FUS is Phosphorylated by DNA-PK and Accumulates in the Cytoplasm after DNA Damage (original) (raw)
Abstract
Abnormal cytoplasmic accumulation of Fused in Sarcoma (FUS) in neurons defines subtypes of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). FUS is a member of the FET protein family that includes Ewing's sarcoma (EWS) and TATA-binding protein-associated factor 2N (TAF15). FET proteins are predominantly localized to the nucleus, where they bind RNA and DNA to modulate transcription, mRNA splicing, and DNA repair. In ALS cases with FUS inclusions (ALS-FUS), mutations in the FUS gene cause disease, whereas FTLD cases with FUS inclusions (FTLD-FUS) do not harbor FUS mutations. Notably, in FTLD-FUS, all FET proteins accumulate with their nuclear import receptor Transportin 1 (TRN1), in contrast ALS-FUS inclusions are exclusively positive for FUS. In the present study, we show that induction of DNA damage replicates several pathologic hallmarks of FTLD-FUS in immortalized human cells and primary human neurons and astrocytes. Treatment with the antibiotic calicheamicin γ1, which causes DNA double-strand breaks, leads to the cytoplasmic accumulation of FUS, TAF15, EWS, and TRN1. Moreover, cytoplasmic translocation of FUS is mediated by phosphorylation of its N terminus by the DNA-dependent protein kinase. Finally, we observed elevated levels of phospho-H2AX in FTLD-FUS brains, indicating that DNA damage occurs in patients. Together, our data reveal a novel regulatory mechanism for FUS localization in cells and suggest that DNA damage may contribute to the accumulation of FET proteins observed in human FTLD-FUS cases, but not in ALS-FUS.
Keywords: amyotrophic lateral sclerosis (ALS), cytoplasmic translocation, DNA damage, frontotemporal lobar degeneration (FTLD), Fused in Sarcoma (FUS), phosphorylation
Introduction
Alterations in RNA metabolism are increasingly thought to be a primary driver of pathogenesis in many cases of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD; Ramaswami et al., 2013). Mutations in the genes FUS and TARDBP, which encode the RNA/DNA-binding protein Fused in Sarcoma (FUS) and TDP-43, respectively, cause familial and sporadic ALS and FTLD (Sreedharan et al., 2008; Van Deerlin et al., 2008; Kwiatkowski et al., 2009; Vance et al., 2009; Lagier-Tourenne et al., 2010). Further, TDP-43 inclusions are now considered a common marker of neuropathology in most ALS cases and in many cases of FTLD (Mackenzie et al., 2010). It is still unclear how mutations in FUS and TDP-43 cause disease (Lagier-Tourenne et al., 2010).
In ALS patients with FUS mutations, FUS accumulates in the cytoplasm as abnormal inclusions in neurons and glia. Most FUS mutations disrupt a C-terminal nuclear localization signal, which reduces binding and nuclear import by transportin-1 (TRN1), leading to increased cytoplasmic levels of FUS (Dormann et al., 2010; Lagier-Tourenne et al., 2010; Ito et al., 2011). It is thought that, over time, the increased levels of cytoplasmic FUS lead to the accumulation of FUS into inclusions (Dormann et al., 2010; Verbeeck et al., 2012).
Intriguingly, FUS-positive inclusions have been found in a subset of FTLD cases that are negative for Tau or TDP-43 inclusions. FTLD-FUS patients do not have FUS mutations and the mechanism leading to FUS pathology is unclear (Ravenscroft et al., 2013). A recent comparison of the neuropathology in ALS-FUS and FTLD-FUS cases has revealed differences. FTLD-FUS inclusions contain all FET members [FUS, Ewing's sarcoma (EWS), and TATA-binding protein-associated factor 2N (TAF15)], along with TRN1. In contrast, ALS-FUS inclusions contain exclusively FUS (Davidson et al., 2012). We have also observed a selective accumulation of FUS, but not EWS, TAF15, or TRN1 (unpublished data), in a mouse model of ALS-FUS (Verbeeck et al., 2012). These data suggest that the pathogenesis of ALS-FUS and FTLD-FUS cases may differ.
FUS, EWS, and TAF15 are multifunctional RNA/DNA-binding proteins that are widely expressed in most cell types and tissues. FUS is predominantly detected in the nucleus, although it can rapidly shuttle back and forth from the nucleus to the cytoplasm (Zinszner et al., 1997). Data from multiple studies suggest that cytoplasmic accumulation of FUS is a critical pathogenic event in FUS-related neurodegeneration (Bosco et al., 2010; Dormann et al., 2010; Gal et al., 2011; Kino et al., 2011; Verbeeck et al., 2012). Here, we report a novel mechanism that regulates the distribution of FUS between the nucleus and cytoplasm. We find that cytoplasmic accumulation of FUS is regulated by phosphorylation of the N terminus of FUS by the DNA-dependent protein kinase (DNA-PK). Further, induction of DNA damage leads to cytoplasmic translocation of FUS, EWS, TAF15, and TRN1, which mimics the pathologic changes that occur in FTLD-FUS cases. Together, these data suggest that DNA damage is a pivotal upstream event that may trigger the pathological changes leading to neurodegeneration and the unique neuropathology found in FTLD-FUS. Therefore, therapeutic strategies to reduce DNA damage or activate DNA repair pathways may be a viable strategy to treat neurodegeneration in FTLD-FUS cases.
Materials and Methods
Cell culture.
Human neuroglioma cells (H4; ATCC) and Human Embryonic Kidney 293T cells (HEK293T; ATCC) were cultured in OPTI-MEM medium plus 5% FBS and 1% penicillin-streptomycin. Human astrocytes and human neurons were purchased from Sciencell and cultured using protocols provided by the manufacturer. GM5849 and GM0637 cells (Henner and Blazka, 1986; Taira et al., 2010) were purchased from ATCC and were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin. M059J and M059K cells were obtained from Dr. Ya Wang in the Department of Radiation Oncology at Emory University (Atlanta, GA) with permission of Dr. Joan Allalunis-Turner in the Department of Oncology, University of Alberta (Edmonton, Alberta, Canada) (Allalunis-Turner et al., 1995; Lees-Miller et al., 1995).
HEK293T or H4 cells were plated in six-well plates and transfected with various cDNA constructs using TransIT LT1 (Mirus Bio) according to the manufacturer's instructions. A series of truncated FUS mutants were generated as described previously (Ito et al., 2011). Artificial genes for wild-type (WT) FUS and FUS mutants were synthesized by GeneArt (Life Technologies). These constructs were sequence optimized and incorporated a twin StrepII tag SA-WSHPQFEK(GGGS)2GGSAWSHPQFEK, followed by a FLAG tag on the N terminus to facilitate purification and detection. All consensus DNA-PK phosphorylation sites, serine or threnonine followed by a glutamine, were mutated to alanines (FUS Ser/Thr to Ala) or to asparates (FUS Ser/Thr to Asp). SiDNA transfection was performed as described previously with the following modification (Croset et al., 2013): HEK293T cells were transfected with 0.2 μm Dbait8H (control) or Dbait32Hc [inducer of the double-strand break (DSB) response and DNA-PK activity] with DharmaFECT (Thermo Scientific) overnight in OPTI-MEM with 5% FBS.
Staurosporine was purchased from Sigma. Calyculin A (Cal) was purchased from Cell Signaling Technology. KU 55933, KU 60019, NU 7441, and NU 7026 were purchased from Tocris Bioscience. Calicheamicin γ1 (CLM) was obtained from Pfizer.
Fractionation of cytoplasmic and nuclear proteins in cell lines and tissue.
H4 cells, HEK293T, and human astrocytes were lysed in 0.5% Triton X-100 lysis buffer (150 mm sodium chloride, 50 mm Tris, pH 8.0, and 0.5% Triton X-100) with 1% protease and phosphatase single-use inhibitor mixture (PPI; Thermo Scientific) on ice for 10 min. The cell suspension was centrifuged at 14,000 rpm for 15 min at 4°C. The supernatant was transferred into a new tube (cytoplasmic fraction). The pellet was washed again with 0.5% Triton X-100 lysis buffer, discarded, and the nuclear pellet was resuspended in RIPA buffer (150 mm sodium chloride 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mm Tris, pH 8.0) with 1% PPI. The suspension was sonicated and vortexed at 4°C. The suspension was centrifuged for 15 min at 14,000 rpm and 4°C. The supernatant (nuclear fraction) was saved. Successful subcellular fractionation was confirmed by absence of GAPDH in the nuclear fraction and absence of H3 in the cytoplasmic fraction. Biochemical fractionation of frozen frontal cortex from control and FTLD-FUS cases was performed using the Subcellular Protein Fractionation Kit for Tissue following the manufacturer's instructions (Thermo Scientific). FTLD-FUS cases were provided by three brain banks and were pathologically defined by the presence of frontotemporal lobar degeneration with inclusions positive for ubiquitin and FUS immunostaining and negative for tau or TDP-43 pathology. Some cases were assigned a subtype of FTLD-FUS: atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions (aFTLD-U), basophilic inclusion body disease (BIBD), or neuronal intermediate filament inclusion disease (NIFID) (Mackenzie et al., 2011). A total of 11 cases were analyzed from Emory University (FTLD-FUS, n = 1; FTLD-FUS/NIFID, n = 2), Harvard University (FTLD-FUS, n = 4), and Mayo Clinic Jacksonville (FTLD-FUS, n = 3; FTLD-FUS/NIFID, n = 1). Control tissue (n = 7) was from patients with no history of neurological disease and absence of other neuropathologies (i.e., infarcts, hemorrhages, amyloid, α-synuclein, tau).
FUS immunoprecipitation.
Cell lysates were immunoprecipitated with mouse anti-FUS (Santa Cruz Biotechnology) coupled to Dynabeads Sheep-Anti Mouse IgG followed the protocol provided by the manufacturer (Life Technologies). Bound material was then subjected to SDS-PAGE and Western blotting.
FUS purification and in vitro phosphorylation assay.
V5-tagged WT FUS was cloned into the pAG3 mammalian expression vector (DeJesus-Hernandez et al., 2010) and transfected into HEK293T cells. After 24 h, cells were lysed with RIPA buffer. Lysate was spin at 14,000 rpm for 10 min. The supernatant was load onto anti-V5 monoclonal antibody magnetic beads (MBL International) and eluted with 150 mm NH4OH, pH 10.5. The elution was concentrated with protein concentrators (PES, 3K MWCO; Pierce). Human recombinant DNA-PK was purchased from Life Technologies. The kinase reactions were performed in a final reaction volume of 18 μl of kinase buffer (5×; Invitrogen) with 2.5 μg/μl DNA-PK activator, 10U/μl DNA PK, 16 ng/μl FUS, 0.2 mm ATP, and 1 mm DTT. DNA-PK inhibitor (NU7026) was 10 μm. The reaction was performed at 37°C for 30 min and terminated by adding a ¼ volume of 4× sample loading buffer followed by heat inactivation.
Western blotting.
Cell lysates and protein samples were analyzed using SDS/PAGE followed by two-channel infrared (LI-COR) quantitative Western blots as described previously (Kukar et al., 2008; Kukar et al., 2011; Chen et al., 2013). Protein concentrations were measured by BCA assay (Pierce). The samples were denatured in 1× sample loading buffer at 70°C for 15 min. Equal amounts of protein were loaded into 12% Mini-PROTEAN TGX Precast Gels (Bio-Rad). After transferring to 0.2 μm nitrocellulose membranes, blots were blocked in blocking buffer (LI-COR) for 1 h and incubated with primary antibodies (diluted in blocking buffer and PBST; PBS plus 0.1% Tween 20) overnight at 4°C. Membranes were washed three times for 10 min in PBST and then incubated with IRDye 680RD or IRDye 800CW goat anti-mouse or anti-rabbit secondary antibodies (LI-COR) for 30 min. Membranes were then washed three times for 10 min and protein expression was visualized with the Odyssey Fc Imaging System (LI-COR). Antibodies to the following proteins were used: α-tubulin (1:20,000; Epitomics), Caspase-3 and cleaved Caspase-3 (1:1000; Cell Signaling Technology), EWS (1:1000; Epitomics), FUS (1:1000; Santa Cruz Biotechnology), FUS (1:2000; Sigma), Flag M2 (, 1:2500; Sigma), GAPDH (1:10,000; Sigma), H2AX (1:2000; Millipore), phospho-H2AX (p-H2AX, 1:2000; Millipore), H3 (1:5000; Millipore), ATM (1:1000, Cell Signaling Technology), Phos-ATM (1:1000; Cell Signaling Technology), Phos-DNA PK (1:1000; Abcam), Phos-P53 (1:1000; Cell Signaling Technology), Phospho-Serine/Threonine (1:500; Abcam), TAF15 (1:2500; Bethyl Lab), TDP-43 (1:2000; Proteintech), TRN1 (1:1000; Abcam), and V5 (1:5000; Invitrogen).
Immunofluorescence.
Cells were fixed with 4% paraformaldehyde for 15 min. After washing with PBS, cells were permeabilized with 0.1% Triton X-100 or 0.5% Tween 20. After blocking with 0.1% BSA, cells were immunostained with the following primary antibodies at the indicated concentrations: FUS (polyclonal, 1:500; Sigma), Flag M2 (monoclonal, 1:1000; Sigma), and MAP2 (polyclonal, 1:5000; Neuromics) at room temperature for 1 h. After washing with PBS, cells were incubated in secondary antibodies conjugated to Cy3 or fluorescein (1:500; Invitrogen) and DAPI (1:2500; Invitrogen). Slides were mounted using Vectashield Hard set. Images were collected with a Zeiss LSM 510 NLO META system or EVOS FL Cell Imaging System (Life Technologies).
Results
FUS is phosphorylated and translocates to the cytoplasm after treatment with staurosporine
Aside from TRN1-mediated import of FUS into the nucleus, little is known about the pathways responsible for shuttling FUS between cellular compartments or how these processes are regulated. To gain more insight into this process, we investigated pathways reported to affect TDP-43 localization because of their structural homology and the fact they are both mutated in ALS. In particular, staurosporine, a nonselective kinase inhibitor, has been reported to cause redistribution of TDP-43 from its nuclear localization to the cytoplasm (Zhang et al., 2007). In agreement with previous data, staurosporine treatment of human H4 neuroglioma cells for 3 h led to increased cytoplasmic levels of TDP-43 and smaller 35 and 25 kDa fragments that result from cleavage by Caspase-3 (Fig. 1C). Similar to TDP-43, we found increased cytoplasmic levels of FUS after staurosporine treatment (Fig. 1A,B). In contrast, we did not see cleavage of FUS; rather, we observed the appearance of a new, higher-molecular-weight, slower-migrating FUS band after Western blot analysis of the same cell lysates (Fig. 1C,D). The increase in the apparent molecular weight of FUS suggested to us that staurosporine treatment induced a posttranslational modification (PTM) on FUS. Because the appearance of this PTM correlated with the cytoplasmic translocation of FUS, we attempted to determine its identity.
Figure 1.

FUS translocates from the nucleus to the cytoplasm and is phosphorylated at serine and threonine residues after staurosporine treatment. Human H4 neuroglioma cells were treated with 1 μm staurosporine for 3 h. After fixing, cells were permeabilized with either Triton X-100 (A) or Tween 20 (B) and stained for FUS. Nuclei were counterstained with DAPI. Tween 20 does not permeabilize the nucleus, leading to exclusive staining of cytoplasmic FUS. A robust increase in staining of cytoplasmic FUS (arrows) after staurosporine treatment is observed with both permeabilization conditions. C, H4 cells were treated with 1 μm staurosporine for 3 h. Cytoplasmic and nuclear fractions were extracted and immunoblotted for FUS, TDP-43, GAPDH, and Histone-3 (H3). Full-length TDP-43 was cleaved into 35 and 25 KDa fragments. A slow-migrating band appeared above the major FUS band (asterisk) after staurosporine treatment (arrow; also see D). Staurosporine significantly increased levels of FUS and TDP-43 in the cytoplasm, as revealed by a decreased ratio of the amounts of either protein in the nucleus (Nuc) compared with the cytoplasm (Cyto). Error bars indicate mean ± SEM (n = 3). *p < 0.05. D, The cytoplasmic fraction of H4 cells was immunoblotted for TDP-43 and FUS. TDP-43 (asterisk) was cleaved into 35 and 25 KDa fragments (arrowheads). A slow-migrating band (arrow) appeared above the major FUS (asterisk) band after staurosporine treatment. E, A lysate from H4 cells treated with staurosporine was immunopreciptated using an anti-FUS antibody. The p-Ser/Thr antibody recognized the slow-migrating FUS band. F, After immnuopreciptation (IP) of FUS from cells treated with staurosporine, beads were incubated with FastAP Alkaline Phosphatase (AP; 100 U/ml) at 37°C for 2 h. AP treatment eliminated immunoreactivity of the p-FUS band with the p-Ser/Thr antibody. G, p-FUS is enriched in the cytoplasm after staurosporine treatment. Both cytoplasmic and nuclear fractions from H4 cells were immunoprecipated for FUS and immunoblotted (IB) with p-Ser/Thr antibody. In addition to p-FUS, the p-Ser/Thr antibody recognizes at least three unknown phosphorylated proteins (asterisks). Scale bar: 10 μm in A and B.
Next, we performed immunoprecipitation after staurosporine treatment to enrich for FUS, followed by a candidate approach to identify the FUS PTM. Ubiquitin is a common PTM involved in protein degradation and FUS inclusions in disease-affected tissue stain for ubiquitin; however, ubiquitin did not colocalize with the higher-molecular-weight FUS band (data not shown). FUS has also been reported to promote Ebp1 sumoylation; however, we found no evidence that staurosporine treatment leads to sumoylation of FUS (Oh et al., 2010; data not shown). Instead, we found that the upper FUS band was robustly colabeled with a phosphoserine/threonine (p-Ser/Thr) antibody, which broadly recognizes proteins phosphorylated at these residues (Fig. 1E). Alkaline phosphatase treatment of immunoprecipitated FUS eliminated colabeling with the p-Ser/Thr antibody, further supporting phosphorylation of FUS (p-FUS) as the PTM responsible for the observed shift in molecular weight (Fig. 1F). The highest levels of p-FUS after staurosporine treatment were found in the cytoplasmic fraction of H4 cells, suggesting that phosphorylation may regulate the translocation of FUS from the nucleus to the cytoplasm (Fig. 1G).
Multiple phosphorylation sites on the N terminus of FUS contribute to FUS translocation
To probe the specificity of FUS phosphorylation in more detail and to address whether this PTM may influence nuclear/cytoplasmic shuttling, we next investigated whether FUS was endogenously phosphorylated using a chemical approach. To locate the predominant sites of phosphorylation on FUS, we transfected a series of truncated FUS mutations into HEK293T cells (Ito et al., 2011). Cells were then treated with calyculin A (Cal), a specific inhibitor of protein phosphatases PP1 and PP2A, lysed, and analyzed by Western blot. WT FUS and seven of the FUS deletion mutations underwent a marked increase in molecular weight after Cal treatment, indicating that they are phosphorylated under these conditions (Fig. 2A,B). FUS immunoprecipitated from cells treated with Cal displayed a marked shift in molecular weight and was robustly immunoreactive with the p-Ser/Thr antibody, both of which could be eliminated by treating samples with alkaline phosphatase (Fig. 2C). These data further verify that the upper band of FUS represents p-FUS. Deletion of the N-terminal domain of FUS was the only mutation that prevented the large shift in molecular weight, implicating this region as the primary site of phosphorylation (Fig. 2B).
Figure 2.

The phosphorylation sites responsible for the shift in FUS molecular weight are located at N-terminal serine/threonine residues and contribute to the translocation of FUS. A, Schematic diagram showing the domain of deletions introduced into FUS-V5 constructs (Ito et al., 2011). B, FUS-V5 constructs were transfected into HEK293T cells and whole-cell lysates were analyzed with indicated antibodies. When cells were treated with Cal (100 nm), a specific inhibitor of protein phosphatases, all mutants except for the N-terminal deletion mutant (ΔNT) showed a near complete shift of FUS to a higher-molecular-weight, slower-migrating species, indicating FUS phosphorylation (arrow). Endogenous TDP-43 showed only a weak shift to a slower migrating species. C, H4 cells were treated with Cal (100 nm), lysed, and immunopreciptated (IP) with an anti-FUS antibody. The anti-p-Ser/Thr anbitody colocalized with the slow-migrating p-FUS band in the input and after IP (arrow). After IP of FUS, beads were incubated with alkaline phosphatase (AP; 100U/ml) at 37°C for 2 h. AP treatment eliminated the p-Ser/Thr reactive band and reversed the shift in FUS migration. D, The FUS ΔNT construct was transfected into H4 cells and treated with Cal (100 nm). The cytoplasmic and nuclear fractions were extracted and analyzed with indicated antibodies. Cal treatment led to robust phosphorylation (p-FUS) and cytoplasmic translocation of endogenous FUS, but not TDP-43. N-terminal deletion of FUS blocked the phosphorylation and translocation of FUS. Statistical comparisons were performed using paired two-tailed t test. Error bars indicate mean ± SEM (n = 3). **p < 0.01.
We next investigated whether phosphorylation of FUS contributes directly to cytoplasmic translocation of FUS. Cal treatment increased levels of FUS in the cytoplasm at the expense of nuclear FUS, whereas deletion of the N terminus of FUS blocked cytoplasmic translocation (Fig. 2D). We noted a slight shift in the migration of TDP-43 after Cal treatment; however, this had no significant effect on the nuclear/cytoplasmic ratio of TDP-43. Together, these data provide further evidence that phosphorylation of FUS on the N terminus regulates cytoplasmic levels of the protein. In contrast, we find that TDP-43 is phosphorylated, in agreement with previous reports; however, phosphorylation of TDP-43 does not appear to play a large role in regulating its cellular localization (Hasegawa et al., 2008; Inukai et al., 2008; Neumann et al., 2009c).
FUS phosphorylation occurs after DNA damage
Our discovery that staurosporine treatment causes phosphorylation of FUS raises an interesting paradox because staurosporine is a broad and nonselective kinase inhibitor (Karaman et al., 2008). Moreover, staurosporine induces apoptosis in many cell lines through multiple mechanisms, including caspase-dependent and caspase-independent pathways, and it was not immediately clear which pathway led to FUS phosphorylation (Belmokhtar et al., 2001). To begin to answer this question, we used a bioinformatics approach to search for consensus phosphorylation sites on the N terminus of FUS. Based on two kinase prediction servers, KinasePhos 2.0 and GPS 2.1, the Ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related protein (ATR), and DNA-PK were ranked as the most likely kinases to phosphorylate serine and threonine residues on FUS (Wong et al., 2007; Xue et al., 2008; Roos and Kaina, 2013). ATM, ATR, and DNA-PK are members of the atypical phosphatidylinositol-3 kinase-related kinase (PIKK) family and are intimately involved in the response to, and repair of, DNA damage (Lempiäinen and Halazonetis, 2009; Roos and Kaina, 2013). The prediction that these kinases can phosphorylate FUS implied that DNA damage may be an initiating event that leads to the molecular weight shift in FUS and redistribution to the cytoplasm that we observed. In support of this idea, staurosporine treatment of SH-SY5Y human neuroblastoma cells has been reported to activate DNA-PK before apoptosis (Chakravarthy et al., 1999).
To test this hypothesis, we investigated whether inducing DNA damage directly would lead to FUS phosphorylation. We treated cells with calicheamicin γ1 (CLM), an antibiotic that cleaves DNA and specifically induces the DNA DSB response (Elmroth et al., 2003; Mårtensson et al., 2003; Muslimović et al., 2009). H4 cells treated with CLM for 3 h showed a potent, dose-dependent shift in FUS migration that correlated with the appearance of a p-Ser/Thr-immunoreactive band that overlapped with the upper FUS band (p-FUS; Fig. 3A). At these doses and time points, CLM treatment led to negligible activation of Caspase-3 or cleavage of TDP-43 (Fig. 3A,B). In contrast, staurosporine treatment led to a dose-dependent activation of Caspase-3 and cleavage of TDP-43 into a 35 kDa fragment, but less effective FUS phosphorylation compared with CLM administration (Fig. 3C,D). Multiple markers of DNA damage are induced after CLM treatment, including phosphorylation of ATM, DNA-PK, P53, and H2AX (Fig. 4). Moreover, at the highest dose, CLM caused a near complete conversion of FUS to p-FUS (Fig. 3A,C). Therefore, phosphorylation of FUS is a downstream event after DNA damage that can be separated from caspase activation and apoptosis.
Figure 3.
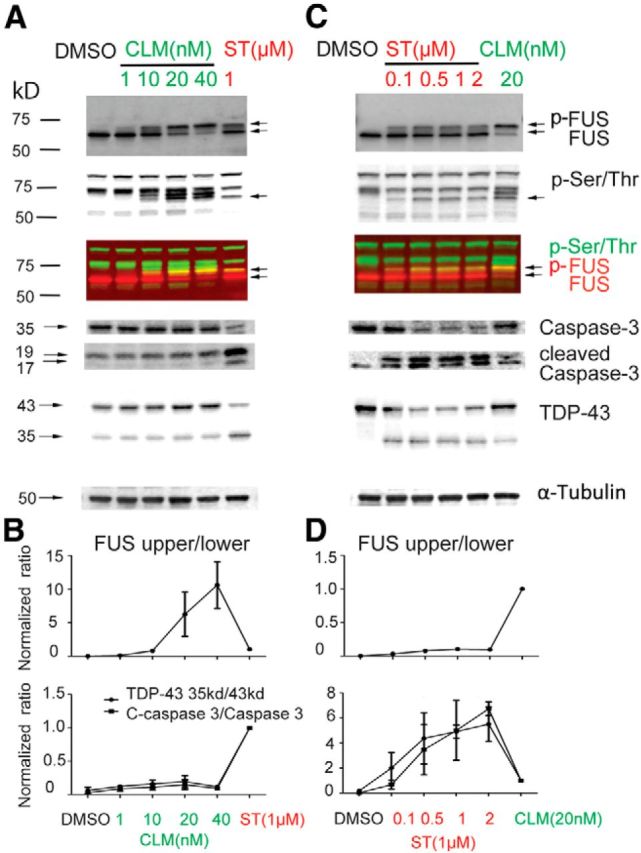
CLM, which causes DNA damage, is a potent inducer of FUS phosphorylation, but does not activate Caspase-3 or induce TDP-43 cleavage. H4 cells were treated with increasing concentrations (nm) of CLM, which causes DNA damage through DNA breaks and staurosporine (ST; μm) for 3 h. A, C, Cell lysates were harvested for SDS-PAGE and analyzed by immunoblotting with indicated antibodies. B, Phosphorylation of FUS (p-FUS; upper band) and cleavage of TDP-43 and Caspase-3 at different concentration of CLM were normalized to ST treatment (1 μm). D, Phosphorylation of FUS and cleavage of TDP-43 and Caspase-3 at different concentration of ST were normalized to CLM treatment (20 nm). Error bars indicate mean ± SEM (n = 3).
Figure 4.

FUS is phosphorylated by DNA-PK. A, Time course of FUS phosphorylation after CLM treatment. HEK293T cells were treated with CLM (10 nm) at indicated time points (minutes), harvested, and protein extracts were immunoblotted for FUS, TDP-43, and GAPDH. Arrow highlights p-FUS. B, FUS phosphorylation (p-FUS) after CLM treatment was not dependent on CHK1 and CHK2 activation. HEK293T cells were preincubated with DMSO (vehicle control), CHK1 inhibitor PF47736, CHK2 inhibitor, ATM inhibitors (KU60019 and KU55933), or DNA-PK inhibitors (NU7441 and NU7026) at the indicated concentration before treatment with CLM (10 nm) for 2 h. Total protein extracts were subjected to SDS/PAGE and analyzed by Western blot with the indicated antibodies. The CHK1 inhibitor PF47736 and a CHK2 inhibitor did not affect the phosphorylation of FUS after CLM treatment. The ATM inhibitor KU 60019 weakly inhibited p-ATM and partially blocked the CLM-induced phosphorylation of FUS, but also displayed nonselective inhibition of p-DNA-PK. The ATM inhibitor KU 55933 was ineffective. In contrast, two selective DNA-PK inhibitors, NU 7026 and NU 7441, robustly inhibited FUS and DNA-PK phosphorylation. Asterisks denote “non-FUS” phosphorylated proteins that are also induced by CLM treatment and detected by the pan p-Ser/Thr antibody. p-FUS migrates immediately below these proteins (asterisk) and is highlighted with an arrow. C, HEK293T cells were pretreated with indicated concentrations of NU7026, a DNA-PK inhibitor, for 30 min and then dosed with 10 nm CLM for 2 h. Cell lysates were harvested and analyzed with indicated antibody. NU7026 leads to dose-dependent inhibition of DNA-PK and blocked phosphorylation of FUS without inhibiting activation of DNA damage (p-H2AX) or ATM. D, V5-tagged FUS was overexpressed in HEK293T cells and purified with V5 magnetic beads. The purified FUS was incubated with recombinant DNA-PKcs, NU7026, or both. DNA-PKcs phosphorylates FUS in vitro and is inhibited by NU7026. E, CLM treatment of ATM-deficient (GM5849; ATM−/−) or control (GM0637; WT) human fibroblasts produces robust phosphorylation of FUS. F, In contrast, there is no phosphorylation of FUS in the DNA-PK-deficient human glioblastoma cell line (M059J; DNA-PK−/−) after CLM compared with a control fibroblast cell line (M059K; WT).
DNA-PK phosphorylates FUS
Although it was evident that treatment with CLM and staurosporine can induce FUS phosphorylation, it was unclear what kinase was responsible for this effect. DNA damage activates the checkpoint kinase signaling network, which leads to widespread phosphorylation of initiating kinases and effector proteins that mediate DNA damage repair (Matsuoka et al., 2007; Summers et al., 2011). We used inhibitors of components of this pathway to determine the primary kinase or kinases that lead to FUS phosphorylation. First, we investigated the time dependence of FUS phosphorylation. After treatment of cells with 10 nm CLM, p-FUS was detectable by 60 min and reached a maximum at 2 h, which was used in all subsequent experiments (Fig. 4A). Incubation with CHK1 or CHK2 kinase inhibitors had no effect on phosphorylation of FUS after DNA damage induced by CLM treatment, suggesting that a kinase further upstream was responsible (Fig. 4B). One ATM inhibitor (KU 60019) partially blocked the CLM-induced phosphorylation of FUS, whereas another (KU 55933) was ineffective (Fig. 4B). Furthermore, when cells were pretreated with KU 60019 followed by CLM, phosphorylation of ATM was only partially blocked and DNA-PK phosphorylation was inhibited, suggesting that it is not selective to ATM kinase. In contrast, two selective DNA-PK inhibitors, NU 7026 and NU 7441, robustly inhibited FUS phosphorylation and the observed shift in molecular weight (Fig. 4B). Inhibition of FUS phosphorylation by NU 7026 was dose dependent and appeared specific to DNA-PK because the levels of ATM phosphorylation remained steady after CLM treatment (Fig. 4C). To determine whether DNA-PK can phosphorylate FUS directly, we performed in vitro kinase assays with V5-tagged FUS purified from HEK293T cells and recombinant DNA-PK. FUS was robustly phosphorylated by recombinant DNA-PK and caused a shift in the molecular weight of FUS (Fig. 4D), similar to the increase in molecular weight we observed in cells treated with CLM (Fig. 4A). Further, the phosphorylation of FUS by DNA-PK was blocked by NU 7441, the same DNA-PK inhibitor that was effective in cell culture. These data demonstrate that FUS can be phosphorylated directly by DNA-PK.
Next, we took a genetic approach to access whether DNA-PK was the primary kinase that phosphorylates FUS after DNA damage in cells. To assess the contribution of ATM to FUS phosphorylation, we used an ATM-deficient fibroblast cell line (ATM−/−; GM5849) derived from an ataxia-telangiectasia patient (Henner and Blazka, 1986; Taira et al., 2010). FUS was phosphorylated at similar levels in response to CLM treatment in both the ATM-deficient and a WT control fibroblast, demonstrating that lack of ATM has no effect on the production of p-FUS after DNA damage (Fig. 4E). Next, we compared the effect of CLM on FUS in a human glioma cell line deficient in DNA-PK activity (DNA-PK−/−; M059J) compared with a control glioma cell line with normal DNA-PK activity (WT; Allalunis-Turner et al., 1995; Lees-Miller et al., 1995). Similar to HEK293T and H4 cells, CLM caused a dose-dependent phosphorylation of FUS in the WT line (Fig. 4F). In contrast, we found no shift in the molecular weight of FUS or production of p-FUS in the DNA-PK−/− cell line after treatment with CLM. DSBs were induced by CLM in DNA-PK−/− cells, as detected by phosphorylation of H2AX and p53, suggesting that it is the lack of DNA-PK activity, not a compromised DNA damage response, that is responsible for the absence of FUS phosphorylation. In total, these data provide compelling evidence that DNA-PK mediates phosphorylation of FUS in cells after DNA damage.
Consensus DNA-PK phosphorylation sites mediate cytoplasmic translocation of FUS
The data presented thus far strongly support the idea that phosphorylation of FUS regulates cytoplasmic translocation; however, they are primarily correlative data. To test the functional impact of FUS phosphorylation on cellular localization directly, we made a series of mutations at predicted DNA-PK phosphorylation sites in the N terminus (1–165 aa) of FUS (Fig. 2A). DNA-PK preferentially phosphorylates serine or threonine residues followed by a glutamine (S/T-Q; Kim et al., 1999). There are a total of 12 S/T-Q sites on the N-terminal region of FUS (1–165 aa; Fig. 5A). To access their role in FUS cytoplasmic translocation, we used gene synthesis to generate FUS constructs with all S/T residues mutated to alanine (FUS-Ala), which should block DNA-PK phosphorylation, or mutated to aspartate (FUS-Asp), to mimic the negative charge state of phosphorylation (Fig. 5A; Pearlman et al., 2011). WT-FUS and the S/T-Q mutant FUS constructs were then transfected into HEK293T cells and treated with CLM. As shown previously, WT-FUS was robustly phosphorylated and shifted in molecular weight (Fig. 5B). In contrast, the FUS-Ala mutant was resistant to the effect of CLM treatment and did not display the large shift in molecular weight characteristic of FUS phosphorylation. Moreover, even without CLM treatment, the FUS-Asp mutant behaved like phosphorylated WT-FUS by migrating at a higher molecular weight on SDS/PAGE gels (Fig. 5B). Importantly, the phosphomimetic FUS (FUS-Asp) accumulated at higher levels in the cytoplasm as measured by both biochemical fractionation and Western blot analysis (Fig. 5C,D) or immunofluorescence (Fig. 5E). CLM treatment did not further shift the molecular weight of FUS-Asp or FUS-Ala, suggesting that the predominant DNA-PK sites had been mutated to aspartate or alanine residues, respectively (Fig. 5B,F). Furthermore, CLM treatment did not significantly alter the nuclear/cytoplasmic ratio of the FUS-Asp or FUS-Ala mutants (Fig. 5F,G). These data strongly suggest that the phosphorylation of serine or threonine residues on the N terminus of FUS by DNA-PK regulates the transport of FUS between the nucleus and cytoplasm directly.
Figure 5.

Mutation of consensus DNA-PK sites on FUS blocks CLM-induced phosphorylation and confirms their role in cytoplasmic translocation of FUS. A, FUS contains 12 consensus DNA-PK (S/T-Q) sites on the N terminus. All serine or threonine residues were mutated to alanine (Ala) or aspartate (Asp). B, The FUS WT, Ala mutation, and Asp mutation were transfected into HEK293T cells. Twenty-four hours after transfection, cells were treated (+) or not treated (−) with 10 nm CLM for 2 h. Exogenous Flag-FUS was detected with anti-Flag antibody. Membrane was reprobed with p-H2AX and H2AX antibodies. FUS-Ala prevents CLM induced phosphorylation. FUS-Asp migrates similar to phosphorylated WT-FUS. C, FUS WT, Ala mutation, and Asp mutation were transfected into HEK293T cells. Twenty-four hours after transfection, cytoplasmic and nuclear fractions were extracted and immunoblotted with Flag antibody and antibodies against the other two FET proteins, EWS and TAF15. GAPDH and H3 were used as cytoplasmic and nuclear markers, respectively, to verify extraction fidelity. D, Samples shown in C were quantified and the ratio of nuclear to cytoplasmic protein calculated as described in Materials and Methods. The FUS-Asp mutation is found basally at higher levels in the cytoplasm (decreased nuclear/cytoplasmic ratio), but has no effect on endogenous EWS and TAF15. Error bars indicate mean ± SEM (n = 3). **p < 0.01, one-way ANOVA. E, WT-FUS and the FUS-Asp mutant were transfected into HEK293T cells. Twenty-four hours after transfection, cells were fixed and permeabilized with Tween 20, which primarily reveals cytoplasmic proteins, and stained with anti-Flag antibody. FUS-Asp is detected at higher levels in the cytoplasm compared with WT-FUS. Scale bar indicates 10 μm. F, The FUS-Ala mutation and FUS-Asp mutation were transfected into HEK293T cells. Twenty-four hours after transfection, cells were treated with 10 nm CLM for 2 h. Cytoplasmic and nuclear fractions were extracted and immunoblotted with Flag antibody. GAPDH and H3 were used as cytoplasmic and nuclear markers, respectively, to verify extraction fidelity. G, Samples shown in F were quantified and the ratio of nuclear to cytoplasmic protein calculated. CLM did not lead to a significant translocation of FUS-Ala or FUS-Asp. Error bars indicate mean ± SEM (n = 3).
CLM treatment increases cytoplasmic translocation of all FET proteins and TRN1
Because the entire FET protein family has been reported to coaggregate with FUS in the pathologic inclusions found in FTLD-FUS cases (Neumann et al., 2011), we wondered whether DNA damage had a similar effect on their cellular localization. Indeed, CLM treatment significantly increased the amount of FUS and TAF15 in the cytoplasm, which was blocked by a DNA-PK inhibitor, NU7026 (Fig. 6A,B). EWS showed a similar trend, but did not translocate as robustly as FUS and TAF15 and did not reach statistical significance. TRN1, the protein that imports the FET proteins into the nucleus and has been reported in FTLD-FUS inclusions (Neumann et al., 2011), also increased in the cytoplasm and decreased in the nucleus after CLM treatment (Fig. 6C). We next investigated whether FET cytoplasmic translocation was due to activation of the DNA damage response and DNA-PK by using a small molecule called Dbait32Hc. Dbait32Hc is a short, stabilized DNA molecule (Dbait) that mimics DNA DSBs, leading to specific and reliable activation of DNA-PK in numerous cell lines (Quanz et al., 2009a; Quanz et al., 2009b). Transfection of Dbait32Hc into HEK293T induced p-H2AX, a marker of DSB, and caused the phosphorylation and cytoplasmic translocation of FUS and TAF15 (Fig. 6D). EWS showed the same trend but did not reach significance. Dbait8H, a control DNA molecule that does not induce the DSB response, had no effect on FET proteins. Therefore, an alternative method to induce the cellular DNA damage response to DSBs, Dbait32Hc, has the same effect on FUS phosphorylation and cytoplasmic translocation of FET proteins.
Figure 6.
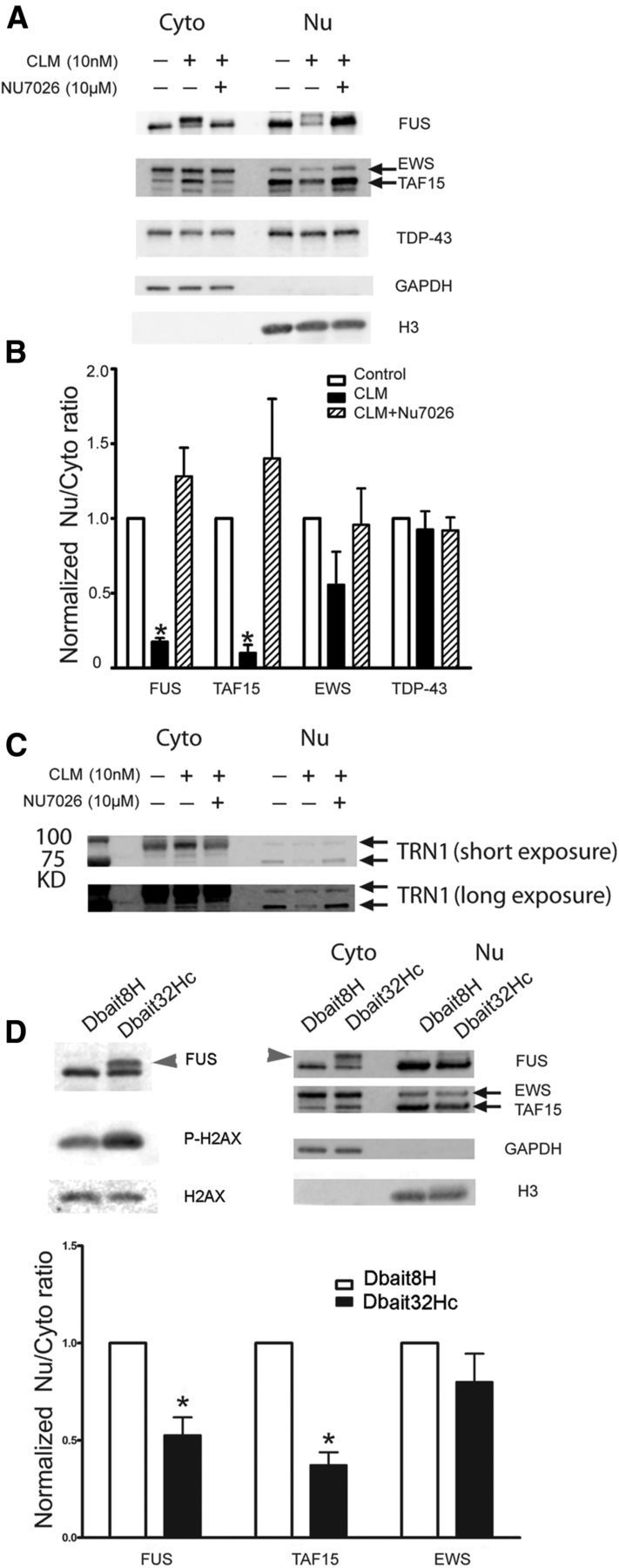
DNA DSBs lead to DNA-PK-dependent translocation of all FET proteins and TRN. A, HEK293T cells were pretreated with DNA-PK inhibitor NU7026 (10 μm, 30 min) before incubation with 10 nm CLM for 2 h. Cytoplasmic and nuclear fractions were extracted and immunoblotted for the FET proteins FUS, TAF15, and EWS. B, CLM significantly decreased the nuclear/cytoplasmic ratio of FUS and TAF15, whereas EWS was decreased but did not reach significance. However, there was no effect on TDP-43 translocation. Error bars indicate mean ± SEM (n = 3). *p < 0.05, one-way ANOVA. C, HEK293T cells were pretreated with the DNA-PK inhibitor NU7026 (10 μm, 30 min) before incubation with 10 nm CLM for 2 h. Cytoplasmic and nuclear fractions were extracted and immunoblotted for TRN1. CLM treatment increased TRN1 in the cytoplasmic fraction and decreased it in the nuclear fraction. This redistribution was blocked by pretreatment with NU7026. D, Dbait32Hc leads to phosphorylation and translocation of FUS and TAF15. HEK293T cells were transfected with 0.2 μm siDNA Dbait8H and Dbait32Hc overnight. Total proteins (left) and the cytoplasmic (Cyto) and nuclear (Nu) fractions were extracted and analyzed with the indicated antibodies. Dbait32Hc treatment led to robust phosphorylation of FUS (arrowhead) and translocation of FUS and TAF15 and EWS showed the same trend. Error bars indicate mean ± SEM (n = 3). *p < 0.05.
CLM treatment leads to FUS phosphorylation and translocation in cultured human astrocytes and primary neurons
Because neurons and glia are the site of FET protein inclusions in FTLD-FUS, we next investigated whether phosphorylation of FET proteins occurred in primary cells from the nervous system. Treatment of human astrocytes with CLM caused a strong phosphorylation of FUS and translocation of both FUS and TAF15 to the cytoplasm (Fig. 7A,B). In primary human cortical neurons, CLM treatment robustly induced phosphorylation of FUS (Fig. 7C). Further, immunofluorescence revealed that FUS translocated from the nucleus to the cytoplasm of the neuronal cell body and dendrites after induction of DNA damage after CLM treatment (Fig. 7D). Next, we investigated how long FUS remained phosphorylated after DNA damage. Human neurons were treated with medium containing CLM for 2 h, and then the medium was removed and replaced with complete medium and neurons were cultured for another 22 h. As observed previously, FUS was phosphorylated after CLM treatment for 2 h; however, 22 h later, the levels of FUS phosphorylation had decreased (Fig. 7C). DNA damage was still evident at this time point based on p-H2AX levels, suggesting that phosphorylation of FUS after DNA damage is a transient event.
Figure 7.

FUS is phosphorylated and translocated to the cytoplasm after DNA damage in human astrocytes and neurons. A, Cultured human astrocytes were treated with DMSO (vehicle) or 10 nm CLM for 2 h. Cell lysates (Input) were immunoprecipitated (IP) with a FUS antibody and samples analyzed by Western blot with an anti-FUS and p-Ser/Thr antibody. p-FUS (arrow) is detected by the p-Ser/Thr antibody and migrates below an unknown phosphorylated protein (asterisk). The quick Western IRDye 680RD (LI-COR) was used as a secondary to eliminate nonspecific detection of heavy and light chains from the antibody used for FUS IP. B, Human astrocytes were treated with CLM (10 nm) and the cytoplasm (Cyto) and nuclear (Nu) fractions were isolated and immunoblotted with indicated antibodies in two independent experiments. FUS and TAF15 showed a robust translocation from the nucleus to the cytoplasm after CLM treatment. C, Primary human neurons were treated with CLM at the indicated times and concentrations and analyzed with the indicated antibodies. FUS showed dose- and time-dependent phosphorylation in response to CLM treatment, as detected by a shift in molecular weight detected with a FUS C-terminal (C-T) or midregion (M-R) antibody. When the cells were treated with CLM for 2 h and cultured an additional 22 h in normal medium without CLM, the p-FUS species collapsed to the lower-molecular-weight band, suggesting that FUS phosphorylation is transient even with persistent p-H2AX activation. No cleavage of TDP-43 was observed. D, Human neurons were treated with 10 nm CLM for 3 h and stained with FUS and MAP-2 antibodies. Nuclei were counterstained with DAPI. CLM treatment increased cytoplasmic distribution (arrow) and dendritic distribution (arrowhead) of FUS. Scale bar, 10 μm.
Evidence of DNA damage in human FTLD-FUS cases
Our data imply that DNA damage could be the pathogenic trigger that initiates the cytoplasmic accumulation of FET proteins that are observed in FTLD-FUS cases. To investigate this possibility, we analyzed the accumulation of FUS and DNA damage in tissue from neuronal intermediate filament inclusion disease (NIFID), which is a subtype of FTLD-FUS (Neumann et al., 2009a). The frontal cortices of two NIFID and two control cases from the Emory Center for Neurodegenerative Disease brain bank were biochemically fractionated into subcellular components and analyzed on a Western blot. FUS was dramatically increased in the insoluble fraction of FTLD-FUS-NIFID cases compared with control, in agreement with previous studies (Fig. 8A; Neumann et al., 2009b; Lashley et al., 2011; Neumann et al., 2011). To determine whether this finding applied to other FTLD-FUS cases, we repeated this analysis on tissue from additional control and FTLD-FUS cases provided by the Harvard and Mayo Clinic brain banks and observed the selective accumulation of FUS in the final, insoluble pellet (Fig. 8A). We did not observe a high-molecular-weight species of FUS equivalent to the heavily phosphorylated species that is induced in cultured cells treated with CLM (Fig. 8A). However, phosphorylated H2AX is significantly increased in FTLD-FUS cases compared with controls (Fig. 8B). Because p-H2AX (also known as γH2AX) is a widely used marker of DNA DSBs, these data supports the hypothesis that DNA damage occurs in FTLD-FUS tissue and may be linked to disease pathogenesis (Sharma et al., 2012).
Figure 8.

Increased insoluble FUS and a marker of DNA damage, p-H2AX, in FTLD-FUS cases. A, Subcellular fractionation of seven controls and 11 FTLD-FUS cases from the Harvard, Mayo Clinic, and Emory University brain banks was performed using the Subcellular Protein Fractionation Kit (Pierce) according to the manufacturer's instructions and analyzed by SDS/PAGE and Western blot. The final “insoluble” pellet from this procedure was probed for FUS, revealing that FUS accumulated in the insoluble fraction in FTLD-FUS cases compared with control (top blot). The chromatin-bound nuclear extract fractions (bottom three blots) were probed with antibodies against p-H2AX (γH2AX) and a total H2AX antibody. The signals for p-H2AX and t-H2AX were separately detected in 800 nm (green) and 700 nm (red) channels, respectively, and were used to calculate the ratio of p-H2AX to t-H2AX (P/T H2AX). B, The ratio of P/T H2AX was significantly increased in FTLD-FUS cases in the chromatin-bound fraction, indicating that these cases may have increased DNA damage. Error bars indicate mean ± SEM. ***p < 0.001.
Discussion
FUS and TDP-43 are related RNA/DNA-binding proteins that have been found recently to abnormally accumulate in ALS, FTLD, and an increasing number of other neurodegenerative conditions (Mackenzie et al., 2010). Immunohistochemically, both FUS and TDP-43 are predominantly observed in the nucleus in normal tissue; however, they accumulate into insoluble neuronal and glial cytoplasmic inclusions in tissue affected by disease. Despite their structural and functional similarity, it is widely thought that FUS and TDP-43 do not co-accumulate in the same inclusions from affected tissue (Mackenzie et al., 2010). Understanding the pathways that regulate the cytoplasmic accumulation of FUS and TDP-43, whether they are shared or unique, is an important goal to help understand the normal biological function of these proteins and to determine what goes wrong in diseases in which they form pathologic inclusions.
In the present study, we report the novel observation that FUS is phosphorylated on the N terminus and that phosphorylation leads to translocation of FUS from the nucleus to the cytoplasm. Using multiple experimental paradigms, we have shown that DNA-PK is the kinase most likely to be responsible for phosphorylation of FUS after DNA damage. There is one report in the literature that FUS can be phosphorylated after DNA damage, supporting our overall conclusion that FUS is phosphorylated and translocated to the cytoplasm in response to DNA damage (Gardiner et al., 2008). However, the investigators of that study concluded that ATM mediates phosphorylation of FUS after ionizing radiation. It is possible that ATM is indeed responsible for the initial phosphorylation of serine 42 on FUS as reported, but DNA-PK phosphorylates additional serine and threonine sites, which cause the dramatic shift in FUS molecular weight that we observed. However, our finding that ATM-deficient cell lines still phosphorylate FUS robustly after DNA damage argues against this possibility (Fig. 4E).
Using site-directed mutagenesis of the DNA-PK sites on the N terminus of FUS, we have generated compelling evidence that phosphorylation of FUS directly influences its distribution between the nucleus and cytoplasm, although the exact molecular mechanism is unclear. TRN1 binding is one well known pathway that leads to the nuclear import of FUS (Dormann et al., 2010). Because TRN1 binds a C-terminal PY motif on FUS, it is not immediately obvious how phosphorylation of the N terminus of FUS would decrease TRN1 import. One possibility is that the negative charge produced after phosphorylation of the N-terminal domain of FUS may impair binding to TRN1, which is negatively charged at physiologic pH (isoelectric point 4.7; Chook and Blobel, 1999). Precedence for this mechanism was reported in a recent study, in which it was shown that arginine methylation of a site adjacent to the C-terminal PY motif impairs TRN1-mediated nuclear import of FUS (Dormann et al., 2012). While this manuscript was in preparation, a recent study suggests another possibility. Han et al. (2012) report that the N terminus of FUS contains a low-complexity domain that can aggregate into fibrils that mimic features of RNA granules. Intriguingly, they show that aggregation of the FUS low-complexity domain can be disrupted by DNA-PK-mediated phosphorylation. Although this result was generated in vitro using only the low-complexity fragment of FUS, one could extrapolate that a similar mechanism operates in cells. Experiments are under way to determine whether FUS phosphorylation disrupts its protein-binding partners or incorporation into RNA granules in cells.
One of the most interesting findings from our work is that DNA damage affects not only FUS, but the whole FET family, including TAF15 and EWS. Earlier work has implied that the FET proteins may play an important role in the response to, and repair of, DNA damage. For example, all FET proteins have homologous DNA-strand-pairing activity in vitro, which could functionally contribute to repair of DNA breaks (Guipaud et al., 2006). Furthermore, FUS knock-out mice have genomic instability, increased sensitivity to radiation, and evidence of impaired DNA repair (Hicks et al., 2000; Kuroda et al., 2000). FUS can also bind long-coding RNA to inhibit translation of cyclin D in response to DNA damage (Wang et al., 2008). Similar to FUS, EWS knock-out mice are hypersensitive to ionizing radiation and EWS has been linked directly to changes in alternative splicing after DNA damage (Li et al., 2007; Paronetto et al., 2011).
More recent studies have found direct evidence that FUS is involved in DNA damage repair. FUS facilitates DNA damage repair in neurons through interaction with histone deacetylase 1 (HDAC1) and recruitment to sites of double-strand DNA damage (Wang et al., 2013). FUS is also recruited to sites of oxidative DNA damage through the activity of poly (ADP-ribose) polymerase (Mastrocola et al., 2013; Rulten et al., 2014). Although these studies globally support our result that FUS, and the FET proteins, respond to DNA damage, they all report a rapid recruitment (seconds to minutes) of FUS to sites of DNA damage. This is in contrast to our study, in which we found phosphorylation and cytoplasmic relocalization of FUS after DNA damage 1–2 h after inducing DNA damage (Fig. 4A). There are a number of potential explanations for the different effects we observe. First, all three groups used laser-induced DNA damage, which can induce many types of DNA lesions, although single-strand DNA breaks are thought to predominate (Reynolds et al., 2013). In contrast, we used the compound, CLM, which is the most selective chemical inducer of DNA DSBs known (Elmroth et al., 2003; Muslimović et al., 2009). Therefore, it seems probable that FUS responds or is regulated differently depending on the DNA lesion. In support of this idea, Mastrocola et al. (2013) reported that FUS did not accumulate at foci induced by ionizing radiation or defined DSBs produced in a cell line using a 4-hydroxytamoxifen-inducible AsiSI restriction endonuclease system. Second, FUS may be initially recruited to sites of DNA damage, where it is phosphorylated by DNA-PK, leading to release from the DNA lesion and export to the cytoplasm. This is supported by the observation that GFP-FUS is rapidly recruited within seconds to DNA damage and slowly decreases over the course of 30 min (Mastrocola et al., 2013). The precise role of FUS, EWS, and TAF15 in DNA damage repair is uncertain, but it is clearly an important area of future research given their link to ALS and FTLD and related neurodegenerative diseases.
Finally, our discovery that all known FET proteins, along with TRN1, are phosphorylated and translocated to the cytoplasm after DNA damage has important implications for the pathologic mechanisms of FTLD-FUS. Detailed comparison of the neuropathology in ALS-FUS and FTLD-FUS suggests that these diseases have different pathogenic mechanisms (Mackenzie and Neumann, 2012; Neumann et al., 2012). Recently, it has been proposed that hypomethylation of arginine residues on FET proteins decreases their binding affinity to TRN1 and is responsible for their accumulation in FTLD-FUS, although it is not clear why or how FET proteins are hypomethylated (Dormann et al., 2012). However, it was recently reported that mutations in the genes encoding protein N-arginine methyltransferases (PRMTs), which mediate FUS methylation, are not found in FTLD-FUS cases, suggesting that PRMT mutations are not the cause of FTLD-FUS (Ravenscroft et al., 2013). Alternatively, a different pathogenic mechanism may be involved. Based on the data presented here, we propose that DNA damage serves as the initial pathogenic trigger that causes phosphorylation and cytoplasmic accumulation of the FET proteins and TRN1 observed in FTLD-FUS cases. In support of this hypothesis, we observed elevated levels of the DNA damage marker p-H2AX in multiple cases of FTLD-FUS (Fig. 8). Although we did not observe a distinct p-FUS species FTLD-FUS tissue extract, it does not rule out that it occurs. For example, p-FUS might be a transient intermediate or of low abundance in the diseased brain, as supported by our observation that FUS phosphorylation is transient after DNA damage despite sustained p-H2AX levels (Fig. 7). Further research is necessary to determine the possible role of FUS/FET phosphorylation in disease and to understand how it is involved in the normal biological function of these proteins. Finally, our data demonstrating that DNA damage could be the upstream trigger of the pathological changes in FTLD-FUS cases suggest that strategies to reduce DNA damage, or activate DNA repair pathways, may be a viable therapeutic route to prevent or treat neurodegeneration in FTLD-FUS.
Footnotes
This work was supported by the National Institutes of Health (Grants P30NS069289, P50AG032362, and R00AG032362 to T.K. and Training Grant T32 “Training and translational research in Neurology” 2T32 NS 007480 to Q.D. and C.J.H.) and the Alzheimer's Association (New Investigator Research Grant to T.K.). We thank Jonathan D. Glass and Deborah S. Cooper at the Emory ADRC/CND Brain and Tissue Bank for kindly providing samples; Marie Dutreix for graciously providing the siDNAs Dbait32H and Dbait8H; John Hedreen and Louis Fernandes of the Harvard Brain Tissue Resource Center, McLean Hospital, Belmont, MA (which is supported in part by National Institutes of Health Grant R24 MH06885) for tissue samples; and members of the Emory Center for Neurodegenerative Disease and the laboratories of Allan Levey and James Lah for helpful discussions.
The authors declare no competing financial interests.
References
- Allalunis-Turner MJ, Zia PK, Barron GM, Mirzayans R, Day RS., 3rd Radiation-induced DNA damage and repair in cells of a radiosensitive human malignant glioma cell line. Radiat Res. 1995;144:288–293. doi: 10.2307/3578948. [DOI] [PubMed] [Google Scholar]
- Belmokhtar CA, Hillion J, Ségal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, Sapp P, McKenna-Yasek D, Brown RH, Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy BR, Walker T, Rasquinha I, Hill IE, MacManus JP. Activation of DNA-dependent protein kinase may play a role in apoptosis of human neuroblastoma cells. J Neurochem. 1999;72:933–942. doi: 10.1046/j.1471-4159.1999.0720933.x. [DOI] [PubMed] [Google Scholar]
- Chen X, Chang J, Deng Q, Xu J, Nguyen TA, Martens LH, Cenik B, Taylor G, Hudson KF, Chung J, Yu K, Yu P, Herz J, Farese RV, Jr, Kukar T, Tansey MG. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J Neurosci. 2013;33:9202–9213. doi: 10.1523/JNEUROSCI.5336-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chook YM, Blobel G. Structure of the nuclear transport complex karyopherin-B2-Ran-GppNHp. Nature. 1999;399:230–237. doi: 10.1038/20375. [DOI] [PubMed] [Google Scholar]
- Croset A, Cordelières FP, Berthault N, Buhler C, Sun JS, Quanz M, Dutreix M. Inhibition of DNA damage repair by artificial activation of PARP with siDNA. Nucl Acids Res. 2013;41:7344–7355. doi: 10.1093/nar/gkt522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson YS, Robinson AC, Hu Q, Mishra M, Baborie A, Jaros E, Perry RH, Cairns NJ, Richardson A, Gerhard A, Neary D, Snowden JS, Bigio EH, Mann DM. Nuclear carrier and RNA-binding proteins in frontotemporal lobar degeneration associated with fused in sarcoma (FUS) pathological changes. Neuropathol Appl Neurobiol, 2012;39:157–165. doi: 10.1111/j.1365-2990.2012.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Kocerha J, Finch N, Crook R, Baker M, Desaro P, Johnston A, Rutherford N, Wojtas A, Kennelly K, Wszolek ZK, Graff-Radford N, Boylan K, Rademakers R. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010;31:E1377–E1389. doi: 10.1002/humu.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, Haass C. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–4275. doi: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmroth K, Nygren J, Mårtensson S, Ismail IH, Hammarsten O. Cleavage of cellular DNA by calicheamicin γ1. DNA Repair. 2003;2:363–374. doi: 10.1016/S1568-7864(02)00235-5. [DOI] [PubMed] [Google Scholar]
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. 2011;32:2323.e27–2323.e40. doi: 10.1016/j.neurobiolaging.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M, Toth R, Vandermoere F, Morrice NA, Rouse J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem J. 2008;415:297–307. doi: 10.1042/BJ20081135. [DOI] [PubMed] [Google Scholar]
- Guipaud O, Guillonneau F, Labas V, Praseuth D, Rossier J, Lopez B, Bertrand P. An in vitro enzymatic assay coupled to proteomics analysis reveals a new DNA processing activity for Ewing sarcoma and TAF(II)68 proteins. PROTEOMICS. 2006;6:5962–5972. doi: 10.1002/pmic.200600259. [DOI] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–779. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henner WD, Blazka ME. Hypersensitivity of cultured ataxia-telangiectasia cells to etoposide. J Natl Cancer Inst. 1986;76:1007–1011. [PubMed] [Google Scholar]
- Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, Van Kaer L, Ruley HE. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24:175–179. doi: 10.1038/72842. [DOI] [PubMed] [Google Scholar]
- Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle FE, Akiyama H, Hisanaga S, Hasegawa M. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582:2899–2904. doi: 10.1016/j.febslet.2008.07.027. [DOI] [PubMed] [Google Scholar]
- Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69:152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucl Acids Res. 2011;39:2781–2798. doi: 10.1093/nar/gkq1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, et al. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Robertson P, Pintchovski SA, Moore B, Bann MA, Ren Z, Jansen-West K, Malphrus K, Eggert S, Maruyama H, Cottrell BA, Das P, Basi GS, Koo EH, Golde TE. Lysine 624 of the amyloid precursor protein (APP) is a critical determinant of amyloid beta peptide length: support for a sequential model of gamma-secretase intramembrane proteolysis and regulation by the amyloid beta precursor protein (APP) juxtamembrane region. J Biol Chem. 2011;286:39804–39812. doi: 10.1074/jbc.M111.274696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, Ron D. Male sterility and enhanced radiation sensitivity in TLS-/- mice. EMBO J. 2000;19:453–462. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley T, Rohrer JD, Bandopadhyay R, Fry C, Ahmed Z, Isaacs AM, Brelstaff JH, Borroni B, Warren JD, Troakes C, King A, Al-Saraj S, Newcombe J, Quinn N, Ostergaard K, Schrøder HD, Bojsen-Møller M, Braendgaard H, Fox NC, Rossor MN, et al. A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain. 2011;134:2548–2564. doi: 10.1093/brain/awr160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS, 3rd, Barron GM, Allalunis-Turner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–1185. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- Lempiäinen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–3073. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Watford W, Li C, Parmelee A, Bryant MA, Deng C, O'Shea J, Lee SB. Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J Clin Invest. 2007;117:1314–1323. doi: 10.1172/JCI31222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Kretzschmar HA, Cairns NJ, Neumann M. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121:207–218. doi: 10.1007/s00401-010-0764-0. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012;1462:40–43. doi: 10.1016/j.brainres.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Mårtensson S, Nygren J, Osheroff N, Hammarsten O. Activation of the DNA-dependent protein kinase by drug-induced and radiation-induced DNA strand breaks. Radiat Res. 2003;160:291–301. doi: 10.1667/0033-7587(2003)160[0291:AOTDPK]2.0.CO%3B2. [DOI] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem. 2013;288:24731–24741. doi: 10.1074/jbc.M113.497974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Muslimović A, Nyström S, Gao Y, Hammarsten O. Numerical analysis of etoposide induced DNA breaks. PLoS One. 2009;4:e5859. doi: 10.1371/journal.pone.0005859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009a;118:605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009b;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009c;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, Roeber S, Kretzschmar HA, Munoz DG, Kusaka H, Yokota O, Ang LC, Bilbao J, Rademakers R, Haass C, Mackenzie IR. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134:2595–2609. doi: 10.1093/brain/awr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Valori CF, Ansorge O, Kretzschmar HA, Munoz DG, Kusaka H, Yokota O, Ishihara K, Ang LC, Bilbao JM, Mackenzie IR. Transportin 1 accumulates specifically with FET proteins but no other transportin cargos in FTLD-FUS and is absent in FUS inclusions in ALS with FUS mutations. Acta Neuropathol. 2012;124:705–716. doi: 10.1007/s00401-012-1020-6. [DOI] [PubMed] [Google Scholar]
- Oh SM, Liu Z, Okada M, Jang SW, Liu X, Chan CB, Luo H, Ye K. Ebp1 sumoylation, regulated by TLS/FUS E3 ligase, is required for its anti-proliferative activity. Oncogene. 2010;29:1017–1030. doi: 10.1038/onc.2009.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, Miñana B, Valcárcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell. 2011;43:353–368. doi: 10.1016/j.molcel.2011.05.035. [DOI] [PubMed] [Google Scholar]
- Pearlman SM, Serber Z, Ferrell JE., Jr A mechanism for the evolution of phosphorylation sites. Cell. 2011;147:934–946. doi: 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quanz M, Chassoux D, Berthault N, Agrario C, Sun JS, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One. 2009a;4:e6298. doi: 10.1371/journal.pone.0006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, Alberti C, Josserand V, Coll JL, Sastre-Garau X, Cosset JM, Larue L, Sun JS, Dutreix M. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin Cancer Res. 2009b;15:1308–1316. doi: 10.1158/1078-0432.CCR-08-2108. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft TA, Baker MC, Rutherford NJ, Neumann M, Mackenzie IR, Josephs KA, Boeve BF, Petersen R, Halliday GM, Kril J, van Swieten JC, Seeley WW, Dickson DW, Rademakers R. Mutations in protein N-arginine methyltransferases are not the cause of FTLD-FUS. Neurobiol Aging. 2013;34:2235.e11–2235.e13. doi: 10.1016/j.neurobiolaging.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds P, Botchway SW, Parker AW, O'Neill P. Spatiotemporal dynamics of DNA repair proteins following laser microbeam induced DNA damage–when is a DSB not a DSB? Mutat Res. 2013;756:14–20. doi: 10.1016/j.mrgentox.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos WP, Kaina B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gómez-Herreros F, Hafezparast M, Caldecott KW. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014;42:307–314. doi: 10.1093/nar/gkt835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. In: Bjergbæk L, editor. DNA repair protocols. New York: Humana; 2012. pp. 613–626. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 Mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers KC, Shen F, Sierra Potchanant EA, Phipps EA, Hickey RJ, Malkas LH. Phosphorylation: the molecular switch of double-strand break repair. Int J Proteomics. 2011;2011:373816. doi: 10.1155/2011/373816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J Biol Chem. 2010;285:4909–4919. doi: 10.1074/jbc.M109.042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeeck C, Deng Q, Dejesus-Hernandez M, Taylor G, Ceballos-Diaz C, Kocerha J, Golde T, Das P, Rademakers R, Dickson DW, Kukar T. Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol Neurodegener. 2012;7:53. doi: 10.1186/1750-1326-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, Chu CH, Huang HD, Ko MT, Hwang JK. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucl Acids Res. 2007;35:W588–W594. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular and Cellular Proteomics. 2008;7:1598–1608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110:1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]