarker being verified, given quantification of the above parameters and the number of non-biomarker candidates (false positives) assessed. The probability of verification for a true biomarker has been estimated when measurement processes have a CV = 15%, with the number of cases equal the number of controls and ranging from 25, 50, 100, to 250, and the signal (Δ SDs) and proportion of cases producing the biomarker p remaining as previously defined. For signals Δ = 1, 2, 3 and 4 SDs of the log-scale mean for cases that produce the biomarker above the control mean; p at 10%, 20%, 30%, 50% and 80%; and the number of candidates being passed to clinical validation stage for measurement by targeted high throughput plasma assays (e.g., ELISAs); C2 = 10% of C1 (2, 5, 10), the probability of success in the verification stage is provided in Table 2 (n2 = number of cases = number of controls = 25, 50, 100 and 250).
The determination of overall sample size and power can be accomplished by the product of probabilities from both discovery and verification stages. For example, a biomarker pipeline has 93% (= 93% × 100%) power, if the marker has a signal = 3.0 SDs in proximal fluids with 50% of cases expressing the marker, a sample size of 50 cases and 50 controls in discovery stage, and 20 candidates to be passed on for verification, followed by a reduced signal of 2 SDs separating cases from controls in plasma in verification stage with sample size of n = 100 cases and n=100 controls.
DISCUSSION
While previous efforts and publications have discussed the topic of statistical design in ‘omics’ studies in general [23, http://iom.edu/Reports/2012/Evolution-of-Translational-Omics.aspx], this manuscript introduces a statistical model applicable to the NCI-CPTAC or other multistage proteomics pipelines specifically for sample size calculation to achieve a reasonable probability of successfully passing a biomarker through discovery and verification to clinical validation. An 80% probability of reaching the clinical validation stage is attained by setting sample sizes to achieve 90% power for discovery and 90% power for verification stages. Factors that affect the required sample size include the separation between cases and controls, the fraction of cases producing the biomarker, and the number of targeted assays to be developed for verification and clinical validation stages. Moreover, we advocate the employment of quantitative criteria at each stage of the biomarker pipeline based on the clinical B/H ratio for the intended use of a diagnostic test. The goal of this approach is to judge whether it may be possible for the candidate to provide clinical utility (with a recommendation to discard candidates that do not pass this test).
Our statistical model takes multiple factors into account, including the distribution of plasma protein concentrations across a wide dynamic range, between person biological variation, measurement variation, etc. The distribution of plasma proteins between subjects based on results reported in the literature for the first 4 orders of magnitude of abundance in plasma protein concentrations has been published [24]. Extrapolating this model across a greater dynamic range provides the basis for simulating the distribution of low abundance plasma proteins. Due to their multiplicity, some proteins will appear to have significantly different distributions between the sample of cases and the sample of controls by chance, giving rise to false positive results within the simulations. For a biomarker with specified separation and proportion of cases producing it in excess, these false positive results can be overcome with (1) a greater number of targeted assays for either verification or validation, thereby increasing the chances the biomarker is passed to the next stage, or with (2) a greater number of case and control samples, thereby reducing the apparent signal in the spuriously identified candidates due to their multiplicity.
The properties of known clinical cancer biomarkers provide guidance on reasonable parameter ranges to expect for the biomarker to be discovered, such as the separation in SDs between cases and controls, and the proportion of cases for which a biomarker is overexpressed. Since known clinical cancer biomarkers are likely to have been detected because of their sufficiently strong signals (separation, % shed), it is assumed that novel cancer biomarkers will likely have less strong signals. Therefore, the parameters from known cancer biomarkers form upper bounds on the parameters for novel cancer biomarkers. With these models, we have simulated discovery and verification stages of a biomarker development pipeline, providing estimates of the probability of detecting a biomarker with a given set of properties as a function of sample sizes in each of the two stages.
If a true plasma biomarker exists with a specified separation of cases from controls, the modeling results show the strong effect of the number of clinical specimens on the probability of successfully passing the biomarker from discovery to verification stages, as shown in Table 1. For discovery, a sample size of n1 = 50 cases and 50 controls is required to have a high chance of detecting a (true) biomarker with a reasonable combination of attributes, expressed in a moderate fraction of cases (> 30%) and separated by ≥ 3 SDs in proximal fluids. The expectation is that new plasma ovarian cancer biomarkers will be expressed in a subset of cases ranging from 25–50%, and the separation between cases and controls will be modest -- on the order of 2–3 SDs for the more highly expressed biomarkers and ≥ 3 SDs for the less expressed biomarkers (e.g., 30% of cases). Under these plausible circumstances, having a high chance of passing a biomarker from discovery to verification requires sample sizes of 50 cases and 50 controls at discovery stage.
For true biomarkers separating cases from controls, the chance that they pass through verification to clinical validation stages is high (> 90%) for most combinations of separation (SDs between cases and controls) and fraction of cases producing the biomarker if the sample size is 250 cases vs. 250 controls, or some additional combinations (Table 2). With the sample size of 250/250, the chance of passing through is only < 90% for biomarkers where cases and controls are ≤ 2 SDs apart and the proportion of cases producing the biomarker is ≤ 10%; or where the cases and controls are very close (1 SD apart) and the proportion of cases producing the biomarker is ≤ 20%. If the separation is ≥ 2 SDs and the proportion of cases producing the biomarker is ≥ 20%, the chance of passing through verification stage exceeds 90%.
Finally, an important area not addressed in this report is replicability, i.e., repeating the same, or similar, experiments to determine if the proposed candidates retain the same signal across replicate measurement. The cost of repeating the same experiment is to reduce the sample size in half, thereby reducing the power of the study. Although there is surely a need for some replicability, it remains an open topic with respect to at which stage of the pipeline an independent repeat of the experiment is warranted. This issue clearly requires attention for each biomarker pipeline developed. We believe that there is a sweet spot between the extremes of ignoring replicability and addressing it at every stage of biomarker development with the consequent implications for workload and budget. This optimal solution is likely to be determined only through extensive experience with successful pipelines.
As a caveat, we advocate against publishing biomarker candidates identified only in the discovery stage with a lack of independent verification studies using an orthogonal technology, an orthogonal biospecimen type, or most appealing, a strongly unbiased sample set of control and case biospecimens. In such a biospecimen set, each sample is obtained prior to its case-control status being ascertained, thereby building unbiasedness into the sample set. However, a practical counterargument to this approach is that promising candidates identified during discovery may not be followed up if subsequent funding first requires publication. This balance must be left to individual investigators and journals. Our concern over publication of candidates identified only from the discovery stage is that the field of biomarker discovery has many published candidates, but very few verified candidates, and a miniscule number of clinically useful biomarkers. We believe that having the field focused on verified (not yet clinically validated) markers may lead to better investigations and more success with identifying clinical biomarkers. Further, the vast numerical gap between the numbers of “discovered” and clinically deployed biomarkers indicates a naïve or overly optimistic field. The proposed caveat may help correct this imbalance. Finally, clinical validation should not be claimed without first identifying an optimal panel, generating a classifier, locking down both panel and classifier, and applying the classifier to a separate biospecimen set obtained from subjects distinct from those whose biospecimens were obtained to form the classifier.
In conclusion, this statistical approach, as presented in the ovarian cancer case study, provides probabilities in Table 1 and Table 2, the product of which give the probability of true biomarkers passing through from discovery to verification, and ultimately to clinical validation. Given reasonable estimates on (1) the number of candidates examined at each stage; (2) the separation between cases and controls (SDs ranging from 1.0 to 5.0), and (3) the proportion of cases producing the biomarker (10%, 20%, 30%, 50%, 80%), the power of a study design for a biomarker to reach clinical validation is the product of the probabilities in Table 1 and Table 2. Any biomarker pipeline will have constraints on the number of candidates passed from one stage to the next, and the number of biospecimens capable of being interrogated at each stage. The method described herein assumes values for these parameters reasonable for current proteomic technologies with a detection level of ~ 8,000 proteins in the discovery stage, 20, 50, or 100 candidates measured in the verification stage, and 2, 5, or 10 candidates measured in the clinical validation stage. As technologies continue to improve, these parameters will likely increase, and the same simulation methodologies can be used to estimate probabilities for passing a candidate through the entire pipeline at each stage. The ultimate goal of this approach is to identify the appropriate number of biospecimens required to have a high probability of the biomarker reaching clinical validation stage.
Supplementary Material
Supplemental Methods
Figure 2. Distribution of Biological CV by Concentration Decade.

The biological CV, denoted by σ, is plotted against the concentration decade for the table of blood protein tests in Ricós, et al. [25]. A statistical regression model estimates the increasing expected level (blue line) and increasing variation (red lines 1 SD and 2 SDs) for σ as a function of concentration decade on the log scale. The model provides estimates for the variation of plasma proteins across the nine decades of concentration simulated for the power calculations.
Figure 4. Distribution and Separation of Cases versus Controls of CA125 in Blood.

For CA125, the typical median measurement in controls is 15 U/mL, while the typical median measurement is 100 U/mL in cases at diagnosis of late stage disease, providing a 6-fold increase, or an increase of 1.9 = log(100/15) on the log scale. With CA125 having an inter-person SD of 0.50 (~CV of 50%), this difference corresponds to a signal of 3.8 SDs. However, CA125 for ovarian cancers is one rare exception where its separation and ubiquity of expression enable it to be detected with relatively small sample sizes. The detection of other protein biomarker candidates would likely require an examination of the impact of sample sizes on discovery and verification of a signal ranging from 1, 2, 3, 4 and 5 SDs.
Abbreviations
NCI-CPTC
NCI’s Clinical Proteomic Technologies for Cancer initiative
CPTAC
Clinical Proteomic Tumor Analysis Consortium
NHLBI
National Heart, Lung and Blood Institute
AACC
American Association of Clinical Chemistry
FDA
US Food and Drug Administration
ESI
Electrospray Ionization
MRM-MS
Multiple Reaction Monitoring Mass Spectrometry
CID
Collisionally-induced Dissociation
SISCAPA
Stable Isotope Standard Capture with Anti-Peptide Antibodies
B/H
Benefits-to-Harms Ratio
ELISA
Enzyme-linked Immunosorbent Assay
IVD
In Vitro Diagnostics
RUO
Research Use Only
IUO
Investigational Use Only
PTMs
Post-translational Modifications
CLIA
Clinical Laboratory Improvement Amendments
SOP
Standard Operating Procedures
SD
Standard Deviation
CV
Coefficient of Variation
ReqSP
Required Specificity
PPV
Positive Predictive Value
References
- 1.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 2.Boja E, Rivers R, Kinsinger C, Mesri M, Hiltke T, Rahbar A, Rodriguez H. Restructuring proteomics through verification. Biomark Med. 2010;4(6):799–803. doi: 10.2217/bmm.10.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Regnier FE, Skates SJ, Mesri M, Rodriguez H, Tezak Z, Kondratovich MV, Alterman MA, Levin JD, Roscoe D, Reilly E, Callaghan J, Kelm K, Brown D, Philip R, Carr SA, Liebler DC, Fisher SJ, Tempst P, Hiltke T, Kessler LG, Kinsinger CR, Ransohoff DF, Mansfield E, Anderson NL. Protein-Based Multiplex Assays: Mock Presubmissions to the US Food and Drug Administration. Clin Chem. 2010;56(2):165–171. doi: 10.1373/clinchem.2009.140087. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez H, Tezak Z, Mesri M, Carr SA, Liebler DC, Fisher SJ, Tempst P, Hiltke T, Kessler LG, Kinsinger CR, Philip R, Ransohoff DF, Skates SJ, Regnier FE, Anderson NL, Mansfield E. Workshop Participants. Analytical Validation of Protein-Based Multiplex Assays: A Workshop Report by the NCI-FDA Interagency Oncology Task Force on Molecular Diagnostics. Clin Chem. 2010;56(2):237–243. doi: 10.1373/clinchem.2009.136416. [DOI] [PubMed] [Google Scholar]
- 5.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics. 2010;9(1):184–196. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Chan DW. The road from discovery to clinical diagnostics: lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiol Biomarkers Prev. 2010;19(12):2995–2999. doi: 10.1158/1055-9965.EPI-10-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bast RC, Jr, Skates S, Lokshin A, Moore RG. Differential diagnosis of a pelvic mass: improved algorithms and novel biomarkers. Int J Gynecol Cancer. 2012;22 (Suppl 1):S5–8. doi: 10.1097/IGC.0b013e318251c97d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gail MH, Pfeiffer RM, Wheeler W, Pee D. Probability that a two-stage genome-wide association study will detect a disease-associated SNP and implications for multistage designs. Ann Hum Genet. 2008;72(Pt 6):812–820. doi: 10.1111/j.1469-1809.2008.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satagopan JM, Venkatraman ES, Begg CB. Two-stage designs for gene-disease association studies with sample size constraints. Biometrics. 2004;60(3):589–597. doi: 10.1111/j.0006-341X.2004.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obuchowski NA, Hillis SL. Sample size tables for computer-aided detection studies. AJR Am J Roentgenol. 2011;197(5):W821–828. doi: 10.2214/AJR.11.6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obuchowski NA, Zhou XH. Prospective studies of diagnostic test accuracy when disease prevalence is low. Biostatistics. 2002;3(4):477–492. doi: 10.1093/biostatistics/3.4.477. [DOI] [PubMed] [Google Scholar]
- 12.Obuchowski NA. Sample size tables for receiver operating characteristic studies. AJR Am J Roentgenol. 2000;175(3):603–608. doi: 10.2214/ajr.175.3.1750603. [DOI] [PubMed] [Google Scholar]
- 13.Alonzo TA, Pepe MS, Moskowitz CS. Sample size calculations for comparative studies of medical tests for detecting presence of disease. Stat Med. 2002;21(6):835–852. doi: 10.1002/sim.1058. [DOI] [PubMed] [Google Scholar]
- 14.Boja ES, Jortani SA, Ritchie J, Hoofnagle AN, Težak Ž, Mansfield E, Keller P, Rivers RC, Rahbar A, Anderson NL, Srinivas P, Rodriguez H. The journey to regulation of protein-based multiplex quantitative assays. Clin Chem. 2011;57(4):560–567. doi: 10.1373/clinchem.2010.156034. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez H, Rivers R, Kinsinger C, Mesri M, Hiltke T, Rahbar A, Boja ES. Reconstructing the pipeline by introducing multiplexed multiple reaction monitoring mass spectrometry for cancer biomarker verification: an NCI-CPTC initiative perspective. Proteomics Clin Appl. 2010;4(12):904–914. doi: 10.1002/prca.201000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witkowska HE, Hall SC, Fisher SJ. Breaking the bottleneck in the protein biomarker pipeline. Clin Chem. 2012;58(2):321–323. doi: 10.1373/clinchem.2011.175034. [DOI] [PubMed] [Google Scholar]
- 17.Ransohoff D. How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J Clin Epidemiol. 2007;60(12):1205–1219. doi: 10.1016/j.jclinepi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 18.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham AJ, Keshishian H, Hall SC, Allen S, Blackman RK, Borchers CH, Buck C, Cardasis HL, Cusack MP, Dodder NG, Gibson BW, Held JM, Hiltke T, Jackson A, Johansen EB, Kinsinger CR, Li J, Mesri M, Neubert TA, Niles RK, Pulsipher TC, Ransohoff D, Rodriguez H, Rudnick PA, Smith D, Tabb DL, Tegeler TJ, Variyath AM, Vega-Montoto LJ, Wahlander A, Waldemarson S, Wang M, Whiteaker JR, Zhao L, Anderson NL, Fisher SJ, Liebler DC, Paulovich AG, Regnier FE, Tempst P, Carr SA. A Multi-site assessment of precision and reproducibility of multiple reaction monitoring-based measurements by the NCI-CPTAC network: toward quantitative protein biomarker verification in human plasma. Nat Biotechnol. 2009;27(7):633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallstrom G, Anderson KS, LaBaer J. Biomarker discovery for heterogeneous diseases. Cancer Epidemiol Biomarkers Prev. 2013;22(5):747–755. doi: 10.1158/1055-9965.EPI-12-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Addona TA, Shi X, Keshishian H, Mani DR, Burgess M, Gillette MA, Clauser KR, Shen D, Lewis GD, Farrell LA, Fifer MA, Sabatine MS, Gerszten RE, Carr SA. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat Biotechnol. 2011;29:635–643. doi: 10.1038/nbt.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I, Yan P, Schoenherr RM, Zhao L, Voytovich UJ, Kelly-Spratt KS, Krasnoselsky A, Gafken PR, Hogan JM, Jones LA, Wang P, Amon L, Chodosh LA, Nelson PS, McIntosh MW, Kemp CJ, Paulovich AG. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol. 2011;29(7):625–634. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerszten RE, Asnani A, Carr SA. Status and prospects for discovery and verification of new biomarkers of cardiovascular disease by proteomics. Circ Res. 2011;109(4):463–474. doi: 10.1161/CIRCRESAHA.110.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidal M, Chan DW, Gerstein M, Mann M, Omenn GS, Tagle D, Sechi S. Workshop Participants. The human proteome - a scientific opportunity for transforming diagnostics therapeutics and healthcare. Clin Proteomics. 2012;9(1):6–17. doi: 10.1186/1559-0275-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hortin GL, Sviridov D, Anderson NL. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin Chem. 2008;54(10):1608–1616. doi: 10.1373/clinchem.2008.108175. [DOI] [PubMed] [Google Scholar]
- 25.Ricós C, Alvarez V, Cava F, García-Lario JV, Hernández A, Jiménez CV, Minchinela J, Perich C, Simón M. Current databases on biological variation: pros, cons and progress. Scand J Clin Lab Invest. 1999;59(7):491–500. doi: 10.1080/00365519950185229. [DOI] [PubMed] [Google Scholar]
- 26.Hoofnagle AN, Becker JO, Wener MH, Heinecke JW. Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin Chem. 2008;54(11):1796–1804. doi: 10.1373/clinchem.2008.109652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJ, Pearson TW, Paulovich AG, Carr SA. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics. 2012;11(6):M111.013854. doi: 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 29.Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS, Kapp EA, Moritz RL, Chan DW, Rai AJ, Admon A, Aebersold R, Eng J, Hancock WS, Hefta SA, Meyer H, Paik YK, Yoo JS, Ping P, Pounds J, Adkins J, Qian X, Wang R, Wasinger V, Wu CY, Zhao X, Zeng R, Archakov A, Tsugita A, Beer I, Pandey A, Pisano M, Andrews P, Tammen H, Speicher DW, Hanash SM. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5(13):3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 30.Polanski M, Anderson NL. A list of candidate cancer biomarkers for targeted proteomics. Biomark Insights. 2007;1:1–48. [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson NL, Polanski M, Pieper R, Gatlin T, Tirumalai RS, Conrads TP, Veenstra TD, Adkins JN, Pounds JG, Fagan R, Lobley A. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol Cell Proteomics. 2004;3(4):311–326. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Haab BB, Geierstanger BH, Michailidis G, Vitzthum F, Forrester S, Okon R, Saviranta P, Brinker A, Sorette M, Perlee L, Suresh S, Drwal G, Adkins JN, Omenn GS. Immunoassay and antibody microarray analysis of the HUPO Plasma Proteome Project reference specimens: systematic variation between sample types and calibration of mass spectrometry data. Proteomics. 2005;5(13):3278–3291. doi: 10.1002/pmic.200401276. [DOI] [PubMed] [Google Scholar]
- 33.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmström J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10(9):M110.006353. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skates SJ, Pauler DK, Jacobs IJ. Screening based on the risk of cancer calculation from Bayesian hierarchical change-point and mixture models of longitudinal markers. J Am Stat Assoc. 2001;96(454):429–439. [Google Scholar]
- 35.Menon U, Gentry-Maharaj A, Hallett R, Ryan A, Burnell M, Sharma A, Lewis S, Davies S, Philpott S, Lopes A, Godfrey K, Oram D, Herod J, Williamson K, Seif MW, Scott I, Mould T, Woolas R, Murdoch J, Dobbs S, Amso NN, Leeson S, Cruickshank D, McGuire A, Campbell S, Fallowfield L, Singh N, Dawnay A, Skates SJ, Parmar M, Jacobs I. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancer: results of the prevalence screen of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS) Lancet Oncol. 2009;10(4):327–340. doi: 10.1016/S1470-2045(09)70026-9. [DOI] [PubMed] [Google Scholar]
- 36.Cordwell SJ, Edwards AV, Liddy KA, Moshkanbaryans L, Solis N, Parker BL, Yong AS, Wong C, Kritharides L, Hambly BD, White MY. Release of tissue-specific proteins into coronary perfusate as a model for biomarker discovery in myocardial ischemia/reperfusion injury. J Proteome Res. 2012;11(4):2114–2126. doi: 10.1021/pr2006928. [DOI] [PubMed] [Google Scholar]
- 37.Sedlaczek P, Frydecka I, Gabryś M, Van Dalen A, Einarsson R, Harłozińska A. Comparative analysis of CA125, tissue polypeptide specific antigen, and soluble interleukin-2 receptor alpha levels in sera, cyst, and ascitic fluids from patients with ovarian carcinoma. Cancer. 2002;95(9):1886–1893. doi: 10.1002/cncr.10917. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs I, Bast RC., jr The CA125 tumour-associated antigen: a review of the literature. Hum Reprod. 1989;4(1):1–12. doi: 10.1093/oxfordjournals.humrep.a136832. [DOI] [PubMed] [Google Scholar]
- 39.Altar CA, Amakye D, Bounos D, Bloom J, Clack G, Dean R, Devanarayan V, Fu D, Furlong S, Hinman L, Girman C, Lathia C, Lesko L, Madani S, Mayne J, Meyer J, Raunig D, Sager P, Williams SA, Wong P, Zerba K. A Prototypical Process for Creating Evidentiary Standards for Biomarkers and Diagnostics. Clin Pharmacol Ther. 2008;83(2):368–371. doi: 10.1038/sj.clpt.6100451. [DOI] [PubMed] [Google Scholar]
- 40.Jacobs IJ, Skates SJ, MacDonald N, Menon U, Rosenthal AN, Davies AP, Woolas R, Jeyarajah AR, Sibley K, Lowe DG, Oram DH. Screening for ovarian cancer: a pilot randomized controlled trial. Lancet. 1999;353(9160):1207–1210. doi: 10.1016/S0140-6736(98)10261-1. [DOI] [PubMed] [Google Scholar]
- 41.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 42.Wu L, Han DK. Overcoming the dynamic range problem in mass spectrometry-based shotgun proteomics. Expert Rev Proteomics. 2006;3(6):611–619. doi: 10.1586/14789450.3.6.611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods